


Abstract
Replicative holoenzymes exhibit rapid and processive primer extension DNA synthesis, but inefficient strand displacement DNA synthesis. We investigated the bacteriophage T4 and T7 holoenzymes primer extension activity and strand displacement activity on a DNA hairpin substrate manipulated by a magnetic trap. Holoenzyme primer extension activity is moderately hindered by the applied force. In contrast, the strand displacement activity is strongly stimulated by the applied force; DNA polymerization is favoured at high force, while a processive exonuclease activity is triggered at low force. We propose that the DNA fork upstream of the holoenzyme generates a regression pressure which inhibits the polymerization-driven forward motion of the holoenzyme. The inhibition is generated by the distortion of the template strand within the polymerization active site thereby shifting the equilibrium to a DNA-protein exonuclease conformation. We conclude that stalling of the holoenzyme induced by the fork regression pressure is the basis for the inefficient strand displacement synthesis characteristic of replicative polymerases. The resulting processive exonuclease activity may be relevant in replisome disassembly to reset a stalled replication fork to a symmetrical situation. Our findings offer interesting applications for single-molecule DNA sequencing.
INTRODUCTION
Replicative DNA polymerases are responsible for faithfully copying genomic DNA in both prokaryotic and eukaryotic systems. They polymerize new DNA strands by adding nucleotides to the 3′ terminus of the primer strand that are complementary to the original DNA template strand. DNA replication must be extremely accurate to avoid deleterious mutations in the genetic information. Replicative polymerases typically have a 3′ to 5′ exonuclease or proofreading activity, which increases the fidelity of DNA replication. If an incorrect nucleotide is incorporated by the polymerase, the faulty primer strand is transferred from the polymerization (pol) site to the exonuclease (exo) site where the erroneous nucleotide is excised. After hydrolysis at the exo site, the trimmed primer strand is transferred back to the pol site and DNA synthesis is continued.
DNA polymerase activity can be tested in two different DNA-polymerase configurations. In a primer extension assay, DNA synthesis is monitored while the polymerase extends a primer hybridized on a single-stranded (ss) DNA molecule. In a strand displacement assay, a DNA fork is used as the substrate requiring the polymerase to open the double-stranded (ds) DNA in order to extend a primer. While many polymerases exhibit rapid and processive primer extension activity, inefficient strand displacement activity seems to be a general feature of replicative polymerases (1–4). As a consequence, replicative helicases are required to open the duplex DNA and facilitate the advancement of the leading-strand polymerase in the context of a replisome (5). Mechanistic insights on the coupling between unwinding and replication for the T4 helicase-holoenzyme system are given in the companion article (6).
Bacteriophages have been widely used as model systems in the study of DNA replication (5). The replicative polymerase of bacteriophage T4 is the gp43 polymerase belonging to polymerase family B (7). In isolation, the T4 gp43 polymerase is not processive; processivity is conferred through loading of the gp45 clamp protein by the gp44/62 clamp-loader complex to form the holoenzyme (8,9). The bacteriophage T7 gp5 polymerase belongs to polymerase family A and together with Escherichia coli thioredoxin is responsible for processive replication of the T7 genome (10). Although T4 gp43 and T7 gp5 belong to different polymerase families, they are structurally similar resembling a right hand with the active polymerization site in the palm, the dNTP-binding region in the fingers and 3′ to 5′ exonuclease domains. Previous ensemble studies with the T4 holoenzyme have reported efficient primer extension activity and very weak strand displacement activity (4). While single-molecule data have been obtained for the T7 holoenzyme acting to extend primers (11,12), a detailed analysis of the strand displacement activity of either holoenzyme is still missing.
In this work we employed magnetic tweezers manipulation to pull on the extremities of a DNA hairpin used as a DNA template and investigate the mechanism of DNA synthesis by the T4 and T7 holoenzymes, both in primer extension (open hairpin) and strand displacement (partially closed hairpin) configurations. We found that depending on the holoenzyme activity (primer extension versus strand displacement) the applied tension controlled the partitioning of a single DNA-holoenzyme complex between the polymerization and exonuclease conformations differently. At low applied mechanical forces, primer extension DNA synthesis preceded at the maximum rate, whereas strand displacement DNA synthesis was completely inhibited leading to a processive exonuclease activity. We propose that the DNA fork in front of the holoenzyme generates a regression pressure preventing the polymerization-driven forward motion of the holoenzyme, thereby shifting the equilibrium to a DNA–protein exonuclease conformation. The polymerase stalling induced by the holoenzyme’s inability to efficiently maintain the DNA–protein polymerase conformation explains the lack of strand displacement DNA synthesis by most replicative polymerases. Our findings have led to the development of a cyclic polymerase assay (CPA) that is the basis for a single-molecule version of the Sanger sequencing method (13).
