


Abstract
With a dataset of more than 600 million small RNAs deeply sequenced from mouse hippocampal and staged sets of mouse cells that underwent reprogramming to induced pluripotent stem cells, we annotated the stem–loop precursors of the known miRNAs to identify isomoRs (miRNA-offset RNAs), loops, non-preferred strands, and guide strands. Products from both strands were readily detectable for most miRNAs. Changes in the dominant isomiR occurred among the cell types, as did switches of the preferred strand. The terminal nucleotide of the dominant isomiR aligned well with the dominant off-set sequence suggesting that Drosha cleavage generates most miRNA reads without terminal modification. Among the terminal modifications detected, most were non-templated mono- or di-nucleotide additions to the 3′-end. Based on the relative enrichment or depletion of specific nucleotide additions in an Ago-IP fraction there may be differential effects of these modifications on RISC loading. Sequence variation of the two strands at their cleavage sites suggested higher fidelity of Drosha than Dicer. These studies demonstrated multiple patterns of miRNA processing and considerable versatility in miRNA target selection.
INTRODUCTION
MicroRNAs (miRNAs) are ∼21 nt non-coding RNAs that serve as key post-transcriptional regulators of gene expression in the specialized cells of multi-cellular organisms (1–3). In animals, miRNAs are typically transcribed from the genomes as primary (pri-miRNA) transcripts by RNA polymerase II (4), and less often as mirtrons from spliceosome-mediated processing (5,6). The pri-miRNAs fold into hairpins and are cleaved at their base into pre-miRNAs by the RNase III family member, Drosha (7–9). Pre-miRNAs are ∼70 nt stem-loop structures, which are exported to the cytoplasm (10–12), where another RNase III family member, Dicer, cleaves the pre-miRNA loop to make a 21-bp duplex (9,13,14) that associates tightly with Argonaute (Ago) proteins (15,16). miR-451 is processed in a Dicer-independent alternative pathway in which the pre-miRNAs are cleaved by AGO2 and then are processed to maturity by exonucleolytic cleavage (17–19). Generally, one strand of the duplex is preferentially selected for incorporation into the RNA-induced silencing complex (RISC) (14,20) where it represses gene expression through interaction with the 3′-untranslated regions (3′-UTRs) of mRNAs [reviewed in (1,3)] and the other strand is released and degraded (21,22). The preferred strand has been called the guide strand or the mature miRNA and non-preferred strand has been called the passenger or the star strand (miRNA*). In general, the retained strand is the one with a less stably base-paired 5′-end in the miRNA/miRNA* duplex (2,20,21,23). The passenger strand may also be incorporated into a functional RISC (24–27).
Depending upon the depth of sequencing low copy number by-products of miRNA processing can be detected (28–31). Importantly, the complex processing of miRNAs greatly expands the targeting potential due to the utilization of both strands of the miRNA:miRNA* duplex, length heterogeneity, sequential Dicer cleavage and RNA editing.
We collected small RNA deep sequencing data from two large groups of cell and tissue types and annotated each known mouse miRNA to reveal general patterns of miRNA biogenesis and expression. Based on the large scale and depth of the integrated data, we were able to collect phased reads and annotate extensively the stem-loop precursors of each known mouse miRNA expressed in these samples. The snapshot quantification of pre-miRNA fragments suggests a high differential stability of the fragment categories—5p strand, 3p strand, loop regions, moRs, as well as extensive isoform variation that may contribute to functional versatility of miRNA function.
MATERIALS AND METHODS
Small RNA samples and datasets
The small RNA samples and datasets are listed in Supplementary Table S1. Six hippocampus samples were used for total RNA isolation with mirVana™ miRNA Isolation kit (Ambion, Austin, TX) following the manufacturer’s protocol. Four samples came from four different mice and were used to isolate small RNAs (designated Group I: Hippocampus-total RNA samples). Two samples from the same source were separately immunoprecipitated with three different Ago antibodies, AGO1, AGO2 and 28 A [generously donated by Z. Mourelatos (32) designated Hippocampus-AGO-IP data] by using Dynabeads protein G, kit (Invitrogen #100.03D). Eight samples came from mouse cells at various stages of reprogramming with four factors (33,34) (designated Group II: Reprogramming-total RNA samples). These samples included uninfected Oct4-GFP MEFs (mouse embryonic fibroblasts), Thy1- cells harvested from Day 5 post-transduction cultures, SSEA1+ cells [identified by APC-conjugated anti-SSEA1 (R&D Systems, Minneapolis, MN; Cat.FAB2155A)], GFP+ iPSC colonies purified by cell sorting, and induced pluripotent stem cell lines (ipS cells). Another three sets of cells were mouse embryonic cells (mES) and two partially induced pluripotent stem cell lines (piPS_4 and piPS_5). The libraries were prepared with the SOLiD™ Small RNA Expression Kit and sequenced by SOLiD™ sequencer version 3 and 4. The samples were sequenced at a length of 25 bp for some datasets and then sequenced at 35 bp.
Small RNA mapping analysis
Using the SOLiD™ small RNA Analysis pipeline (http://solidsoftwaretools.com), the raw data was filtered for tRNA, rRNA and adaptor sequences and then aligned against miRNA precursors and the mouse genome. To identify the adaptor sequences at the 3′-end of reads, the first 18 nt with a maximum of two or three mismatches were aligned to find the candidate matches. Candidate matches were then extended to identify the best alignments and locate adaptors, producing high quality data for the exact size of the small RNAs (Supplementary Figure S1). Reads were first mapped to known mouse miRNAs (miRBse version 16), and then reads that did not align were mapped against the mouse genome for further analysis. A sequencing error rate of <1% was calculated by determining the percentage of tags that did not match the first 10 nt of the mature miRNA.
