


Abstract
As key epigenetic regulators, polycomb group (PcG) proteins are responsible for the control of cell proliferation and differentiation as well as stem cell pluripotency and self-renewal. Aberrant epigenetic modification by PcG is strongly correlated with the severity and invasiveness of many types of cancers. Unfortunately, the molecular mechanism of PcG-mediated epigenetic regulation remained elusive, partly due to the extremely limited pool of experimentally confirmed PcG target genes. In order to facilitate experimental identification of PcG target genes, here we propose a novel computational method, EpiPredictor, that achieved significantly higher matching ratios with several recent chromatin immunoprecipitation studies than jPREdictor, an existing computational method. We further validated a subset of genes that were uniquely predicted by EpiPredictor by cross-referencing existing literature and by experimental means. Our data suggest that multiple transcription factor networking at the cis-regulatory elements is critical for PcG recruitment, while high GC content and high conservation level are also important features of PcG target genes. EpiPredictor should substantially expedite experimental discovery of PcG target genes by providing an effective initial screening tool. From a computational standpoint, our strategy of modelling transcription factor interaction with a non-linear kernel is original, effective and transferable to many other applications.
INTRODUCTION
Originally discovered in Drosophila as the regulators of homeotic (HOX) genes, polycomb group (PcG) proteins are well-conserved epigenetic modifiers that repress the expression of thousands of target genes in a given genome (1–12). These target genes are essential for many fundamental, evolutionarily conserved processes including development, cell-fate determination, proliferation, stem-cell pluripotency and self-renewal (1,4,7,8,13–16). Mutations of PcG proteins are implicated in defects in stem-cell fates and their abnormal levels exhibit a striking correlation with the severity and invasiveness of a number of cancer types including prostate cancer and breast cancer (1,4,7,8,13–16).
PcG proteins impose gene silencing through their interactions with polycomb response elements (PREs) that are present on the promoter regions of polycomb target genes (31). This interaction is mediated by three types of multiprotein complexes, polycomb repressive complex 1 and 2 (PRC1 and PRC2) and a recently discovered PhoRC that contains the DNA-binding protein Pleiohomeotic (Pho) or Pleiohomeotic-like (PhoL) (17) in Drosophila and Ying and Yang 1 and 2 (YY1 and YY2) in mammals (18–20). The known members of Drosophila PRC1 include Polycomb (PC), Polyhomeotic (PH), Posterior sex combs (PSC) and dRing, whereas Drosophila PRC2 contains at least three core components: Enhancer of zeste (E(z)), Extra sex comb (Esc) and Suppressor of zeste 12 (Su(z)12) (18). Since none of these PRC1 and PRC2 proteins can bind to DNA directly, a hierarchical recruitment model has been proposed stating that DNA-binding transcription factors including Pho and PhoL first bind to PREs on the target genes and recruit the PRC2 complex to trimethylate the lysine 27 residue of histone H3 (H3K27me3) that is later bound by the PRC1 complex for maintenance (21). Besides Pho and PhoL, the best studied Drosophila transcription factors contributing to PRC2 recruitment include GAGA factor (GAF)/Pipsqueak (PSQ) (22,24,26), Zeste (25), Dorsal switch protein (DSP) (23,28), Grainyhead (Grh) (28) and Sp1/KLF (29), (reviewed in 30). In addition, several Drosophila PREs have been identified through both computational and experimental analyses (31–40). More recently, the first two mammalian genomic regions have been discovered to confer PcG responsiveness, one in the human HOXD cluster (41) and the other in the regulatory region of the mouse MafB gene (9).
Recent advances in high-throughput techniques such as chromatin immunoprecipitation in conjunction with microarray (ChIP-on-chip), DNA adenine methyltransferase identification (DamID) and ChIP-sequencing (ChIP-seq), have greatly enriched our knowledge on the scale of genes regulated by PcG (1,4–8,10,11,13–16,42–49). However, the rather low overlaps of target genes identified in separate ChIP studies, at ∼30% for three ChIP studies on Drosophila melanogaster (4,14,15), stress the need for additional experimental and computational verifications of individual PcG target genes. Ideally, a powerful computational method that is able to predict/screen, with a reasonable accuracy, PcG target genes in a given genome would drastically expedite experimental verification of these genes.
In the literature, there are considerable efforts in developing computational methods to predict PRE sequences and to locate the genes regulated by PcG based upon their adjacency to PREs. For instance, Ringrose et al. investigated the combinatorial pattern of transcription factors known to be involved in PcG recruitment and assigned to each genomic region of interest a score equalling the weighted sum of the occurrence of every possible transcription factor pairs (40). Fiedler and Rehmsmeier extended this idea and developed jPREdictor for PRE prediction (50). Hauenschild and colleagues used the latest version of jPREdictor to perform a genome-wide prediction on D. melanogaster and predicted 201 PREs together with 243 associated genes (51). They also incorporated the aspect of comparative genomics and expanded their prediction to 285 PREs with 322 associated genes. More recently, Liu et al. integrated data from a ChIP study and transcription factor binding analysis to predict a set of PcG target genes in mouse embryonic stem cells (52). Despite these efforts, however, due to the plasticity of PRE sequences, developing a reliable computational PRE predictor remains a difficult task. For example, the overlaps between the top target genes predicted by jPREdictor and those shown in the three recent ChIP studies in D. melanogaster (4,14,15) are strikingly low (at ∼8–20%).
We have addressed this challenge by developing a novel computational approach, EpiPredictor, to predict PcG target genes via the identification of PRE sites. With the incorporation of novel features including the use of a support vector machine (SVM)-based classifier, global sequence information, conservation analysis and comparative genomics, our approach was able to predict PcG target genes in the D. melanogaster genome with substantially improved accuracy. Most of the predicted PcG target genes are transcription factors involved in key biological processes such as development, neurogenesis and cell fate determination. Our results suggest that multiple transcription factor networking at the cis-regulatory elements is critical for PcG recruitment, and high GC content and high conservation level are also important features of PcG target genes.
MATERIALS AND METHODS
Selection of motifs
In Drosophila, several transcription factors responsible for PcG recruitment have been identified, which, together with the consensus sequences of their DNA binding sites, are collectively referred to as motifs hereafter. We used seven motifs corresponding to four transcription factors, GAF (G, G10), Pho (PS, PM, PF), engrailed-1 (EN1) and Zeste (Z), all of which are known to be instrumental for PcG recruitment (Supplementary Table S1). The same motif set was also used in jPREdictor (40,51). Though a few other transcription factors, e.g. DSP, Grh, Sp1/KLF, are also implicated in PcG recruitment in some studies, we did not include them in our current system because doing so did not lead to any performance improvement (data not shown) and also may not allow a fair comparison with jPREdictor.
Construction of the validation sets
In order to validate our prediction of PcG target genes in an objective way, we used the gene lists reported in three recent ChIP studies in D. melanogaster, where Schwartz et al. (4) used ChIP-on-chip technique on S2 cultured cell line with antibodies to PC, E(z), PSC and H3K27me3; Tolhuis et al. (15) used DamID approach on Kc cells to identify binding sites of PC, Esc, Sex combs extra (Sce) and H3K27me3; while Schuettengruber et al. (14) applied ChIP-on-chip on Drosophila embryos and employed antibodies to PC, PH and H3K27me3. Different choices of cell lines and antibodies all had an impact on the results of these experiments that differed from one another at varying degrees. Since our in silico PRE prediction is independent of any experimental conditions, we expected that a comparison of our results with these three well-annotated studies, which as a whole investigated a range of antibodies and cell types, would provide a comprehensive evaluation of our system. To ensure that the validation gene lists to be used were as reliable and up-to-date as possible, we performed a post-processing procedure on the published data using the following stringent selection criteria. For all three validation sets, we used the gene lists published by the authors as input and removed duplicates if there was any. We also eliminated the genes that were withdrawn in the newer release of the gene annotation to ensure that the validation gene sets are up-to-date. In particular, in processing Schwartz’s data (4), we only selected the target genes with strong PcG binding signals to all of the four PcG proteins [PC, PSC, PH and Su(z)12] simultaneously as defined by the authors. As a result, we obtained three lists consisting of 176 (Schwartz), 225 (Tolhuis) and 215 (Schuettengruber) predicted PcG target genes, respectively (Supplementary Table S2). Among them, 38 genes appeared in all of the three validation sets, denoted as Intersection, making the degree of overlap in the range of ∼17–22%.
Construction of the training set
Our PRE classifier is a supervised learner. Therefore we needed to provide it with a training set of good quality. This consisted of two steps: (i) construction of a PRE/non-PRE sequence collection and (ii) construction of the training set containing examples of both PRE sites (positive) and non-PRE sites (negative).