MATERIALS AND METHODS
Proteins
The T4 proteins, wild-type (wt) and exonuclease-deficient gp43 polymerase (14), gp45 clamp (15) and gp44/62 clamp-loader complex (16), were purified as previously described. The T7 DNA polymerase (T7 gp5 in a 1:1 complex with thioredoxin) was purchased from New England Biolabs.
DNA hairpin substrate
The 1.2 Kbp long DNA hairpin (LH) substrate was constructed as previously described (17). Similarly, a 100 bp short DNA hairpin (SH) substrate containing three locked nucleic acid (LNA) nucleotides to inhibit the polymerization reaction of the holoenzymes was constructed. Briefly, a fork structure formed by two partially annealed oligos (A-1 was 5′-biotinylated to allow for attachment to the magnetic bead and A-3) and a short hairpin oligo (C) were annealed and ligated to the compatible ends of an 80 bp DNA fragment formed by annealed oligos (B-1 and B-2); oligo sequences are given in Supplementary Table S1. The digoxigenin label was incorporated by annealing a primer (oligo A-2) to the template strand and filling in the overhang with Klenow fragment (3′ → 5′ exo-) (New England Biolabs) in the presence of dUTP-digoxigenin (Roche). The hairpin products were purified with NucleoSpin Extract II Kits (Clontech).
The mechanical stability of the DNA hairpin constructs was characterized by measuring the extension of the substrate as a function of the pulling force along a force-cycle in which the force is first increased and then relaxed (Supplementary Figure S1). We observed stably folded hairpin substrates below 14 pN and mechanical unfolding of the hairpins above ∼16 pN.
Single-molecule assay
We used a PicoTwist magnetic tweezers instrument (www.picotwist.com) to manipulate individual DNA hairpin molecules tethered between a glass surface at the 3′ end and a magnetic bead at the 5′ end. The glass surface was treated with anti-digoxigenin antibody (Roche) and passivated with bovine serum albumin. Streptavidin-coated Dynal magnetic beads (Invitrogen) were either 1 µm or 2.8 µm in diameter. T4 holoenzyme (10 nM polymerase, clamp loader, and clamp trimer) or T7 polymerase (100 units/mL) was flowed into the chamber diluted in the reaction buffer (25 mM TrisOAc pH 7.5, 150 mM KOAc, 10 mM Mg(OAc)2, 1 mM dithiothreitol, 0–100 μM dNTPs and 2.5 mM ATP only for the T4 holoenzyme) at 37°C.
The DNA hairpins were manipulated by capturing the bead in a magnetic trap generated by a pair of permanent magnets. The applied force was controlled by varying the distance of the magnets from the sample. Video-microscopy was used to track the position of the magnetic bead in three dimensions with nanometre resolution at 30 Hz, from which the extension of the DNA molecule and the strength of the stretching force was deduced (18). A calibration curve of the applied force versus the vertical position of the magnets was used to exert forces with 10% error on the DNA molecules.
Single-molecule data analysis
Raw data, corresponding to the real-time evolution of the DNA extension in nm, was converted into the number of base pairs synthesized using a calibration factor determined from the elastic properties of ssDNA and dsDNA (Supplementary Figure S2). Instantaneous enzymatic rates were obtained from a linear fit to the traces filtered with a third-order Savitzky-Golay filter over a sliding time window of varying size depending on the applied force. Separate velocity distributions for primer extension activity and strand displacement activity were determined from the histogram of the instantaneous rate measured during the respective region of each data trace.
For each force, the primer extension velocity distribution was fit to a sum of two Gaussians. The mean of the Gaussian functions centred at V > 0 and V = 0 represented the primer extension rate and enzyme pausing, respectively. The primer extension rate 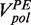 was computed as the average of the instantaneous rates measured during the primer extension mode excluding pausing.
was computed as the average of the instantaneous rates measured during the primer extension mode excluding pausing.