The coordinates of known miRNA loci from miRBase were used to make a miRNA precursor database from the UCSC genome browser, and 50 nt were added at both ends to identify moR sequences. Reads were aligned against the precursor database to obtain matches for 5p strands, 3p strands, moRs and loop regions. Only perfect matches were used for further analysis. The most frequently sequenced isomiRs of both arms were chosen as a reference to classify other isomiRs. IsomiRs with no more than a 3 nt difference at either end from the most frequent isomiR were counted and included in Supplementary Datasets (Supplementary Datasets S1, S2 and S3). moRs were identified by surveying clustered reads outside the dominant isomiRs; a gap of 3 nt at most between moRs and dominant isomiRs was allowed. Only very rarely did reads span the cleavage sites of the hairpin. Most of the analysis was done with in-house written Perl scripts. Z-tests were used to determine the significance of isomiR variation.
Non-templated nucleotides at the termini of mature miRNAs were identified by checking sequences before adaptors that did not exactly match the genome. The accuracy of adaptor identification was very important for correctly identifying non-templated nucleotides. Therefore, only data with 35-bp reads, where much of the adaptor was sequenced, were used.
RESULTS
Deep sequencing of mouse small RNAs
A total of 34 datasets from 17 samples and more than 626 million (M) reads were analyzed (see Materials and Methods section and Supplementary Table S1). The 188M reads matched known mouse miRNA precursors from miRBase (Figure 1) and an additional 128 M matched exons, introns and intergenic regions in the mouse genome. Reads that matched multiple miRNAs were rejected and only perfect matches were kept for downstream analysis. miRBase version 16 contains 582 well-authenticated mouse miRNA species and we identified 526 miRNA species in our samples (416 for Group I and 489 for Group II) by requiring detection of both strands with at least ten reads on the non-preferred strand. Those miRNAs not validated in our samples may be expressed in tissues not used in our analysis. miRNA expression in Group I and Group II have distinct and overlapping patterns of miRNA expression (Supplementary Figure S2, Supplementary Tables S2 and S3). The miRNAs in AGO-IP data from the mouse hippocampus (Supplementary Table S4) correlated very well with total hippocampus small RNA profiling data, whereas groups I and II miRNAs were not significantly correlated. With a cutoff of >0.1% of miRNA percent in the sample, half of the expressed known miRNAs overlapped between Group I and II.
Figure 1.
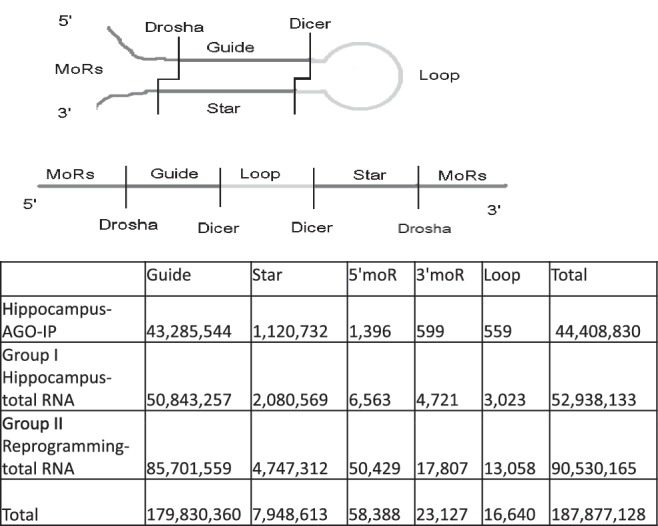
Total phased reads of miRNA/miRNA*, moRs and loops.
Abundance of 5p and 3p strands
Of those reads that perfectly matched miRNA precursors, 180 M reads corresponded to the more abundant strands and 8 M reads to the less abundant strands from each duplex. The preferred strand was considered the one with more reads. Strands were also named 5p or 3p without regard to the preferred strand. Mature miRNAs can be processed from either arm of the pre-miRNA hairpins, but in most cases, the discrimination of the preferred strand was very high. Although a bias toward preferential stabilization of the 5′-arm in mammalian species has been reported (35,36), our data show more equal strand stabilization (Supplementary Figure S3) as seen in Caenorhabditis elegans (37).
The products of the preferred strand were 25 times that of the non-preferred strand in Group I and 18 times in Group II cells. We calculated the strand ratio for the 416 miRNAs with more than 50 reads on both arms. Sixty-five miRNAs had preferred strand reads that were < 2-fold higher than the non-preferred strand including some highly expressed miRNAs such as miR-292, miR-126, let-7d and miR-22. A total of 200 miRNAs had >10-fold difference between the two strands of the hairpin structures. The expression of 5p and 3p strands did not correlate well (R2 = 0.093) and large variations existed among different miRNAs (Supplementary Figure S4).
In the AGO-IP samples, the preferred strand was 39 times as abundant as the opposite strand. The correlations of the preferred/non-preferred ratio in the hippocampus between the AGO-IP samples and the total small RNAs data (Supplementary Figure S5) indicated that most miRNAs had higher ratios in the AGO-IP samples. Thus preferred strands were relatively more enriched in the AGO-IP sample, a finding, which supports the preferred strand bias for entry to the RISC. Nevertheless, non-preferred strands were often highly represented and may either associate with AGO, or alternatively, even with relatively high reads, fail to enter the RISC. If they are captured by a RISC, they could play a physiological role. A total of 90% of the miRNAs, which aligned to miRBase version 16 had more than 10 reads from the non-preferred strand and 71% of them had more than 50 reads from the non-preferred strand. miR-7a, miR-9, miR-129, miR-136, miR-132 miR-126 and let-7d in the mouse hippocampus (Supplementary Table S2), and miR-24, miR-199, miR-130 and the miRNA cluster on chromosome 7 that includes miR-292, miR-293, miR-294 and miR-295 in Group II cells all had both strands highly represented (Supplementary Table S3).