First, we constructed a sequence collection containing 12 known PRE sequences and 23 control (non-PRE) sequences. Among them, the 12 PRE sequences and 16 control sequences were the same as those used by Ringrose and colleagues (40,51). The 12 PRE sequences had solid evidence to support the existence of PRE site(s) within, whereas the 16 control sequences included promoters of genes regulated by GAF and Zeste but not by PcG proteins (40). To reflect the most recent progress in the field, we followed the same methodology used by Ringrose (40) and collected seven extra control sequences (Supplementary Table S3) for our training set that were bound by GAF, Pho and Zeste but did not have any enrichment for PC, PH or H3K27me3 in a genome-wide ChIP study (14). They were obtained by examining whether a given locus bound by GAF, Pho and Zeste was in the proximal promoter region of any gene, i.e. −1000 to +1000 base pairs (bps) with respect to the gene’s transcription start site (TSS). If so, we retained the locus and the gene, otherwise, we discarded them. To ensure that our control sequences did interact with GAF, we consulted another list of GAF target genes by an independent study (53). If the genes associated with any retained loci under investigation were not included in the second study, the loci were eliminated from our list. It was evident that, despite the addition of seven new control sequences in our study, the size of the sequence collection remained rather small.
A PRE sequence containing PRE site(s) is much larger than an actual PRE site. Due to the limited resolution of the experimental verification process, most known PRE sequences included in our sequence collection spanned thousands of bps long whereas the core-PRE sites are usually much shorter (<200 bps) (37). In other words, in addition to core PRE sites, a known PRE sequence might also contain non-PRE sites. Thus it was prudent to identify the loci that were most likely the bona fide PRE sites. For this purpose, we scanned each PRE/non-PRE sequence in our collection with a sliding window of 200 bps that incrementally moved downstream with a constant step of 20 bps. For each PRE sequence, we chose the window(s) with the highest sum of motif occurrence (calculated by the Motif Analyzer in the following section) as PRE sites. For every control (non-PRE) sequence, all the windows from scanning a control sequence were kept to ensure that the classifier was to be trained under very stringent condition.
Our new system EpiPredictor
Our system consisted of six primary components including Motif Analyzer, PRE Classifier, GC Analyzer, PRE-to-gene Mapper, Conservation Level Analyzer and Comparative Genomics Analyzer (Figure 1A). With the exception of PRE-to-gene Mapper, which was a utility module, each component rendered a unique perspective of investigating the genomic sequence or gene of interest. The first three units were centred around the prediction of PRE sites (Figure 1A and B), whereas the last three were focused on analyses at the gene level (Figure 1A).
Figure 1.

Our EpiPredictor system. (A) Architecture of EpiPredictor. The modules of Motif Analyzer, PRE Classifier and GC Analyzer are dedicated to the prediction of PRE sites and those of PRE-to-gene Mapper, Conservation Level Analyzer and Comparative Genomics Analyzer are focused on the prediction of PcG target genes. (B) Flowchart of the PRE site prediction modules of EpiPredictor.
Prediction of PRE sites
Motif Analyzer
We employed a sliding window of 200 bps and a step size of 20 bps to scan the whole genome where the DNA sequence overlapping with the window was captured and analyzed at any given time by the Motif Analyzer. Using a set of n motifs of transcription factors that were known to be involved in PcG recruitment, denoted by M1, M2, …, Mn, the Motif Analyzer constructed a profile for each window sequence/locus (denoted by Si) and represented it by a feature vector Fi = (fi1, fi2, …, fin), where fij denoted the occurrence frequency of motif Mj in sequence Si. This feature vector was then analyzed by the pre-trained PRE classifier (below) that predicted whether the test window/locus was a PRE or not (Figure 1B).
SVM-based PRE Classifier
Ringrose et al. (40) examined the occurrence of paired motifs at the putative PRE sites and observed that the weighted sum of the occurrence frequencies of all possible motif pairs were far more effective than a linear sum up of the occurrence frequency of single motifs. This suggested that the pattern of transcription factor interactions at the PRE sites be combinatorial. In order to abstractly model the multifaceted interactions among transcription factors at PRE sites, we incorporated an SVM-based PRE classifier, which is a powerful supervised learning method for handling classification tasks. SVM has achieved prominent success in a spectrum of biological applications including gene selection (54,55), protein classification (56–58), cancer tissue characterization (59,60), outperforming many other classic machine learning techniques such as neural network, decision tree, k-nearest neighbour (61,62).
There are four basic kernel functions in SVM, including linear, polynomial, radial basis function (RBF) and sigmoid. Given the context of PRE prediction, we provided a further annotation to SVM coupled with some of these kernels. For instance, in the case of a polynomial kernel, the parameter d corresponded to the degree of motif combinations, e.g. when d = 1 (equivalent to a linear kernel), only single motif occurrence was noted; when d = 2 (quadratic kernel), the occurrence of motif pairs was considered; whereas when d = 3 (cubic kernel), the occurrence of motif triplets was analyzed. In the case of the RBF kernel, the data was mapped to an infinite dimensional Hilbert space where intuitively speaking, all the motifs were mapped to a circle/hypersphere. Taken together, we expected the polynomial (d > 1) and RBF kernels to be best for modelling transcription factor interaction at the PRE sites. While the windows/loci classified to be non-PREs were discarded, those classified to be PREs had to undergo further scrutiny by GC Analyzer (below).
GC Analyzer
Previous studies indicated that native DNA sequence features, such as GC content, are associated with epigenetic modification activities such as DNA methylation and PcG binding (63–65). In particular, the work of Ku et al. (5) suggested that CpG islands influence recruitment of PcG. Furthermore, GC-rich sequence elements have been shown to recruit PRC2 in mammalian embryonic stem cells (66) and high-CpG-density promoters are associated with highly regulated key developmental genes and are enriched with the H3K27me3 marks (67). Therefore, we implemented a GC Analyzer to further scrutinize the output from the PRE Classifier. For a sequence window/locus Si that was positively predicted by the PRE classifier, our GC Analyzer compared its GC ratio Ri with a threshold value RT and discarded Si if Ri < RT (Figure 1B). To decide on an appropriate threshold for a region of 200-bps window size that we used, we examined six experimentally verified PRE sequences where short core PRE segments were identified (37). The lowest GC ratio of these core PRE segments was 44%. We then chose this lower bound of the GC ratio as our threshold in order to ensure that all the verified PRE segments satisfy this GC ratio cut-off so that they can pass the GC Analyzer’s scrutiny. We also compared the cut-off values of 44, 42 or 40% on EpiPredictor, and found 44% yielded the best performance. Therefore, we used the 44% threshold in our subsequent analysis. Only the ones that passed the GC content test were considered as the potential PRE loci. Each locus was given a numerical score SCi by the Motif Analyzer that equalled the sum of motif occurrence in the sequence Si, i.e. SCi = 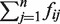 (Figure 1B).
(Figure 1B).
Uncertainty measurement
To characterize the probability of a predicted PRE site being the real PRE site, we performed a non-parametric analysis on 100 randomly generated genomes whose size and nucleotide distribution (A: 29%, C: 21%, T: 29%, G: 21%) are the same as the D. melanogaster genome. We used our software to predict PRE sites on these random genomes and for a given score s we counted Os that denoted the occurrence of a score that is higher or equal to s. We then calculated Es, i.e. the E-value of score s by Os/100 and the corresponding P-value would be Es/100.
Genome-wide prediction of PcG target genes
PRE-to-gene Mapper
From our genome-wide PRE site prediction results, the PRE-to-gene Mapper first mapped all of the predicted PRE sites to their genomic coordinates on the genome. When several windows/loci adjacent to each other were all predicted to be PRE sites, they were all combined into a longer PRE. The Mapper then analyzed every locus that had a positive PRE score S, located its most adjacent gene G and credited G a score equalling S. If the locus was positioned closely in between two genes and if the second closest gene G2 was within 4000 bps away, the Mapper granted G2 a score equalling S as well.
Conservation Level Analyzer
Due to their roles in regulating key developmental processes, PcG target genes were expected to be evolutionarily conserved. The Conservation Level Analyzer considered six Drosophila genomes that are close to D. melanogaster according to the phylogenetic tree (68), including D. simulans, D. sechellia, D. yakuba, D. erecta, D. pseudoobscura and D. ananassae. For each annotated D. melanogaster gene, it queried the Flybase database (www.flybase.org) to locate its orthologues in any of the six related genomes. If a gene failed to have any orthologue, it was eliminated from the eligible gene list. That is, the Analyzer excluded the genes that did not have any orthologue in any related species from the pool of candidate genes to be considered as PcG targets. Eventually all the remaining genes were ranked according to the genes’ associated PRE scores. The version of our EpiPredictor up to this point was termed as EpiPredictor-Basic.