Likewise, the strand displacement velocity distribution was fit to a sum of three Gaussians. The mean of the Gaussian functions centred at V > 0, V = 0 and V < 0 represented the synthesis rate 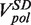 , enzyme pausing and the degradation rate
, enzyme pausing and the degradation rate 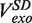 , respectively. The mean strand displacement rate,
, respectively. The mean strand displacement rate, 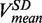 , was computed as the average of the instantaneous rates including polymerization, exonuclease and pausing behaviour. Pauses were detected as data segments satisfying
, was computed as the average of the instantaneous rates including polymerization, exonuclease and pausing behaviour. Pauses were detected as data segments satisfying 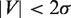 , where
, where  was the width of the Gaussian centred at V = 0. Data segments between pauses were identified as phases of synthesis (V > 0) or degradation (V < 0). The segment length associated with pausing, synthesis and degradation were used to compute the distribution of the residence pause times and the transition times for switching between pol and exo, as well as, the mean lifetime of each state used to estimate the kinetic rates of the primer transfer reaction.
was the width of the Gaussian centred at V = 0. Data segments between pauses were identified as phases of synthesis (V > 0) or degradation (V < 0). The segment length associated with pausing, synthesis and degradation were used to compute the distribution of the residence pause times and the transition times for switching between pol and exo, as well as, the mean lifetime of each state used to estimate the kinetic rates of the primer transfer reaction.
The occupancy of the paused and moving states of the holoenzyme was defined as the fraction of time the polymerase spent in that state. The occupancy of each state was calculated as the area under the Gaussian peak for that state (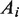 ) divided by the total area of the velocity distribution (
) divided by the total area of the velocity distribution (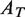 ); i.e.
); i.e. 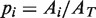 . Along the elongation branch, the occupancy of the pause state i (I or ip) was computed as:
. Along the elongation branch, the occupancy of the pause state i (I or ip) was computed as: 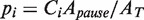 , where
, where 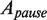 corresponds to area under the Gaussian peak centered at V = 0 and
corresponds to area under the Gaussian peak centered at V = 0 and 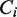 stands for the weight of the exponential function associated with the distribution of the residence time of the pause state i.
stands for the weight of the exponential function associated with the distribution of the residence time of the pause state i.
RESULTS
Observation of strand displacement synthesis and primer extension by single holoenzymes
To investigate the enzyme activity of individual T4 or T7 holoenzymes, we used magnetic tweezers to apply mechanical tension to DNA hairpin substrates attached between magnetic beads and a glass surface (Figure 1A and Supplementary Figure S1B) and monitored the end-to-end distance change as the DNA hairpin was opened and replicated to dsDNA. Increasing the applied mechanical force served as a means to either assist the holoenzyme in the strand displacement configuration by destabilizing the upstream DNA fork or hinder its advancement in the primer extension configuration by strongly stretching the ssDNA template.
Figure 1.

Observation of strand displacement synthesis and primer extension by single holoenzymes. (A) Schematic representation of the experimental set-up. A DNA hairpin substrate was tethered between the glass surface and a magnetic bead held in a magnetic tweezers. A holoenzyme loaded at the 3′ end of the primer may display strand displacement synthesis activity (light green), exonuclease activity (yellow) or primer extension activity (dark green). Experimental traces corresponding to wt T4 holoenzyme activity (B), exo-T4 holoenzyme activity (C) and wt T7 holoenzyme activity (D) are shown. The total change in extension observed in the two phases correspond to the polymerization of 1200 nt. The right axis shows the extension of the replicated substrate as computed in Supplementary Figure S2.
Due to the hairpin design of our DNA substrates, we were able to observe the DNA holoenzymes operating in both strand displacement and primer extension modes. During strand displacement synthesis, the enzyme needed to open one base pair of the DNA hairpin in order to incorporate one nucleotide resulting in a large increase in molecular extension, ∼0.8 nm for each nucleotide incorporated at 10 pN of applied force (Supplementary Figure S2). Once the enzyme synthesized through the hairpin loop to completely open it (or the hairpin substrate was mechanically opened with an applied force > 16 pN), the holoenzyme operated in the primer extension mode converting the ssDNA template into its dsDNA counterpart. This conversion resulted in a smaller positive (at F < 6 pN) or negative (at F > 6 pN) change in molecular extension depending on the applied force (11,12). Thus at F = 10 pN the molecular extension decreased by ∼0.1 nm per nucleotide incorporated (Supplementary Figure S2). The number of incorporated nucleotides was obtained by dividing the observed change in DNA extension by the expected change in DNA extension resulting from the incorporation of a single nucleotide.