Characteristically, miRNAs are differentially expressed among tissue types. Most miRNAs showed a consistent strand preference across the cell and tissue types. The ratio of levels of 5′- to 3′-arms was well correlated between groups I and II (Figure 2). However, some miRNAs switched the preferred strand when comparing the two sample groups (Table 1). Most strand switches occurred among those miRNAs with read counts that did not differ greatly between the arms in the two sets. For example, hippocampal miR-22 had 5′-read counts of 69 071 and 3′-read counts of 49 670; however, in Group II cells the reads were 41 172 and 60 029 respectively. On the other hand, major strand switching occurred in the case of miR-1-2, which had 8368 and 22 642 reads on 5′-and 3′-arms in hippocampus and the corresponding reads in Group II cells were 10 356 and 903 (Table 1).
Figure 2.

Correlation of 5p expression and 3p expression between Group I and II. The ratio of expressed 5′-strand and 3′-strand for each miRNA in each group were calculated and the log values plotted.
Table 1.
miRNAs that switched their dominant strands
| miRNAs | Group I, Hippocampus-total RNA |
Hippocampus-AGO-IP |
Group II, Reprogramming-total RNA |
||||||
|---|---|---|---|---|---|---|---|---|---|
| 5p | 3p | Log(5/3) | 5p | 3p | Log(5/3) | 5p | 3p | Log(5/3) | |
| mmu-mir-335 | 27 358 | 5796 | 2.24 | 3798 | 3179 | 0.26 | 17 249 | 17 808 | −0.05 |
| mmu-let-7d | 99 119 | 33 204 | 1.58 | 68 496 | 48 751 | 0.49 | 85 006 | 86 817 | −0.03 |
| mmu-mir-1981 | 2740 | 966 | 1.50 | 1507 | 415 | 1.86 | 1578 | 3734 | −1.24 |
| mmu-mir-1839 | 52 292 | 19 723 | 1.41 | 28 259 | 4664 | 2.60 | 17 913 | 22 600 | −0.34 |
| mmu-mir-22 | 49 670 | 19 396 | 1.36 | 31 206 | 23 627 | 0.40 | 41 172 | 60 029 | −0.54 |
| mmu-mir-411 | 32 728 | 19 146 | 0.77 | 33 580 | 12 712 | 1.40 | 30 959 | 32 499 | −0.07 |
| mmu-mir-154 | 34 822 | 21 917 | 0.67 | 56 741 | 27 785 | 1.03 | 14 577 | 36 736 | −1.33 |
| mmu-mir-582 | 39 319 | 25 954 | 0.60 | 24 241 | 3883 | 2.64 | 8735 | 9454 | −0.11 |
| mmu-mir-431 | 5283 | 6511 | −0.30 | 1923 | 3824 | −0.99 | 22 977 | 3513 | 2.71 |
| mmu-mir-1-2 | 8368 | 22 642 | −1.44 | 26 | 6377 | −7.94 | 10 356 | 903 | 3.52 |
| mmu-mir-490 | 1127 | 3575 | −1.67 | 955 | 2719 | −1.51 | 339 | 158 | 1.10 |
| mmu-mir-1943 | 37 | 134 | −1.86 | 21 | 164 | −2.97 | 2631 | 805 | 1.71 |
| mmu-mir-501 | 8920 | 4491 | 0.99 | 2373 | 2168 | 0.13 | 6874 | 10 638 | −0.63 |
moRs and miRNA loop sequences
For each miRNA in our samples we determined the reads for each of the five phased products of miRNA processing: 5′-and 3′-moRs, loops, 5′-and 3′-miRNA strands. The sum of the lengths of the 5′-and 3′-miRNA strand and the loop sharply peaked in the range 55–65 nt (Supplementary Figure S6) and corresponded to the length distribution of the sequences in miRBase. A total of 65 000 reads matched moRs sequences, and 20 000 reads matched loop sequences within miRNA precursors (Figure 1). Relative to total small RNA profiling samples, reads from the mature miRNAs dominated (Figure 1).
Reads of moRs were very common for most of the miRNAs (Figure 3, Supplementary Tables S2, S3, S4, Supplementary Datasets S1, S2 and S3). Although more highly expressed miRNAs tended to have higher loop and moR reads, their levels were not strictly correlated with the expression level of mature miRNAs. A total of 77% of known miRNAs had >10 moR reads. Relatively fewer moR reads were sequenced in Ago-IP samples, compared to data from total small RNA profiling (Figure 1). Reads of 5′-moR reads exceeded 3′-moRs by > 3-fold in Group I and 5-fold in Group II samples. 5′-moRs were also reported to be more abundant than 3′-moRs in Drosophila (30), and therefore moR processing may be a highly conserved feature of the miRNA biogenesis pathway. The levels of 5′-moRs and 3′-moRs did not correlate (R2 = 0.025) suggesting some degree of independence in their generation. In Group II cells, miR-27b, miR-503, miR-21, miR-24 and miR-16 had the most abundant 5′-moRs, while miR-294 and miR-30e had the most 3′-moRs. In the hippocampus, miR-134 and miR-503 had abundant 5′-moRs (Supplementary Table S2, Supplementary Datasets S1, S2 and S3). Generally, the dominant moRs aligned perfectly with the dominant isomiRs (e.g. for a 3′-miR, the last base of the dominant isomiR was the base preceding the first base of the dominant moR), thereby suggesting that Drosha products without terminal modifications predominate among the phased fragments (Figure 3, Supplementary Datasets S1, S2 and S3). These findings also suggest that Drosha generates the ends of the 5′-and 3′-moRs that are adjacent to the pre-miR stem.
Figure 3.

Phased read distributions of (A) miR-125b and (B) miR-191.