Comparative Genomics Analyzer
We investigated the value of incorporating comparative genomics (69–71) into PcG target gene prediction. For this, we constructed a variant version of EpiPredictor, hereafter referred to as EpiPredictor-CG, which integrated analyses on three well-annotated Drosophila organisms (D. simulans, D. yakuba and D. pseudoobscura) that are close to D. melanogaster in the phylogenetic tree (68).
Our tactic in implementing EpiPredictor-CG was to construct an ensemble system that employed the top-ranked genes provided by our original EpiPredictor-Basic as the base set and incorporated the information obtained from our comparative genomics study for rank adjustment when necessary. To be more specific, if our ultimate goal was to retrieve N genes that were most likely the PcG targets, we started our process with a gene list containing the top M genes ranked by EpiPredictor-Basic (M = 1.5N) and reordered the genes based upon the scores of the candidate genes’ orthologues in different Drosophila species.
In order to achieve this, we applied EpiPredictor-Basic onto each of these three Drosophila genomes. For each genome, all annotated genes were evaluated and ranked according to their predicted PRE scores. If a gene is orthologous to a D. melanogaster gene, the rank of that gene was linked to its D. melanogaster gene orthologue. Therefore, for any D. melanogaster gene included in the top list, up to four ranks could be obtained, each representing the rank of the gene (or its orthologue) in the respective species, i.e. D. melanogaster, D. simulans, D. yakuba and D. pseudoobscura. A final rank was calculated by averaging all the ranks. The gene list was then re-sorted accordingly.
BART-based PRE classifier
BART (Bayesian Additive Regression Trees) is a nonparametric regression method that can also be used as a binary classifier. As a comparison to the SVM-based PRE Classifier in our system, we used BART as an alternative classifier to evaluate whether a given locus is actually the PRE site. This was achieved by using the R package (BayesTree) by Hugh Chipman and Robert McCulloch.
Computational complexity
The primary computational complexity of our EpiPredictor model came from the component of the SVM-based PRE classifier. During the training phase, the complexity of the SVM was O(Ns3 + (Ns2)l + NsdLl) where Ns denoted the number of support vectors, l denoted the number of training points and dL denoted the dimension of input data. During the testing phase, the complexity of the SVM was O(MNs) where M was O(dL). In our experiments, Ns = 21.
On a regular Dell desktop (Intel Duo CPU 3.00 GHz, 1 G memory), our system spent 63 ms in training. During the prediction phase, it took about 30 min to process the entire D. melanogaster genome of 137 million bps and used around 5 MB memory. Due to the integration of SVM, it was necessary to store a substantial amount of feature vectors onto a text file. This Input/Output process was responsible for the majority of the execution time.
Importantly, our system is an automated program in which the components such as Motif Analyzer, SVM-based PRE Classifier and GC Analyzer were run sequentially requiring no human intervention after the genome sequence under study is input, and is readily scalable. For example, we used our software to predict the PcG target genes on the entire human genome and obtained complete results within three and a half hours on the same PC.
In marked contrast, when we used BART as an alternative to SVM to classify PRE, we noticed that BART required substantial computational resources. It was impossible to complete the prediction of D. melanogaster genome on the same PC. On an Intel Xeon computer cluster which contains 134 SunFire x4150 nodes from Sun Microsystems, the computation took about 33 h to complete. The average usage of memory was 16 GB.
Immunoprecipitation of crosslinked chromatin from D. melanogaster S2 cells
Drosophila melanogaster Schneider S2 cells were cultured in 1× Schneider’s medium (Invitrogen) supplemented with 20% fetal bovine serum, 100 U/ml Penicillin and 100 µg/ml Streptomycin at room temperature. Cells were passaged at 1:4 ratio every two days to keep logarithmic growth. Crosslinking, immunoprecipitation with anti-E(z) antibody and quantitative PCR (qPCR) were done as described previously (72). In brief, 5 µg anti-E(z) (Santa Cruz Biotechnology, Inc) or anti-FLAG mock antibody (Sigma) were added to 4 × 108 crosslinked S2 cells to immunoprecipitate protein/DNA complexes. The antibody–protein/DNA complexes were then purified using 50 µl protein A Sephorose 4 Fast Flow Beads (GE Healthcare). DNA was extracted from the purified antibody-protein/DNA complexes by phenol–chlorophorm extraction. Purified DNA was subjected to qPCR using primer pairs designed to amplify DNA of ∼250 bps using a SYBER green detection mix (Applied Biosystems). All experiments were carried out in triplicates.
RESULTS
Empirical analysis of the SVM-based PRE classifier
In order to identify the most appropriate kernel for the SVM-based PRE classifier, we performed an empirical analysis on the training set to gauge how well a certain kernel distinguished known PRE sequences. This is done using three runs of 10-fold cross validation so as to avoid any potential over-fitting problem. With the default parameters provided by LibSVM (73), the performance of all four basic kernel methods was analyzed by sensitivity and specificity (Table 1). As we expected, the non-linear kernels such as polynomial worked very well in distinguishing PRE sequences from control sequences, further confirming the advantage of modelling the motif interaction in a combinatorial manner. Among them, the polynomial (d = 2 and d = 3) kernels (also called the quadratic kernel and cubic kernel, respectively) achieved the best results in terms of specificity and sensitivity when both the average and standard deviation are taken into account, implicating that at the PRE sites, multiple transcription factors interact with each other that as a whole serves as the platform for PcG recruitment. Although the cubic kernel did not significantly outperform the quadratic kernel, it is still the best model given all the parameters considered. Therefore, we used the cubic kernel on the SVM throughout our analyses.
Table 1.
SVM kernel evaluation
| Metric | Kernel |
||||
|---|---|---|---|---|---|
| Linear | Polynomial (d = 2) | Polynomial (d = 3) | RBF | Sigmoid | |
| Sensitivity | 0.80 ± 0.05 | 0.80 ± 0.05 | 0.82 ± 0.03 | 0.60 ± 0.05 | 0.00 ± 0.00 |
| Specificity | 0.91 ± 0.01 | 0.96 ± 0.01 | 0.96 ± 0.01 | 0.99 ± 0.02 | 0.84 ± 0.03 |
Sensitivity = TP/(TP + FN); Specificity = TN/(TN + FP),
where TP, TN, FP, FN correspond to true positive, true negative, false positive and false negative, respectively. We performed three independent runs of 10-fold cross validation on the training collection and reported the average sensitivity/specificity and the standard deviation. The kernel with the best performance in both sensitivity and specificity is highlighted in bold. This is also the kernel we used throughout our analyses.
Test of the training set
To compare our new sequence collection with the original one used by Ringrose and colleagues, we ran independently EpiPredictor-Basic using the modules (a, b, c) on both training sets and the jPREdictor (static) on our new training set (Supplementary Table S4). Also included in Supplementary Table S4 is the result of jPREdictor (static) with Ringrose’s training set as originally reported (51). We found virtually no difference in performance when using different training sets. Therefore, we elected to use our training set throughout the analyses.
Performance evaluation of EpiPredictor components
We tested our classifier on the D. melanogaster genome that contains roughly 137 million bps and 13 000 genes. Each chromosome was scanned with a sliding window of 200 bps and a step size of 20 bps (parameters determined by empirical analysis), and each window was analyzed by the Motif Analyzer component and represented by a seven-dimensional feature vector (each corresponding to one of the seven motifs we used). The performance of the system was evaluated by the matching ratios between our top predicted genes and those of the three validation sets derived from ChIP studies (4,14,15) together with their intersection set (Intersection) (Table 2). In order to examine whether the performance of our system is sensitive to different window size and step sizes, we also varied the values of these parameters (Supplementary Table S5). It is clear that with different window and step sizes, the performance of our system did vary slightly but the change was not very substantial. Overall, the parameter setting of window size = 200, step size = 20 produced the best results. Therefore, we used the window size of 200 bps and step size of 20 bps throughout.
Table 2.