We have investigated the activity of three different replicative polymerase units on a 1.2 Kbp DNA hairpin (LH) including the wt T4 holoenzyme (gp43 polymerase and gp45 trimeric clamp), a mutant T4 holoenzyme (gp43exo polymerase and gp45 trimeric clamp), which lacks the exonuclease activity, and the wt T7 holoenzyme (gp5 polymerase and E. coli thioredoxin). The primer extension activity for each holoenzyme proceeded at a relatively constant speed with some pausing that depended on the particular holoenzyme and the applied force (Figure 1B–D). On the other hand, the strand displacement activity for each holoenzyme varied. The strand displacement activity of wt T4 and T7 holoenzymes displayed periods of DNA synthesis (an increase in molecular extension) and DNA degradation due to exonuclease activity (a decrease in molecular extension) with intermittent periods of enzyme inactivity or pausing (Figure 1B and D). The strand displacement activity of the mutant T4 holoenzyme demonstrated only periods of DNA synthesis and enzyme pausing, as expected for an enzyme lacking exonuclease activity (Figure 1C).
Applied force hinders the primer extension activity of T4 and T7 holoenzymes
At moderate forces (<13 pN) replication of the second part of the hairpin (after the apex of the hairpin’s loop) proceeded almost continuously and only short sporadic pausing was observed for both the T4 and T7 holoenzymes (Supplementary Figures S3 and S4). At larger forces, pausing became more predominant. The primer extension rate 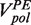 was determined from the slope of the data traces displaying primer extension activity without pauses. The primer extension rate was sensitive to the concentration of dNTPs and to the applied force (Supplementary Figures S3 and S4). At low applied force and saturating dNTPs concentration, primer extension by the T4 and T7 holoenzymes reached a maximum velocity of ∼300 nt/s and ∼600 nt/s, respectively, coinciding with their reported average velocities in bulk assays at 37°C (19,20). As the applied force increased, the primer extension rate
was determined from the slope of the data traces displaying primer extension activity without pauses. The primer extension rate was sensitive to the concentration of dNTPs and to the applied force (Supplementary Figures S3 and S4). At low applied force and saturating dNTPs concentration, primer extension by the T4 and T7 holoenzymes reached a maximum velocity of ∼300 nt/s and ∼600 nt/s, respectively, coinciding with their reported average velocities in bulk assays at 37°C (19,20). As the applied force increased, the primer extension rate 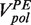 decreased, an effect generated by the difference in the elastic properties between ssDNA and dsDNA. The dependence of the primer extension activity on the applied force can be used to estimate the polymerase step size [(11,12) and Supplementary Materials]. Our results for both the T4 and T7 holoenzymes are best described by an enzyme with a step size of one nucleotide (Supplementary Figures S3 and S4), as expected for a polymerase incorporating one nucleotide at a time.
decreased, an effect generated by the difference in the elastic properties between ssDNA and dsDNA. The dependence of the primer extension activity on the applied force can be used to estimate the polymerase step size [(11,12) and Supplementary Materials]. Our results for both the T4 and T7 holoenzymes are best described by an enzyme with a step size of one nucleotide (Supplementary Figures S3 and S4), as expected for a polymerase incorporating one nucleotide at a time.
Strand displacement synthesis by T4 holoenzyme includes DNA synthesis, pausing and DNA degradation
To investigate the strand displacement activity of the T4 holoenzyme, we first generated a partially replicated substrate by applying a large force (Fopen ∼ 16 pN) to open the hairpin and allow initiation of DNA replication in the primer extension mode. Next, we decreased the force to a desired value (Ftest) and observed the behaviour of the T4 holoenzyme (Figure 2A). The generation of the partially replicated substrate was needed for creating a situation where both DNA synthesis and degradation could occur. Depending on the value of Ftest, the strand displacement activity of the holoenzyme changed dramatically (Figure 2B). If the assisting force was substantial (Ftest > 10 pN), the holoenzyme replicated DNA with a rate close to the rate of primer extension. Upon decreasing Ftest to ∼9 pN, pausing became the predominant behaviour observed. An even further decrease in the assisting force (Ftest < 8 pN) triggered a processive exonuclease activity. The holoenzyme stopped degrading the primer strand when the non-palindromic sequence of the DNA molecule was reached, i.e. when the hairpin had completely rehybridized. The holoenzyme switched back to synthesizing DNA when the assisting force was increased.
Figure 2.