Although >90% of mouse miRNA precursors found in miRBase version 16 (31) had detectable loop regions, only 15% of the miRNAs had loop read numbers that exceeded 10. Therefore, loop sequences were the most unstable among all of the phased fragments. For comparably expressed mature miRNAs, the loop levels could differ greatly in different tissues. For example, some miRNAs, highly expressed in Group II had thousands of loop reads, e.g. such as miR-34a (2927 loop reads, 0.26% of the dominant strand), miR-106b (2359 loop reads, 5.34% of miRNAs from the dominant strand), miR-182 (2285 loop reads, 0.21% of the dominant strand) and miR-27a (2189 loop reads, 0.57% the dominant strand), whereas in the mouse hippocampus these miRs were also highly expressed, but had less than 10 loop reads (Supplementary Tables S2, S3, Supplementary Datasets S1 and S2). In mouse hippocampus, miRNAs with the most abundant loops were miR-138-2 (1018 loop reads, 0.06% of the dominant strand), let-7c-1 (745 loop reads, 0.21% of the dominant strand) and miR-219-2 (380 loop reads, 0.43% of the dominant strand).
Variation in canonical miRNA isomiRs
Deep sequencing revealed variation in the Drosha and Dicer cleavage sites, which generate pre-miRNAs and RNA duplexes. These products are termed isomiRs (28,38). Some highly expressed miRNA genes had over a hundred isomiRs. The dominant isomiR was considered the one with the most reads. IsomiRs in our data set that exceed the length of the dominant isomiR are unlikely to be explained by the addition of non-templated nucleotides that fortuitously match the genome sequence (39–41). IsomiRs that are shorter than the dominant isomiR may arise in part due to exonucleolytic cleavage, as occurs in plants (42), but not, as yet, observed in animals (43). The variation is unlikely due to degradation or sequencing error (44,45), because species that were shorter than the dominant isomiR were approximately as frequent as those that were longer (39) and their numbers exceeded the rate of sequencing error. When considering the most abundant isomiRs, they differed by only 1 or 2 nt at the 5′-or 3′-end of their sequences. Consistent with a previous proposal (46), the distribution of isomiRs in the cell is probably not random. However in our samples, the typical size distribution of isomiRs was nearly normal, especially for those highly expressed miRNAs (Figure 4A). When the isomiRs were grouped by only their 5′-ends or by their 3′-ends, they remained normally distributed (data not shown). This pattern was more obvious for highly expressed miRNAs. Our data showed that both strands exhibited higher 5′-fidelity, which agrees with previous data in several other species (30,36). Figure 4B shows the distributions of all isomiRs grouped by their 5′-ends and 3′-ends, rather than by the preferred strand. We further observed that both the 5′- and 3′-ends of the preferred strands showed a higher percentage of dominant isomiRs, which indicated less variation than occurs on the non-preferred strand (Figure 4B).
Figure 4.

Distributions of isomiRs. (A) miR-124 isomiRs. IsomiRs were listed according their 5′-ends, and for each group with the same 5′-end, up to five most abundant isomiR with different 3′-ends are shown. (B) The distributions of all isomiRs grouped either by their 5′-end or 3′-end for both strands. IsomiRs were grouped by their distance of 5′-end or 3′-end compared to the dominant ones (X-axis). At most 3 nt variations at either end were allowed.
Much of the isomiR variability can be explained by variability in Dicer and Drosha cleavage positions. Variation in the termini of isomiRs produced by Dicer and Drosha reflect the fidelity of Dicer and Drosha (Figure 5) quantified as the percent of a set of isomiRs represented by the dominant isomiR. We grouped all the isomiRs based on their locations on the precursor hairpins as isomiRs derived from either the 5′- or 3′-arms, and compared the sequence variation of the enzyme cleavage positions. Because variation differed greatly at the 5′-ends compared to the 3′-ends of isomiRs, in order to compare directly the fidelity of Drosha to Dicer, we set up comparisons for both ends produced by the two enzymes. We found that the dominant isomiRs were 95.2% of the total isomiRs at the 5′-end of the 5p strand (Drosha cleavage), compared to 91.3% the 5′-end of the 3p strand (Dicer1 cleavage) (Z-value = 1004.4 and P-value < 0.0001). The dominant isomiRs were 60.5% of the total isomiRs at the 3′-end of the 3p strand (Drosha cleavage) compared to 56.2% at the 3′-end of the 5p strand (Dicer1 cleavage) (Z-value = 592.0 and P-value < 0.0001). Overall, for both 5′- and 3′-ends of isomiRs, the variations on Drosha cleavage positions were less than Dicer (Figure 5), a finding, which implies a higher fidelity of Drosha than Dicer. As a result, more variation in the cleavage sites occurs near the loop than at the base of the stem in the pre-miRNA hairpins. Although Dicer functions as a precise ‘measuring stick’ from the Drosha cleavage site, several possible RNA folding structures with variation in the nucleotide at the terminus of the stem could serve as possible Dicer substrates. An earlier report in mouse suggested greater fidelity of Dicer (36); however, those authors restricted their analysis to cases in which Dicer produced the 5′-terminus of the miRNA and was determined by the percent of offset reads. A study in C. elegans (37) showed approximately equal fidelity of Dicer and Drosha after they removed a single miRNA (miR-58) with an abundance that greatly exceeded all the other miRNAs.
Figure 5.

End variations of isomiRs at Drosha and Dicer cleavage sites. Two comparisons were performed: one was based on variations at the 5′-end of isomiRs and the other was based on variation at the 3′-ends. Both comparisons showed Drosha had a higher chance to match the sequences with the dominant isomiRs.