Evaluation of the performance of individual EpiPredictor components against three genome-wide ChIP studies in D. melanogaster and their intersection
| Number of top genes | EpiPredictor Components | Schwartz et al.a | Tolhuis et al.b | Schuettengruber et al.c | Intersectiond |
|---|---|---|---|---|---|
| 243 e | (a) | 14.20%f | 5.33% | 12.09% | 2.63% |
| (a,b) | 22.73% | 9.78% | 19.53% | 23.68% | |
| (a,b,c) | 26.14% | 10.22% | 25.12% | 23.68% | |
| (a,b,c,d)g | 27.27% | 10.67% | 26.05% | 26.32% | |
| 322 h | (a,b,c,d) | 32.39% | 14.22% | 30.70% | 34.21% |
| (a,b,c,d,e)i | 35.80% | 15.11% | 33.02% | 44.74% |
(a): Motif Analyzer; (b): SVM Classifier; (c): GC Analyzer; (d): Conservation Level Analyzer; (e): Comparative Genomics Analyzer.
aOverlap with the genes predicted by Schwartz et al. (4).
bOverlap with the genes predicted by Tolhuis et al. (15).
cOverlap with the genes predicted by Schuettengruber et al. (14).
dOverlap with the genes intersected by Schwartz et al., Tolhuis et al., and Schuettengruber et al.
eThe number of top genes retrieved from EpiPredictor-Basic analysis.
fSuppose the validation set includes V genes. Among the top N genes predicted by our system, W genes matched the validation set, the overlap was represented as W/V.
gThe EpiPredictor-Basic module.
hThe number of top genes retrieved from EpiPredictor-CG analysis.
iThe EpiPredictor-CG module. The results corresponding to the EpiPredictor-Basic and EpiPredictor-CG models are highlighted in bold.
Our system contains multiple components. The effect of each component was evaluated by sequentially adding each component onto the Baseline system that used only the Motif Analyzer.
Baseline system
To thoroughly evaluate the merit of each component of EpiPredictor, we constructed a baseline system that did not incorporate SVM or any other subsequent component but instead only used the Motif Analyzer that calculated the sum of the motif occurrence frequency. The baseline system achieved a moderate performance, having the matching ratios of 14.20, 5.33, 12.09, 2.63%, with the three validation sets and their intersection, respectively (Table 2). It is noteworthy that to perform a fair comparison with jPREdictor that reported their top 243 genes, we also retrieved the top 243 genes from our system to obtain the aforementioned results.
SVM-based PRE Classifier
In order to estimate the merit of SVM, we then integrated SVM into the baseline system. The application of SVM drastically enhanced the performance of our system when compared to the baseline system, with matching ratios of 22.73, 9.78, 19.53, 23.68%, respectively (Table 2).
GC Analyzer
Subsequently we incorporated the GC Analyzer into our program. The prediction performance of EpiPredictor was further improved to 26.14, 10.22, 25.12, 23.68%, respectively (Table 2), demonstrating that the bona fide PcG sites tend to have relatively high GC content.
Uncertainty measurement
The non-parametric tests conducted on 100 random genomes indicated that a PRE score of 12.7 corresponded to a P-value of 0.01. In our prediction, the top 190 predicted PRE sites had a PRE score of higher than 12.7, with the highest score being 39.2. We also corrected the issue of multiple comparisons using Bonferroni correction, and found that a PRE score of 17.3 corresponded to a P-value of 0.0001 (0.01/100). In our prediction, the top 73 predicted PRE sites had a PRE score of 17.3 or higher. Thus these top 73 predicted PRE sites are regarded as predictions with significant confidence, even under such a stringent condition.
Conservation Level Analyzer
The integration of the Conservation Level Analyzer slightly enhanced our system’s performance to 27.27, 10.67, 26.05, 26.32%, respectively (Table 2). At this point, we completed the construction of the basic version of our system, EpiPredictor-Basic. A complete list of the top genes thereby generated is provided in Supplementary Table S6.
It is worth mentioning that an attempt of using the base-by-base evolutionary conservation score compiled on D. melanogaster genome in comparison to 14 insects (71) failed to produce any improvement in the prediction performance (data not shown). Taken together, this suggested that the bona fide PcG target genes be most likely evolutionarily conserved; however, their positions might be more flexible in the course of evolution.
Comparative Genomics Analyzer
In order to evaluate the performance of EpiPredictor-CG, which integrated the Comparative Genomics Analyzer, we retrieved the top 322 predicted genes, which was the same number as generated by our counterpart jPREdictor (dynamic) (Supplementary Tables S7 and S8). Due to the integration of comparative genomics, some of the genes with lower scores were boosted up into the top list (Supplementary Table S9) and yielded an improved performance of 35.80, 15.11, 33.02, 44.74%, respectively (Table 2), in comparison to the performance of EpiPredictor-Basic in predicting 322 genes: 32.39, 14.22, 30.70, 34.21% (Table 2).
It is worth noting that the Intersection set obtained by intersecting all the three validation sets derived from ChIP studies (4,14,15) did have very high matching ratio with our EpiPredictor-CG prediction (Table 2), consistent with the expectation that it is the highest confidence set of the target genes.
Performance comparison between SVM-based and BART-based PRE classifier
Besides SVM, several other statistical models including BART (74) are also able to capture nonlinear interactions among the sequence features. For instance, Liu et al. (52) used BART to predict polycomb target genes with a good performance. Therefore we compared our system’s performance using SVM-based or BART-based PRE classifier (Table 3). It is clear that the SVM-based classifier consistently outperformed the BART-based counterpart.
Table 3.
Evaluation of the performance of our system using SVM-based PRE classifier vs BART-based PRE classifier
| Method | EpiPredictor components | Schwartz et al.a | Tolhuis et al.b | Schuettgurber et al.c | Intersectiond |
|---|---|---|---|---|---|
| SVM | (a, b) | 22.73% | 9.78% | 19.53% | 23.68% |
| (a, b, c) | 26.14% | 10.22% | 25.12% | 23.68% | |
| BART | (a, d) | 21.59% | 8.44% | 19.07% | 21.05% |
| (a, d, c) | 22.73% | 9.33% | 22.79% | 21.05% |
(a): Motif Analyzer; (b): SVM-based Classifier; (c): GC Analyzer; (d): BART-based Classifier.
aOverlap between the top 243 predicted genes with the genes predicted by Schwartz et al. (4).
bOverlap between the top 243 predicted genes with the genes predicted by Tolhuis et al. (15).
cOverlap between the top 243 predicted genes with the genes predicted by Schuettengruber et al. (14).
dOverlap between the top 243 predicted genes with the genes intersected by Schwartz et al., Tolhuis et al. and Schuettengruber et al. The results of the SVM-based classifier are highlighted in bold.
Comparative analysis of EpiPredictor and jPREdictor
We conducted a comparative analysis of EpiPredictor and jPREdictor (Table 4) by using the matching ratios as well as the receiving operating characteristics (ROC) curve as our evaluation metrics. The former metric indicates the overall accuracy of prediction while the latter one depicts the trade-off between sensitivity and specificity, which focuses on evaluating the ranking scheme. In terms of the matching ratio, EpiPredictor-Basic outperformed jPREdictor (static) by 6.25, 2.67, 6.05, 5.27%, respectively, against the three validation sets and their intersection set and the improvement is statistically significant (P < 0.05 in one-tailed Students’ t-test). In addition, EpiPredictor-CG surpassed the performance of jPREdictor (dynamic) by 7.96, 2.67, 10.23, 18.42%, respectively (P < 0.05). In terms of the area under curve (AUC) of ROC curve, EpiPredictor-Basic achieved comparable results with jPREdictor (static), whereas EpiPredictor-CG outperformed jPREdictor (dynamic) in three out of the four cases (Figure 2). It is worth noting that the AUCs of EpiPredictor-Basic, EpiPredictor-CG and jPREdictor (static) were all significantly larger than 0.5 (random guess) (P < 0.05) but it was not the case for jPREdictor (dynamic). Furthermore, using the AUCs as a measure, neither EpiPredictor nor jPREdictor’s advanced version significantly outperformed their basic counterpart.
Table 4.
Comparison of the overlaps between the PRE genes predicted by EpiPredictor and jPREdictor and three genome-wide ChIP studies in D. melanogaster and their intersection
| Scheme | Approach | Schwartz et al.a | Tolhuis .et al.b | Schuettengruber et al.c | Intersectiond |
|---|---|---|---|---|---|
| Original (243 genes) | EpiPredictor-Basic f | 27.27% | 10.67% | 26.05% | 26.32% |
| jPREdictor (static)e | 21.02% | 8.00% | 20.00% | 21.05% | |
| Comparative genomics (322 genes) | EpiPredictor-CG f | 35.80% | 15.11% | 33.02% | 44.74% |
| jPREdictor (dynamic)e | 27.84% | 12.44% | 22.79% | 26.32% |
aOverlap with the genes detected by Schwartz et al. (4).
bOverlap with the genes detected by Tolhuis et al. (15).
cOverlap with the genes detected by Schuettengruber et al. (14).
dOverlap with the genes intersected by Schwartz et al., Tolhuis et al. Schuettengruber et al.
eData reported in the original publication (51).
fThe results of EpiPredictor-Basic and EpiPredictor-CG are highlighted in bold.