Weighted average size of nucleotide variation
Although most miRNAs showed typical symmetrical distributions of isomiRs, the proportion of sub-dominant isomiRs could vary greatly for different miRNA loci. Alternative miRNA isomiRs expressed at sufficiently high levels could direct the repression of a distinct target set (36). Therefore, we identified miRNAs with relatively abundant secondary isomiRs and miRNAs with differentially expressed isomiRs in different samples. As shown previously (37,46), the extent of variation at the ends of isomiRs varied among different miRNAs. Some miRNAs had high variation at their sequence ends with nearly equal proportions of their isomiRs. For example, the dominant isomiR of miR-1982 was only 49.55% of the total reads and the secondary isomiR, which differed by 2 nt, accounted for 45.67% of total reads. To measure the isomiR variation, we calculated the weighted average size of nucleotide variation (WAZNV) according to the dominant sequences as:
where pi was the percentage of isomer i in total observed isomiRs and di was the relative distance of ends of isomiR i compared to the dominant one. WAZNV can be calculated based on either the 5′- or 3′-end for both the preferred and non-preferred strands. Because the miRNA targeting depends heavily on the 5′-end, we mainly focused on WAZNVs based on the 5′-end of miRNAs. WAZNV measured the variation among isomiRs. The top 10 sequences, whether preferred or non-preferred strands, with high isomiR variation based on all data sets are shown in Figure 6A. At the top of the list is miR-485-3p, which had shown significant variation in the literature (47–49). In our dataset, the dominant isomiR (isomiR I) was on the 3′-arm, which accounted 48.9% of miR-485-3p reads. The secondary and tertiary isomiRs accounted 28.1% (isomiR II) and 19.7% (isomiR III) of total reads, which shifted the mature sequence 2 and 3 nt in the 5′-direction. Although the isomiR distribution around the dominant isomiR is usually normal, some isomiR distributions shift the secondary and tertiary isomiRs 2 and 3 nt from the dominant isomiR (Figure 6B).
Figure 6.

miRNAs with high isomiR variation. (A) Distributions of preferred and non-preferred isomiRs with high variation grouped by their 5′-ends. These miRNAs have second isomiRs with significant abundance, which usually shifted 2 or 3 nt’s compared to the dominant isomiR. (B) The distributions of three principle isomiRs of miR-495-3p positioned on the hairpin structure of miR-495 precursors. The dominant isomiR switched in Group II cells.
By comparing isomiR variation between different samples, WAZNV can detect differentially expressed isomiRs. By defining the position of dominant isomiRs within the whole data set, we calculated the WAZNV of miRNAs in both Group I and II samples as well as the AGO-IP sample separately. Switching of dominant isomiRs was uncommon between hippocampus and Group II cells (Supplementary Datasets S1 and S2), and also very rare between AGO-IP and total small RNA samples. However, we identified several cases that miRNAs switched the dominant isomiRs between hippocampus and Groups II cells such as miR-485-3p, or had significant differences in isomiR distributions (Table 2 and Figure 7). In addition to miR-485-3p (Figure 7A), miR-543-5p (Figure 7B), miR-592-5p (Figure 7C) and mu-miR-137-3p (Figure 7D) also switched dominant isomiRs between Group I and II cells. miR-125b-2-3p (Figure 7E) was highly expressed in hippocampus, in which the dominant isomiR accounted for 78.9% of total reads; in contrast, the secondary isomiR, which shifted 2 nt in the 5′-direction compared to the dominant isomiR, accounted for 40.0% of the total 3261 reads of miR-125b-2-3p in Group II cells. A few cases showed large differences between AGO-IP and total small RNA samples. miR-690 had 1010 reads, 3162 reads and 2196 in AGO-IP, Group I and Group II cells respectively, but its dominant isomiRs in AGO-IP samples shifted 2 nt in the 3′-direction compared to the location of dominant isomiR in the other two groups which showed identical distributions of isomiRs (Figure 7F). The dominant isomiR of miR-34a-3p in AGO-IP samples shift 1 nt compared the dominant ones in the other two samples. These examples could indicate an isomiR preference of the AGO complex to maintains greater uniformity in the mature miRNA that is guided to the mRNA.
Table 2.
Top 10 miRNAs with large isomiR variation
| miRNAs | Group I, Hippocampus-total RNA |
Hippocampus-AGO-IP |
Group II, Reprogramming-total RNA |
|||
|---|---|---|---|---|---|---|
| WAZNV | Reads | WAZNV | Reads | WAZNV | Reads | |
| mmu-mir-485-3p | 0.66 | 13 022 | 0.40 | 9292 | 1.67 | 29 513 |
| mmu-mir-543-5p | 1.39 | 1371 | 1.34 | 909 | 0.45 | 2415 |
| mmu-mir-384-3p | 0.99 | 26 284 | 1.01 | 10 710 | 0.21 | 923 |
| mmu-mir-551b-3p | 0.1 | 93 143 | 0.01 | 52 706 | 0.87 | 15 963 |
| mmu-mir-10b-5p | 1 | 3050 | 0.19 | 325 | 0.30 | 30 7739 |
| mmu-mir-485-5p | 0.13 | 5314 | 0.35 | 655 | 0.74 | 623 |
| mmu-mir-345-3p | 0.91 | 1205 | 0.72 | 252 | 0.32 | 19 172 |
| mmu-mir-125b-2-3p | 0.5 | 10 323 | 0.38 | 4729 | 1.04 | 3261 |
| mmu-mir-592-5p | 0.41 | 4205 | 0.13 | 2062 | 0.80 | 1099 |
| mmu-mir-137-3p | 0.52 | 85 173 | 0.3′- | 52 369 | 0.90 | 1176 |
Figure 7.

Distributions of isomiRs in different groups of samples for miRNAs with isomiR variation. (A) miR-485-3p which switched the dominant isomiR in Group II samples. (B) miR-543-5p; (C) miR-592-5p; (D) miR-137-3p; (E) miR-125b-2-3p which did not switch the dominant isomiR, but the second isomiR increased in Group II; (F) miR-690-3p which showed the difference between AGO-IP and total small RNA samples.