Figure 2.

ROC curves of the PRE genes predicted by EpiPredictor and jPREdictor. Shown are overlaps with the genes predicted by Schwartz et al. (A), Tolhuis et al. (B), Schuettengruber et al. (C) and the genes intersected by all three sets (D). The AUCs on the four validation sets are 0.61, 0.61, 0.58 and 0.60, respectively, for EpiPredictor-Basic, 0.62, 0.57, 0.62 and 0.53, respectively, for EpiPredictor-CG, 0.64, 0.56, 0.59 and 0.67 for jPREdictor (static), 0.56, 0.49, 0.55 and 0.59 for jPREdictor (dynamic).
Annotation of EpiPredictor prediction
To reveal the major function enrichment of the genes predicted by our system and jPREdictor, we used the DAVID bioinformatics tool (75) to perform a gene ontology analysis on the genes uniquely predicted by either EpiPredictor-CG or jPREdictor (dynamic), as well as those predicted by both EpiPredictor-CG and jPREdictor (dynamic) (Figure 3). Most of the highly represented gene functions were related to the regulation of transcription, development, pattern specification, morphogenesis and cell-fate commitment, consistent with the expected roles of PcG in regulating key developmental processes of an organism (4,6–8,13–16). The consensus genes predicted by both EpiPredictor-CG and jPREdictor (dynamic) made up about 28% of the top 322 genes and their corresponding gene ontology analysis presented good consistency with experimental studies.
Figure 3.

Gene ontology analysis of genes predicted by EpiPredictor and jPREdictor. Shown are the top 10 gene ontology terms related to the genes predicted by: (A) EpiPredictor-CG; (B) EpiPredictor-CG but not jPREdictor (dynamic); (C) jPREdictor (dynamic); (D) jPREdictor (dynamic) but not EpiPredictor-CG; (E) both EpiPredictor-CG and jPREdictor (dynamic); (F) EpiPredictor-CG except the seven annotated genes.
By cross-referencing existing literature, we found experimental evidence for seven genes, which were uniquely identified by EpiPredictor-CG and also matched at least one of the three ChIP studies, of their critical roles in key developmental processes (Table 5). To exemplify, the inv locus was recently found to harbour one PRE site which has been experimentally verified (38) and its role in regulating Drosophila hindgut development is well established (76). The wg locus belongs to the important Wg/Wnt signal transduction pathway that directs a variety of cell fate decisions in developing animal embryos (86). In Drosophila, wg alone directs a wide range of cell fate and patterning decisions (77). The nub locus is involved in embryogenesis and neurogenesis (78–80). The pdm2 locus is responsible for a variety of cell fate decision in the Drosophila development (81). The dac is an essential part of a complex that functions to induce ectopic eye development (82). The Gsc mediates effective repression in Drosophila blastoderm embryos (83). The tup has a key function in the development of imaginal disc (84) and is also a key component in early cardiogenesis (85). Interestingly, a recent ChIP study (7) revealed that the human homologues of wg (WNT1), dac (DACH-1), Gsc (GSC) and tup (ISL1) are all targeted by PcG. In particular, WNT1 is known to be involved in embryogenesis and cancer development (86). The functions of the genes uniquely identified by our system but excluding the abovementioned seven genes are shown by a gene ontology analysis using DAVID (Figure 3F).
Table 5.
Annotation of a set of seven genes uniquely identified by EpiPredictor-CG
| Gene | Verified function in Drosophila | Vertebrate homologue |
|---|---|---|
| inv | A newly experimentally validated PRE was found to exist in the inv locus (38). It is important for hindgut development (76) | |
| wg | Embryogenesis (Wingless/Wnt signaling pathway) (77) | WNT1: predicted as PcG target in human (7); involved in embryogenesis and cancer (86) |
| nub | Embryogenesis, neurogenesis (78–80) | |
| pdm2 | Important for a variety of cell fate decisions in development (81) | |
| dac | Induce ectopic eye development (82) | DACH-1: predicted as PcG target in human (7) |
| Gsc | groucho-dependent repression in embryo (83) | GSC: predicted as PcG target in human (7) |
| tup | Imaginal disc development (84), key component in early cardiogenesis (85) | ISL1: predicted as PcG target in human (7) |
To further validate our prediction, we also cross-referenced our gene list with the 27 PcG target genes confirmed by ChIP-qPCR in the work of Ringrose and colleagues (40), of which EpiPredictor-CG correctly predicted 19 genes (70%), exhibiting a good correlation.
Experimental validation of EpiPredictor prediction
In order to experimentally validate EpiPredictor prediction, ChIP-qPCR was used to investigate the enrichment of 15 predicted PRE sites that were randomly selected from the top 150 predictions (Supplementary Table S10) using anti-E(z) antibody. For positive controls we used three known PREs, bxd, iab2, and en_DM, as established in the literature (87) along with four sequences from Ringrose et al. (40), hth, unc-4, idgf4, and cato, for which ChIP-qPCR experiments have been done using anti-PC antibody. Three housekeeping genes with no previous evidence as PRE or of polycomb related activity, hsp22, hsp26 and Pc, were selected as negative controls (40). Primers for qPCR are listed in Supplementary Table S11.
The results of ChIP-qPCR showed that there are more than two-fold enrichments for 12 out of the 15 tested PRE sites (Figure 4). Among them, five showed enrichment greater than the averaged value of 5.66 for the seven positive controls, indicating a higher degree of confidence for their potential as PcG target genes. Our E(z)-ChIP derived data and Ringrose’s PC data are scaled roughly to the same level (Supplementary Table S12) with the exception of idgf4 which exhibited enrichment in our data but not in Ringrose’s (40). However, this discrepancy is not completely unexpected given the fact that on the whole genome scale PC and E(z) do not always align well (4).
Figure 4.

ChIP-qPCR verification of EpiPredictor prediction. Shown are the enrichment of each genomic region (predicted PRE site) in S2 cell ChIP samples using anti-E(z) versus the use of anti-FLAG mock antibodies. The horizontal line shows an enrichment of 1 (no enrichment). The gene symbols listed are those of the genes closest to the tested genomic regions. For specific coordinates please refer to Supplementary Table S10.
By mapping the positively enriched sequences onto their closest genes, we found that all 12 corresponding genes are of crucial importance to Drosophila embryonic development, since the knockout of each of these genes conferred serious body morphological changes. The antp and abd-A are Drosophila HOX genes (88), while bxd is expressed directly upstream of and is known to directly influence the behavior of ubx, another HOX gene (89). Furthermore, both disco and eve regulate the localization or expression of HOX genes, conversely, salm and bab2 are directly regulated by HOX genes (90–93), while unc-4 is a homeobox-containing protein and a paralogue of the HOX genes with similar functions (94). Finally, both noc and pnr are critical for proper eye formation (95,96), grn has importance in multiple organ development (97) and immune response in the midgut (98), and zfh1 is essential to cell differentiation of lateral mesodermal derivative lineages and in neurogenisis (99). The critical importance of these genes and the computational prediction of them being PcG target genes highlight the importance of understanding how sequence influences PcG binding in order to properly understand embryonic development in Drosophila.
PcG target genes are essentially free of transposons
Transposons are mobile genetic elements that can cause mutations and change the amount of DNA in the genome (105). Given their critical importance in cellular functions, we predicted that PcG target genes in D. melanogaster should have a minimal presence of transposons, generally termed as transposon-free regions (TFRs). We performed a whole-genome search and identified 1400 TFRs of > 10 000 bps in length, of which 1232 overlapped with at least one gene’s TSS. In the top 322 putative PcG target genes predicted by our system, 319 of them (99%) had TSS overlapping with one or more TFRs. Thus, as we expected, the D. melanogaster PcG target genes are indeed essentially free of identifiable transposon-derived sequences. This is a novel finding in Drosophila and corroborates well with several recent mammalian studies that revealed strong correlations between TFRs and genes encoding developmental regulators (100), as well as the H3K27me3 marks (101).
DISCUSSION
Sequence ambiguity and multi-motifs in EpiPredictor
Given the ambiguity in the consensus sequence of motifs, our system considered different versions of the same motifs (for instance, PS, PM and PF for Pho) as well as allowed the existence of ambiguity codes and mutations (Supplementary Table S1). In addition, by using an SVM with non-linear kernel as a PRE classifier, our program abstractly models how multiple motifs interact with each other at the genomic site of interest. These two considerations are similar to the options of position-specific probability matrices and multi-motifs in jPredictor.
Transcription factor networking is important for PcG recruitment
To the best of our knowledge, this is the first application of SVM to PRE prediction. With the integration of a non-linear kernel, our system EpiPredictor succeeded in modelling the spatial relationship and combinatorial interaction among transcription factors that are involved in PcG recruitment. This strategy offers a higher level of abstraction over any other approaches that use a linear function. The fully automated process of constructing the classifier in SVM also reduces the level of bias in the analysis.
Our novel computational strategy also offers new insights into the interactions among transcription factors at the cis-regulatory elements in vivo. The outstanding performance of the non-linear kernels indicates that multiple transcription factors are networking at the cis-regulatory elements for efficient recruitment of PcG proteins. However, the details of such networking remain to be illustrated in future studies.
High GC content and conservation level are important features of PcG target genes
Among the array of perspectives that we used in EpiPredictor, SVM classifier, high GC content and comparative genomics all led to substantial performance improvements (Table 2). The success of integrating GC analysis suggested that relatively high GC content be an important feature of PcG target genes, consistent with previous studies that hyper-conserved CpG domains underlie polycomb-binding sites (65). In addition, given their critical importance in cellular functions, PcG target genes are not surprisingly highly conserved in evolution.
PcG target genes are essential for transcription and development
The gene ontology analysis on the genes predicted by our system revealed that the target genes of PcG are mainly regulators of transcription activities and are crucial for key developmental processes. Some genes uniquely predicted by our system are confirmed by several independent experimental studies to be essential for normal development and patterning. These observations further support the fundamental roles of PcG proteins in development and cellular functions.
Prediction of TrxG target genes
Trithorax group (TrxG) proteins methylate histone 3 lysine 4 to reverse the repression imposed by PcG proteins (18,102). There exists substantial evidence that Trithorax response elements (TREs) and PREs co-localize. For example, several major TrxG proteins bind at essentially all known or presumptive PREs, suggesting that the regulatory platforms are switchable (18,103). In mouse embryonic stem cells, large bivalent domains were found to contain chromatin modifications generated by both PcG and TrxG, suggesting the co-presence of PcG and TrxG in developmental genes (101). A recent genetic study on Drosophila also revealed that PcG repression is dynamic and that ASH1 (absent, small or homeotic discs 1), the histone methyltransferase belonging to the TrxG complex, is critical for the active state of Polycomb target genes (102). Taken together, accumulating evidence suggests that the epigenetic regulations mediated by PcG and TrxG are likely to be closely intertwined and that the approach that accurately predicts PcG target genes will also shed new light on TrxG target genes. Thus, it is fully expected that some of the PcG target genes we predicted here will turn out to be TrxG target genes.
CONCLUSIONS
Despite a large number of genome-wide ChIP studies of PcG target genes (1,4–8,10,12,14–16) recently appeared in the literature that substantially enriched our knowledge of the scales of PcG-mediated epigenetic modification and their roles in normal cellular functions and in cancer development, our mechanistic understanding of this process remains extremely poor. To exemplify, up to date, there are only two mammalian PREs (9,41) and a dozen of Drosophila PREs (31–40) that have been experimentally verified. In addition, there are only nine Drosophila transcription factors confirmed to be involved in PcG recruitment, among which only two have mammalian homologues (20,104). The extremely limited pools of confirmed PREs and their interacting transcription factors are the main restraints for the relatively mediocre performance of computational methods such as EpiPredictor and jPredictor, with 20–30% matching ratios with genome-wide ChIP data. Although our EpiPredictor has substantially outperformed jPredictor (by up to >10% in matching ratio), we expect a much better performance if we had had more knowledge on PREs and their interacting transcription factors. Thus, the more accurate computational method such as EpiPredictor will provide a very useful tool for initial screening of PcG target genes from ChIP studies so as to identify the most likely candidates for labour-intensive experimental verifications. The enhanced knowledge of PREs will in turn improve the performance of these computational methods, and ultimately leads to a comprehensive understanding of PcG-mediated gene repression in normal cellular functions as well as in epigenetic dysregulation. Thus, our new EpiPredictor program reported in this study represents an important step toward this ultimate goal in the field of epigenetics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables S1–S11.
FUNDING
Collaborative Advances in Biomedical Computing from The John and Ann Doerr Fund for Computational Biomedicine at Rice University (to J.M. and Q.W.); training fellowship from the Keck Center Computational Cancer Biology Training Program of the Gulf Coast Consortia (CPRIT Grant No. RP101489 to J.Z.); training fellowship from the NLM Training Program of the Keck Center of the Gulf Coast Consortia (NIH Grant No. 5 T15LM07093-17 to B.D.K.); the National Institutes of Health (R01-GM067801), the National Science Foundation (MCB-0818353); the Welch Foundation (Q-1512) to J.M. Funding for open access charge: Start-up funds from Baylor College of Medicine (to Q.W.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Drs Leonie Ringrose and Marc Remsmeier for providing valuable details regarding their publications. We also thank Kit Menlove for his input on the project. The source code of EpiPredictor is available upon request.
REFERENCES
- 1.Squazzo SL, O'Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, Margueron R, Reinberg D, Green R, Farnham PJ. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006;16:890–900. doi: 10.1101/gr.5306606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanzuolo C, Roure V, Dekker J, Bantignies F, Orlando V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol. 2007;9:1167–1174. doi: 10.1038/ncb1637. [DOI] [PubMed] [Google Scholar]
- 3.Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 2004;38:413–443. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz YB, Kahn TG, Nix DA, Li XY, Bourgon R, Biggin M, Pirrotta V. Genome-wide analysis of Polycomb targets in Drosophila melanogaster. Nat. Genet. 2006;38:700–705. doi: 10.1038/ng1817. [DOI] [PubMed] [Google Scholar]
- 5.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 9.Sing A, Pannell D, Karaiskakis A, Sturgeon K, Djabali M, Ellis J, Lipshitz HD, Cordes SP. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell. 2009;138:885–897. doi: 10.1016/j.cell.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Choi JH, Li Y, Guo J, Pei L, Rauch TA, Kramer RS, Macmil SL, Wiley GB, Bennett LB, Schnabel JL, et al. Genome-wide DNA methylation maps in follicular lymphoma cells determined by methylation-enriched bisulfite sequencing. PLoS One. 2010;5:e13020. doi: 10.1371/journal.pone.0013020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwong C, Adryan B, Bell I, Meadows L, Russell S, Manak JR, White R. Stability and dynamics of polycomb target sites in Drosophila development. PLoS Genet. 2008;4:e1000178. doi: 10.1371/journal.pgen.1000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuettengruber B, Ganapathi M, Leblanc B, Portoso M, Jaschek R, Tolhuis B, van Lohuizen M, Tanay A, Cavalli G. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol. 2009;7:e13. doi: 10.1371/journal.pbio.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tolhuis B, de Wit E, Muijrers I, Teunissen H, Talhout W, van Steensel B, van Lohuizen M. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat. Genet. 2006;38:694–699. doi: 10.1038/ng1792. [DOI] [PubMed] [Google Scholar]
- 16.Ke XS, Qu Y, Rostad K, Li WC, Lin B, Halvorsen OJ, Haukaas SA, Jonassen I, Petersen K, Goldfinger N, et al. Genome-wide profiling of histone h3 lysine 4 and lysine 27 trimethylation reveals an epigenetic signature in prostate carcinogenesis. PLoS One. 2009;4:e4687. doi: 10.1371/journal.pone.0004687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mihaly J, Mishra RK, Karch F. A Conserved Sequence Motif in Polycomb-Response Elements. Mol. Cell. 1998;1:1065–1066. doi: 10.1016/s1097-2765(00)80107-0. [DOI] [PubMed] [Google Scholar]