Non-templated nucleotide addition
Sequence alterations of miRNAs can occur by addition of non-templated nucleotides to miRNA termini (38,40,49). Most miRNAs in our data set did not have non-templated nucleotide modifications. Of the total reads, 16.00%, 14.10% and 12.67% were extended by 1 nt in the Ago-IP, Group I, and Group II samples, respectively. Of the total reads, 7.81%, 6.85% and 3.18% were extended by 2 nt in the Ago-IP, Group I and Group II samples, respectively. The nucleotides most frequently added to murine miRNAs were U and A (Figure 8 and Table 3). However, in the Ago-IP sample from hippocampus the addition of C was most common; C also showed a small increase in the Group I samples compared to the Group II samples. Di-nucleotide extensions not infrequently consisted of two different nucleotides (Supplementary Table S5). Half of the miR-143-3p reads in our samples were extended similarly to the very high proportion of extended reads previously described for this miRNA (36). Among highly expressed miRNAs, 32% of the miR-124 reads were extended and 33% of the miR-24 reads were extended (Supplementary Table S6).
Figure 8.

Untemplated nucleotide addition rate at the 3′-ends of miRNA/miRNA*.
Table 3.
Non-templated nucleotide addition at the 3′-ends of miRNA/miRNA* between 5p and 3p
| Group I, Hippocampus- total RNA (%) | Hippocampus- AGO-IP (%) | Group II, Reprogramming- total RNA (%) | ||
|---|---|---|---|---|
| A | 5p | 5.84 | 4.59 | 6.02 |
| 3p | 3.83 | 2.42 | 4.33 | |
| C | 5p | 4.18 | 4.45 | 1.71 |
| 3p | 3.61 | 4.66 | 1.66 | |
| G | 5p | 1.10 | 1.31 | 0.83 |
| 3p | 1.56 | 2.40 | 0.79 | |
| U | 5p | 3.28 | 4.17 | 1.84 |
| 3p | 4.88 | 7.44 | 8.21 | |
Nucleotide addition can occur on the mature miRNA (50) or on the precursor species (43,51). For our entire data set nucleotide addition on the 5′-end of both the 5p (0.32%) and 3p (0.31%) strand was infrequent. The pattern of non-templated extension differs between Group I hippocampal tissue and Group II cultured cells undergoing re-programming (Figure 8). Group II cells also have a greater tendency toward 3′-mono-uridylation on the 3p arm relative to the 5p arm in Group II compared to Group I (Table 3, 4.88%/3.88% in Group I and 8.21%/1.84% in Group II). In general, most of the nucleotide additions occurred on the 3′-end with 19.85% on the 3p strand and 16.07% on the 5p strand. Whether the bias toward the 3′-end of the 3p strand represents nucleotide addition on the pre-miRNA or a bias to the 3p strand after Dicer cleavage cannot be discerned from these data. A further breakdown by the specific nucleotide added shows addition of A, U and C, but very low values of G as previously noted (46). The specific extended nucleotide at the 3′-ends shows the expected preference for mono-uridylation on the 3p arm, i.e. the extreme end of the precursor; however, the 5p arm has a bias toward the addition of adenine (Table 3). One possible reason for this asymmetry is a nucleotide specific tendency to extend the pre-miRNA versus the mature miRNA. There is a hint in the data that the choice of U or A may have an effect on promoting or hindering RISC entry. Comparing total miRNAs from the hippocampus (Group I) to miRNAs Ago IP’ed from the hippocampus there is a relative increase in mono U extension and a relative decrease in mono A extension in the Ago-IP fraction.
DISCUSSION
Several studies have used deep sequencing for detailed miRNA annotations in a range of model organisms including D. melanogaster (30), C. elegans (37) and mouse (29,36). By sampling diverse tissues and cells and sequencing at a great depth, several novel observations emerge here and other findings were confirmed. More than 90% of mouse miRNAs had more than 10 reads from both arms of the precursor, and therefore both strands could potentially play a biological role in target repression. Taking advantage of the tight association between miRNAs and AGO, we compared total small RNA (Group I) and a pan-Ago IP both from the mouse hippocampus to profile miRNAs which entered RISC. These data showed fewer non-preferred strands present in the RISC even in cases with high read counts. Nevertheless, not infrequently, the non-preferred strand also associated with AGO, and therefore possibly with the RISC. It seems likely that in some cases, a pool of pre-miRNAs sort their preferred and non-preferred strands into different RISCs without degradation of one of the strands. Although usually the preferred strand is readily recognizable by a many-fold enrichment, some miRNAs show quite similar read counts from both the preferred and non-preferred strands. 10% of miRNA loci showed a comparable expression of both strands with <2-fold difference.
Data from several large-scale miRNA sequencing studies have demonstrated that the arm from which the preferred miRNA is processed can switch in different tissues and at different developmental times (36,38,52–55). Among a variety of human samples, Cloonan et al. (56) found that 12.9% of miRNAs switched the dominant strand in at least one tissue. Utilization of opposite strands for target binding would result in significant changes in the miRNA targeting. However, often miRNA loci with comparable expression levels of both arms show switching of the preferred strand in different tissues, and in these cases both strands may be used in different proportions rather than a more dramatic switch in the target field if one or the other strand were used exclusively. When strand choice is closer to equilibrium different tissues appear to tilt the balance toward one strand or the other. The use of both strands among a sizable fraction of miRNAs is consistent with the demonstration of selection at the 5′-end of both strands (57). Other reports proposed that star sequences would not be excluded from functional complexes because they are present in substantial levels or sorted to different Argonaute complexes, in which the dominant arm directs translational repression (by means of Ago1) and the miR* sequence directs transcriptional degradation by means of Ago2 (25–27,36,38,58–63). Star sequences, such as miRNAs like miR-19* (64) and miR-223* (65), can have a detectable impact on target networks in Drosophila and mammals (25–27,54,66).