- 18.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome Regulation by Polycomb and Trithorax Proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen N, Zhang X, Olashaw N, Seto E. Molecular cloning and functional characterization of the transcription factor YY2. J. Biol. Chem. 2004;279:25927–25934. doi: 10.1074/jbc.M402525200. [DOI] [PubMed] [Google Scholar]
- 20.Brown JL, Mucci D, Whiteley M, Dirksen ML, Kassis JA. The Drosophila Polycomb group gene pleiohomeotic encodes a DNA binding protein with homology to the transcription factor YY1. Mol. Cell. 1998;1:1057–1064. doi: 10.1016/s1097-2765(00)80106-9. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell. 2004;14:637–646. doi: 10.1016/j.molcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Strutt H, Cavalli G, Paro R. Co-localization of Polycomb protein and GAGA factor on regulatory elements responsible for the maintenance of homeotic gene expression. EMBO J. 1997;16:3621–3632. doi: 10.1093/emboj/16.12.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Decoville M, Giacomello E, Leng M, Locker D. DSP1, an HMG-like Protein, Is Involved in the Regulation of Homeotic Genes. Genetics. 2001;157:237–244. doi: 10.1093/genetics/157.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodgson JW, Argiropoulos B, Brock HW. Site-specific recognition of a 70-base-pair element containing d(GA)(n) repeats mediates bithoraxoid polycomb group response element-dependent silencing. Mol. Cell. Biol. 2001;21:4528–4543. doi: 10.1128/MCB.21.14.4528-4543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dejardin J, Cavalli G. Chromatin inheritance upon Zeste-mediated Brahma recruitment at a minimal cellular memory module. EMBO J. 2004;23:857–868. doi: 10.1038/sj.emboj.7600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagstrom K, Muller M, Schedl P. A Polycomb and GAGA dependent silencer adjoins the Fab-7 boundary in the Drosophila bithorax complex. Genetics. 1997;146:1365–1380. doi: 10.1093/genetics/146.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dejardin J, Rappailles A, Cuvier O, Grimaud C, Decoville M, Locker D, Cavalli G. Recruitment of Drosophila Polycomb group proteins to chromatin by DSP1. Nature. 2005;434:533–538. doi: 10.1038/nature03386. [DOI] [PubMed] [Google Scholar]
- 28.Blastyak A, Mishra RK, Karch F, Gyurkovics H. Efficient and specific targeting of Polycomb group proteins requires cooperative interaction between Grainyhead and Pleiohomeotic. Mol. Cell. Biol. 2006;26:1434–1444. doi: 10.1128/MCB.26.4.1434-1444.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown JL, Grau DJ, DeVido SK, Kassis JA. An Sp1/KLF binding site is important for the activity of a Polycomb group response element from the Drosophila engrailed gene. Nucleic Acids Res. 2005;33:5181–5189. doi: 10.1093/nar/gki827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ringrose L, Paro R. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development. 2007;134:223–232. doi: 10.1242/dev.02723. [DOI] [PubMed] [Google Scholar]
- 31.Kassis JA. Unusual properties of regulatory DNA from the Drosophila engrailed gene: three “pairing-sensitive” sites within a 1.6-kb region. Genetics. 1994;136:1025–1038. doi: 10.1093/genetics/136.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gindhart JG, Jr, Kaufman TC. Identification of Polycomb and trithorax group responsive elements in the regulatory region of the Drosophila homeotic gene Sex combs reduced. Genetics. 1995;139:797–814. doi: 10.1093/genetics/139.2.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Americo J, Whiteley M, Brown JL, Fujioka M, Jaynes JB, Kassis JA. A complex array of DNA-binding proteins required for pairing-sensitive silencing by a polycomb group response element from the Drosophila engrailed gene. Genetics. 2002;160:1561–1571. doi: 10.1093/genetics/160.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bloyer S, Cavalli G, Brock HW, Dura JM. Identification and characterization of polyhomeotic PREs and TREs. Dev. Biol. 2003;261:426–442. doi: 10.1016/s0012-1606(03)00314-2. [DOI] [PubMed] [Google Scholar]
- 35.Gruzdeva N, Kyrchanova O, Parshikov A, Kullyev A, Georgiev P. The Mcp element from the bithorax complex contains an insulator that is capable of pairwise interactions and can facilitate enhancer-promoter communication. Mol. Cell. Biol. 2005;25:3682–3689. doi: 10.1128/MCB.25.9.3682-3689.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeVido SK, Kwon D, Brown JL, Kassis JA. The role of Polycomb-group response elements in regulation of engrailed transcription in Drosophila. Development. 2008;135:669–676. doi: 10.1242/dev.014779. [DOI] [PubMed] [Google Scholar]
- 37.Kozma G, Bender W, Sipos L. Replacement of a Drosophila Polycomb response element core, and in situ analysis of its DNA motifs. Mol. Genet. Genomics. 2008;279:595–603. doi: 10.1007/s00438-008-0336-3. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham MD, Brown JL, Kassis JA. Characterization of the polycomb group response elements of the Drosophila melanogaster invected Locus. Mol. Cell. Biol. 2010;30:820–828. doi: 10.1128/MCB.01287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horard B, Tatout C, Poux S, Pirrotta V. Structure of a polycomb response element and in vitro binding of polycomb group complexes containing GAGA factor. Mol. Cell. Biol. 2000;20:3187–3197. doi: 10.1128/mcb.20.9.3187-3197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ringrose L, Rehmsmeier M, Dura JM, Paro R. Genome-wide prediction of Polycomb/Trithorax response elements in Drosophila melanogaster. Dev. Cell. 2003;5:759–771. doi: 10.1016/s1534-5807(03)00337-x. [DOI] [PubMed] [Google Scholar]
- 41.Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell. 2010;140:99–110. doi: 10.1016/j.cell.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L, Zhong K, Dai Y, Zhou H. Genome-wide analysis of histone H3 lysine 27 trimethylation by ChIP-chip in gastric cancer patients. J. Gastroenterol. 2009;44:305–312. doi: 10.1007/s00535-009-0027-9. [DOI] [PubMed] [Google Scholar]
- 44.Araki Y, Wang Z, Zang C, Wood WH, 3rd, Schones D, Cui K, Roh TY, Lhotsky B, Wersto RP, Peng W, et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity. 2009;30:912–925. doi: 10.1016/j.immuni.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miao F, Natarajan R. Mapping Global Histone Methylation Patterns in the Coding Regions of Human Genes. Mol. Cell. Biol. 2005;25:4650–4661. doi: 10.1128/MCB.25.11.4650-4661.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008;40:741–750. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 47.Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, Farnham PJ. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–1605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiedler T, Rehmsmeier M. jPREdictor: a versatile tool for the prediction of cis-regulatory elements. Nucleic Acids Res. 2006;34:W546–550. doi: 10.1093/nar/gkl250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hauenschild A, Ringrose L, Altmutter C, Paro R, Rehmsmeier M. Evolutionary plasticity of polycomb/trithorax response elements in Drosophila species. PLoS Biol. 2008;6:e261. doi: 10.1371/journal.pbio.0060261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Shao Z, Yuan GC. Prediction of Polycomb target genes in mouse embryonic stem cells. Genomics. 2010;96:17–26. doi: 10.1016/j.ygeno.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 53.van Steensel B, Delrow J, Bussemaker HJ. Genomewide analysis of Drosophila GAGA factor target genes reveals context-dependent DNA binding. Proc. Natl Acad. Sci. USA. 2003;100:2580–2585. doi: 10.1073/pnas.0438000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guyon I, Weston J, Barnhill S, Vapnik V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002;46:389–422. [Google Scholar]
- 55.Tang EK, Suganthan PN, Yao X. Gene selection algorithms for microarray data based on least squares support vector machine. BMC Bioinformatics. 2006;7:95. doi: 10.1186/1471-2105-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang SW, Pan Q, Zhang HC, Zhang YL, Wang HY. Classification of protein quaternary structure with support vector machine. Bioinformatics. 2003;19:2390–2396. doi: 10.1093/bioinformatics/btg331. [DOI] [PubMed] [Google Scholar]
- 57.Shamim MT, Anwaruddin M, Nagarajaram HA. Support Vector Machine-based classification of protein folds using the structural properties of amino acid residues and amino acid residue pairs. Bioinformatics. 2007;23:3320–3327. doi: 10.1093/bioinformatics/btm527. [DOI] [PubMed] [Google Scholar]
- 58.Leslie C, Eskin E, Noble WS. The spectrum kernel: a string kernel for SVM protein classification. Pac. Symp. Biocomput. 2002;7:564–575. [PubMed] [Google Scholar]