Although isomiRs are frequently observed, their origin has been attributed to sequencing or alignment artifacts (67–69). However, non-random features of their distributions suggest otherwise (29,70) and recently it was suggested that isomiRs with different repertoires of mRNA targets would distribute the ‘off-target’ hits while still targeting core biological networks (56). Among the non-random features of isomiRs is variation in the choice of the dominant isomiR. Furthermore, a potentially disruptive shift of the dominant isomiR by 2 nt (36) was observed. In particular, variation at 5′-end of the mature isomiR will presumably have greater effects on targets (30,36). Secondary isomiRs could be explained by differential processing of the two paralogous hairpins (36,38), or alternative Drosha and/or Dicer1 cleavages (30). Finally, miRNAs can be modified by the non-templated addition of nucleotides almost always at the 3′-end. Reports in the literature vary on the prevalence of non-templated nucleotide addition among miRNAs (36,71,72) with patterns described in mammals (36,49), worms and flies (38,40). A bias toward mono-uridylation and away from mono-adenylation among miRNAs in Ago IP fractions compared to the total small RNA fraction suggests that these two additions may serve to promote or hinder association with the RISC. The methods here preclude the discovery of long 3′-terminus poly(U)-tailing and those reports that do use deep sequencing to annotate miRNAs also do not report extensive poly(U)-tailing (46).
The origin of moRs is unclear. They may arise by exonucleolytic activity on precursor transcripts (38,73) or double-stranded cleavage of extended hairpin regions on pri-miRNA transcripts via secondary DROSHA processing (30,74). However, the broad length distributions of moRs derived from 5′-and 3′-regions flanking pre-miRNAs are inconsistent with a DROSHA-based cleavage mechanism (72). However, our data show that the ends of both the dominant 5′-and the 3′-moRs fit very well with the dominant termini of the 3p and 5p strands arising from Drosha cleavage. It therefore seems likely that at least one end of the moR sequences is a Drosha product.
ACCESSION NUMBERS
The sequencing data reported in this study can be obtained from the Sequence Read Archive (SRA) at NCBI under accession number SRP010127, SRP010168, SRP010169 and SRP010170.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–6, Supplementary Figures 1–6 and Supplemental Datasets 1–3.
FUNDING
California Institute for Regenerative Medicine (CIRM) (to K.S.K.); National Institutes of Health (to T.M.R.); a postdoctoral fellowship from California Institute for Regenerative Medicine to E.J.L. Funding for open accesss charge: California Institute for Regenerative Medicine and the NIH.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to the Kosik lab members for helpful comments.
REFERENCES
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 3.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 8.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 10.Okada C, Yamashita E, Lee SJ, Shibata S, Katahira J, Nakagawa A, Yoneda Y, Tsukihara T. A high-resolution structure of the pre-microRNA nuclear export machinery. Science. 2009;326:1275–1279. doi: 10.1126/science.1178705. [DOI] [PubMed] [Google Scholar]
- 11.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004;10:185–191. doi: 10.1261/rna.5167604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 14.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 15.Leuschner PJ, Ameres SL, Kueng S, Martinez J. Cleavage of the siRNA passenger strand during RISC assembly in human cells. EMBO Rep. 2006;7:314–320. doi: 10.1038/sj.embor.7400637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 17.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007;131:1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 18.Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 21.Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 22.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 23.Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Takamatsu K, Chuma S, Kojima-Kita K, Shiromoto Y, Asada N, Toyoda A, Fujiyama A, et al. MVH in piRNA processing and gene silencing of retrotransposons. Genes Dev. 2010;24:887–892. doi: 10.1101/gad.1902110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jazdzewski K, Liyanarachchi S, Swierniak M, Pachucki J, Ringel MD, Jarzab B, de la Chapelle A. Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proc. Natl Acad. Sci. USA. 2009;106:1502–1505. doi: 10.1073/pnas.0812591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czech B, Zhou R, Erlich Y, Brennecke J, Binari R, Villalta C, Gordon A, Perrimon N, Hannon GJ. Hierarchical rules for Argonaute loading in Drosophila. Mol. Cell. 2009;36:445–456. doi: 10.1016/j.molcel.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okamura K, Liu N, Lai EC. Distinct mechanisms for microRNA strand selection by Drosophila Argonautes. Mol. Cell. 2009;36:431–444. doi: 10.1016/j.molcel.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghildiyal M, Xu J, Seitz H, Weng Z, Zamore PD. Sorting of Drosophila small silencing RNAs partitions microRNA* strands into the RNA interference pathway. RNA. 2010;16:43–56. doi: 10.1261/rna.1972910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee LW, Zhang S, Etheridge A, Ma L, Martin D, Galas D, Wang K. Complexity of the microRNA repertoire revealed by next-generation sequencing. RNA. 2010;16:2170–2180. doi: 10.1261/rna.2225110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res. 2011;21:203–215. doi: 10.1101/gr.116657.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson PT, De Planell-Saguer M, Lamprinaki S, Kiriakidou M, Zhang P, O'Doherty U, Mourelatos Z. A novel monoclonal antibody against human Argonaute proteins reveals unexpected characteristics of miRNAs in human blood cells. RNA. 2007;13:1787–1792. doi: 10.1261/rna.646007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Yang CS, Nakashima K, Rana TM. Small RNA-mediated regulation of iPS cell generation. EMBO J. 2011;30:823–834. doi: 10.1038/emboj.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 35.Hu HY, Yan Z, Xu Y, Hu H, Menzel C, Zhou YH, Chen W, Khaitovich P. Sequence features associated with microRNA strand selection in humans and flies. BMC Genomics. 2009;10:413. doi: 10.1186/1471-2164-10-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warf MB, Johnson WE, Bass BL. Improved annotation of C. elegans microRNAs by deep sequencing reveals structures associated with processing by Drosha and Dicer. RNA. 2011;17:563–577. doi: 10.1261/rna.2432311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seitz H, Ghildiyal M, Zamore PD. Argonaute loading improves the 5' precision of both MicroRNAs and their miRNA* strands in flies. Curr. Biol. 2008;18:147–151. doi: 10.1016/j.cub.2007.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, Ge H, Bartel DP. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 41.Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, Manjunath N. miRNA profiling of naive, effector and memory CD8 T cells. PLoS One. 2007;2:e1020. doi: 10.1371/journal.pone.0001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramachandran V, Chen X. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science. 2008;321:1490–1492. doi: 10.1126/science.1163728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kai ZS, Pasquinelli AE. MicroRNA assassins: factors that regulate the disappearance of miRNAs. Nat. Struct. Mol. Biol. 2010;17:5–10. doi: 10.1038/nsmb.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pantano L, Estivill X, Marti E. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 2010;38:e34. doi: 10.1093/nar/gkp1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009;37:2461–2470. doi: 10.1093/nar/gkp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman MA, Mani V, Hammond SM. Deep sequencing of microRNA precursors reveals extensive 3' end modification. RNA. 2011;17:1795–1803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sewer A, Paul N, Landgraf P, Aravin A, Pfeffer S, Brownstein MJ, Tuschl T, van Nimwegen E, Zavolan M. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinformatics. 2005;6:267. doi: 10.1186/1471-2105-6-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mineno J, Okamoto S, Ando T, Sato M, Chono H, Izu H, Takayama M, Asada K, Mirochnitchenko O, Inouye M, et al. The expression profile of microRNAs in mouse embryos. Nucleic Acids Res. 2006;34:1765–1771. doi: 10.1093/nar/gkl096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, Zamore PD. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YK, Heo I, Kim VN. Modifications of small RNAs and their associated proteins. Cell. 2010;143:703–709. doi: 10.1016/j.cell.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 52.Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature. 2008;455:1193–1197. doi: 10.1038/nature07415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griffiths-Jones S, Hui JH, Marco A, Ronshaugen M. MicroRNA evolution by arm switching. EMBO Rep. 2011;12:172–177. doi: 10.1038/embor.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ro S, Park C, Young D, Sanders KM, Yan W. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007;35:5944–5953. doi: 10.1093/nar/gkm641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Wit E, Linsen SE, Cuppen E, Berezikov E. Repertoire and evolution of miRNA genes in four divergent nematode species. Genome Res. 2009;19:2064–2074. doi: 10.1101/gr.093781.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cloonan N, Wani S, Xu Q, Gu J, Lea K, Heater S, Barbacioru C, Steptoe AL, Martin HC, Nourbakhsh E, et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011;12:R126. doi: 10.1186/gb-2011-12-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seitz H, Ghildiyal M, Zamore PD. Argonaute loading improves the 5' precision of both MicroRNAs and their miRNA* strands in flies. Curr. Biol. 2008;18:147–151. doi: 10.1016/j.cub.2007.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, Siomi MC. Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc. Natl Acad. Sci. USA. 2008;105:7964–7969. doi: 10.1073/pnas.0800334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, Dus M, Perrimon N, Kellis M, Wohlschlegel JA, Sachidanandam R, et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The regulatory activity of microRNA* species has substantial influence on microRNA and 3' UTR evolution. Nat. Struct. Mol. Biol. 2008;15:354–363. doi: 10.1038/nsmb.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goff LA, Davila J, Swerdel MR, Moore JC, Cohen RI, Wu H, Sun YE, Hart RP. Ago2 immunoprecipitation identifies predicted microRNAs in human embryonic stem cells and neural precursors. PLoS One. 2009;4:e7192. doi: 10.1371/journal.pone.0007192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat. Rev. Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Suzuki H, Hayashizaki Y, Daub CO. Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol. 2011;8:158–177. doi: 10.4161/rna.8.1.14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC. Widespread regulatory activity of vertebrate microRNA* species. RNA. 2011;17:312–326. doi: 10.1261/rna.2537911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuchenbauer F, Mah SM, Heuser M, McPherson A, Rüschmann J, Rouhi A, Berg T, Bullinger L, Argiropoulos B, Morin RD, et al. Comprehensive analysis of mammalian miRNA* species and their role in myeloid cells. Blood. 2011;118:3350–3358. doi: 10.1182/blood-2010-10-312454. [DOI] [PubMed] [Google Scholar]
- 66.Schulte JH, Marschall T, Martin M, Rosenstiel P, Mestdagh P, Schlierf S, Thor T, Vandesompele J, Eggert A, Schreiber S, et al. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res. 2010;38:5919–5928. doi: 10.1093/nar/gkq342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim J, Cho IS, Hong JS, Choi YK, Kim H, Lee YS. Identification and characterization of new microRNAs from pig. Mamm. Genome. 2008;19:570–580. doi: 10.1007/s00335-008-9111-3. [DOI] [PubMed] [Google Scholar]
- 68.Reese TA, Xia J, Johnson LS, Zhou X, Zhang W, Virgin HW. Identification of novel microRNA-like molecules generated from herpesvirus and host tRNA transcripts. J. Virol. 2010;84:10344–10353. doi: 10.1128/JVI.00707-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Hoon MJ, Taft RJ, Hashimoto T, Kanamori-Katayama M, Kawaji H, Kawano M, Kishima M, Lassmann T, Faulkner GJ, Mattick JS, et al. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res. 2010;20:257–264. doi: 10.1101/gr.095273.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fernandez-Valverde SL, Taft RJ, Mattick JS. Dynamic isomiR regulation in Drosophila development. RNA. 2010;16:1881–1888. doi: 10.1261/rna.2379610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo L, Yang Q, Lu J, Li H, Ge Q, Gu W, Bai Y, Lu Z. A comprehensive survey of miRNA repertoire and 3' addition events in the placentas of patients with pre-eclampsia from high-throughput sequencing. PLoS One. 2011;6:e21072. doi: 10.1371/journal.pone.0021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burroughs AM, Kawano M, Ando Y, Daub CO, Hayashizaki Y. pre-miRNA profiles obtained through application of locked nucleic acids and deep sequencing reveals complex 5'/3' arm variation including concomitant cleavage and polyuridylation patterns. Nucleic Acids Res. 2012;40:1424–1437. doi: 10.1093/nar/gkr903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAs arises from pre-miRNA-proximal regions in a simple chordate. Nat. Struct. Mol. Biol. 2009;16:183–189. doi: 10.1038/nsmb.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.