- 59.Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 60.Laufer S, Rubinsky B. Tissue characterization with an electrical spectroscopy SVM classifier. IEEE Trans. Biomed. Eng. 2009;56:525–528. doi: 10.1109/TBME.2008.2003105. [DOI] [PubMed] [Google Scholar]
- 61.McQuisten KA, Peek AS. Comparing artificial neural networks, general linear models and support vector machines in building predictive models for small interfering RNAs. PLoS One. 2009;4:e7522. doi: 10.1371/journal.pone.0007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen L, Tan EC. Reducing multiclass cancer classification to binary by output coding and SVM. Comput. Biol. Chem. 2006;30:63–71. doi: 10.1016/j.compbiolchem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 63.Das R, Dimitrova N, Xuan Z, Rollins RA, Haghighi F, Edwards JR, Ju J, Bestor TH, Zhang MQ. Computational prediction of methylation status in human genomic sequences. Proc. Natl Acad. Sci. USA. 2006;103:10713–10716. doi: 10.1073/pnas.0602949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bock C, Paulsen M, Tierling S, Mikeska T, Lengauer T, Walter J. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 2006;2:e26. doi: 10.1371/journal.pgen.0020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanay A, O'Donnell AH, Damelin M, Bestor TH. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc. Natl Acad. Sci. USA. 2007;104:5521–5526. doi: 10.1073/pnas.0609746104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010;6:e1001244. doi: 10.1371/journal.pgen.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Flybase 2010 [Google Scholar]
- 69.Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, Li H, Blacque OE, Li L, Leitch CC, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 70.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 71.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- 73.Chang C, Lin C. LIBSVM: a library for support vector machines. ACM Transactions on Intelligent Systems and Technology. 2011;2:1–27. [Google Scholar]
- 74.Chipman HA, George EI, McCulloch RE. BART: Bayesian Additive Rgression Trees. Annu. Appl. Stat. 2010;4:266–298. [Google Scholar]
- 75.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 76.Iwaki DD, Lengyel JA. A Delta-Notch signaling border regulated by Engrailed/Invected repression specifies boundary cells in the Drosophila hindgut. Mech. Dev. 2002;114:71–84. doi: 10.1016/s0925-4773(02)00061-8. [DOI] [PubMed] [Google Scholar]
- 77.Jones WM, Bejsovec A. RacGap50C negatively regulates wingless pathway activity during Drosophila embryonic development. Genetics. 2005;169:2075–2086. doi: 10.1534/genetics.104.039735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Billin AN, Cockerill KA, Poole SJ. Isolation of a family of Drosophila POU domain genes expressed in early development. Mech. Dev. 1991;34:75–84. doi: 10.1016/0925-4773(91)90045-8. [DOI] [PubMed] [Google Scholar]
- 79.Lloyd A, Sakonju S. Characterization of two Drosophila POU domain genes, related to oct-1 and oct-2, and the regulation of their expression patterns. Mech. Dev. 1991;36:87–102. doi: 10.1016/0925-4773(91)90075-h. [DOI] [PubMed] [Google Scholar]
- 80.Dick T, Yang XH, Yeo SL, Chia W. Two closely linked Drosophila POU domain genes are expressed in neuroblasts and sensory elements. Proc. Natl Acad. Sci. USA. 1991;88:7645–7649. doi: 10.1073/pnas.88.17.7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Poole SJ. Conservation of complex expression domains of the pdm-2 POU domain gene between Drosophila virilis and Drosophila melanogaster. Mech. Dev. 1995;49:107–116. doi: 10.1016/0925-4773(94)00307-9. [DOI] [PubMed] [Google Scholar]
- 82.Chen R, Amoui M, Zhang Z, Mardon G. Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell. 1997;91:893–903. doi: 10.1016/s0092-8674(00)80481-x. [DOI] [PubMed] [Google Scholar]
- 83.Jimenez G, Verrijzer CP, Ish-Horowicz D. A conserved motif in goosecoid mediates groucho-dependent repression in Drosophila embryos. Mol. Cell. Biol. 1999;19:2080–2087. doi: 10.1128/mcb.19.3.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Navascues J, Modolell J. tailup, a LIM-HD gene, and Iro-C cooperate in Drosophila dorsal mesothorax specification. Development. 2007;134:1779–1788. doi: 10.1242/dev.02844. [DOI] [PubMed] [Google Scholar]
- 85.Mann T, Bodmer R, Pandur P. The Drosophila homolog of vertebrate Islet1 is a key component in early cardiogenesis. Development. 2009;136:317–326. doi: 10.1242/dev.022533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 87.Strutt H, Paro R. The polycomb group protein complex of Drosophila melanogaster has different compositions at different target genes. Mol. Cell. Biol. 1997;17:6773–6783. doi: 10.1128/mcb.17.12.6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lemons D, McGinnis W. Genomic evolution of Hox gene clusters. Science. 2006;313:1918–1922. doi: 10.1126/science.1132040. [DOI] [PubMed] [Google Scholar]
- 89.Petruk S, Sedkov Y, Riley KM, Hodgson J, Schweisguth F, Hirose S, Jaynes JB, Brock HW, Mazo A. Transcription of bxd noncoding RNAs promoted by trithorax represses Ubx in cis by transcriptional interference. Cell. 2006;127:1209–1221. doi: 10.1016/j.cell.2006.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanders LR, Patel M, Mahaffey JW. The Drosophila gap gene giant has an anterior segment identity function mediated through disconnected and teashirt. Genetics. 2008;179:441–453. doi: 10.1534/genetics.107.084988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wagner-Bernholz JT, Wilson C, Gibson G, Schuh R, Gehring WJ. Identification of target genes of the homeotic gene Antennapedia by enhancer detection. Genes Dev. 1991;5:2467–2480. doi: 10.1101/gad.5.12b.2467. [DOI] [PubMed] [Google Scholar]
- 92.Biggin MD, Tjian R. A purified Drosophila homeodomain protein represses transcription in vitro. Cell. 1989;58:433–440. doi: 10.1016/0092-8674(89)90424-8. [DOI] [PubMed] [Google Scholar]
- 93.Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, Carroll SB. The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell. 2008;134:610–623. doi: 10.1016/j.cell.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tabuchi K, Yoshikawa S, Yuasa Y, Sawamoto K, Okano H. A novel Drosophila paired-like homeobox gene related to Caenorhabditis elegans unc-4 is expressed in subsets of postmitotic neurons and epidermal cells. Neurosci. Lett. 1998;257:49–52. doi: 10.1016/s0304-3940(98)00799-x. [DOI] [PubMed] [Google Scholar]
- 95.Singh A, Choi KW. Initial state of the Drosophila eye before dorsoventral specification is equivalent to ventral. Development. 2003;130:6351–6360. doi: 10.1242/dev.00864. [DOI] [PubMed] [Google Scholar]
- 96.Luque CM, Milan M. Growth control in the proliferative region of the Drosophila eye-head primordium: the elbow-noc gene complex. Dev. Biol. 2007;301:327–339. doi: 10.1016/j.ydbio.2006.07.050. [DOI] [PubMed] [Google Scholar]
- 97.Lin WH, Huang LH, Yeh JY, Hoheisel J, Lehrach H, Sun YH, Tsai SF. Expression of a Drosophila GATA transcription factor in multiple tissues in the developing embryos. Identification of homozygous lethal mutants with P-element insertion at the promoter region. J. Biol. Chem. 1995;270:25150–25158. doi: 10.1074/jbc.270.42.25150. [DOI] [PubMed] [Google Scholar]
- 98.Senger K, Harris K, Levine M. GATA factors participate in tissue-specific immune responses in Drosophila larvae. Proc. Natl Acad. Sci. USA. 2006;103:15957–15962. doi: 10.1073/pnas.0607608103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lai ZC, Fortini ME, Rubin GM. The embryonic expression patterns of zfh-1 and zfh-2, two Drosophila genes encoding novel zinc-finger homeodomain proteins. Mech. Dev. 1991;34:123–134. doi: 10.1016/0925-4773(91)90049-c. [DOI] [PubMed] [Google Scholar]
- 100.Simons C, Pheasant M, Makunin IV, Mattick JS. Transposon-free regions in mammalian genomes. Genome Res. 2006;16:164–172. doi: 10.1101/gr.4624306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 102.Aichinger E, Villar CB, Farrona S, Reyes JC, Hennig L, Kohler C. CHD3 proteins and polycomb group proteins antagonistically determine cell identity in Arabidopsis. PLoS Genet. 2009;5:e1000605. doi: 10.1371/journal.pgen.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schwartz YB, Kahn TG, Stenberg P, Ohno K, Bourgon R, Pirrotta V. Alternative epigenetic chromatin states of polycomb target genes. PLoS Genet. 2010;6:e1000805. doi: 10.1371/journal.pgen.1000805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Matharu NK, Hussain T, Sankaranarayanan R, Mishra RK. Vertebrate homologue of Drosophila GAGA factor. J. Mol. Biol. 2010;400:434–437. doi: 10.1016/j.jmb.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 105.McClintock B. The origin and behavior of mutable loci in maize. Proc. Natl Acad. Sci. USA. 1950;36:344–355. doi: 10.1073/pnas.36.6.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.