Strand displacement activity of the wt T4 holoenzyme. (A) Schematic representation of the experimental protocol. Primer synthesis was initiated with the hairpin open at Fopen = 16 pN; the force was then lowered to various Ftest values and strand displacement activity was observed. (B) Traces of strand displacement activity recorded at Ftest values of 13 pN (blue), 11 pN (red), 8.5 pN (green) and 5 pN (magenta). Bars show the extension change corresponding to the synthesis or degradation of 100 nt. (C) Velocity distributions of instantaneous enzyme rates measured during strand displacement activity at different forces (colours as in panel B). The number of molecules (Nmol) analysed was 9, 14, 21 and 15 for 13, 11, 8.5 and 5 pN cases resulting in 82, 131, 115 and 224 number of enzymatic traces (N), respectively. (D) The mean synthesis rate 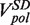 (light green), mean degradation rate
(light green), mean degradation rate 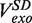 (orange) and mean strand displacement rate
(orange) and mean strand displacement rate 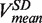 (purple) measured for the wt T4 holoenzyme shown as a function of the applied force. Error bars are the s.e.m. The number of molecules and enzymatic traces analysed varies between Nmol = 9–27 and N = 75–253, respectively, depending on the condition.
(purple) measured for the wt T4 holoenzyme shown as a function of the applied force. Error bars are the s.e.m. The number of molecules and enzymatic traces analysed varies between Nmol = 9–27 and N = 75–253, respectively, depending on the condition.
The holoenzyme displayed numerous pauses at every applied force. Duration of the pausing events increased with decreasing holoenzyme concentration (<1 nM), suggesting that holoenzyme dissociation could occur during strand displacement (Supplementary Figure S5). However, at protein concentrations above 1 nM, the duration of the pausing events did not change. Moreover, protein binding occurs very fast at the concentrations used in our single-molecule assays (10 nM), demonstrating that the holoenzyme unbinding/rebinding dynamics were sufficiently fast and did not cause the pauses. Control experiments with polymerase alone showed constant pausing, thus eliminating the possibility that polymerase and not the holoenzyme was the active species (data not shown). We thus conclude that the observed pausing events represent holoenzyme–DNA complexes in a transient inactive conformation.
At each force, a histogram was constructed of the instantaneous rates measured from the slope of the recorded traces of strand displacement activity. Three different phases, synthesis, degradation and pausing, could easily be identified in the velocity distributions (Figure 2C). The mean synthesis rate 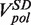 depended on the applied force reaching ∼200 nt/s at high forces, whereas the mean degradation rate
depended on the applied force reaching ∼200 nt/s at high forces, whereas the mean degradation rate 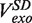 was approximately −100 nt/s and independent of the applied force, leading to an overall strand displacement rate
was approximately −100 nt/s and independent of the applied force, leading to an overall strand displacement rate 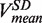 , which included synthesis, degradation and pausing behaviour, that was also force dependent (Figure 2D). The ratio between the three phases was strongly dependent on the applied force, indicating that the force significantly altered the equilibrium between the holoenzyme–DNA conformations responsible for DNA synthesis, DNA degradation and pausing.
, which included synthesis, degradation and pausing behaviour, that was also force dependent (Figure 2D). The ratio between the three phases was strongly dependent on the applied force, indicating that the force significantly altered the equilibrium between the holoenzyme–DNA conformations responsible for DNA synthesis, DNA degradation and pausing.
Minimal model for the T4 holoenzyme strand displacement activity
Insight into a model for the T4 holoenzyme strand displacement activity may be gained from an investigation of the pauses associated with inactive holoenzyme conformations. We constructed a histogram of the length of all the pauses at each applied force. The distribution of residence pause times at a given applied force deviated significantly from a single-exponential fit to the data indicating the presence of multiple inactive holoenzyme–DNA conformations. The different inactive intermediates could be on-pathway, meaning conformations visited when switching from pol to exo or from exo to pol or off-pathway, meaning an inactive conformation linked to one of the two active states, pol or exo.
To distinguish between the different scenarios we performed a more extensive analysis of the pause length. We separated our experimental traces into two branches: the elongation branch corresponding to regions of DNA synthesis with intermittent pauses (regions shown in green in Figure 3A) and the degradation branch corresponding to regions of exonuclease activity with intermittent pauses (regions shown in orange in Figure 3A). We then analyzed the residence time of the pauses for each branch, 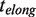 and
and 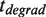 , as well as the transition time to switch from one branch to the other,
, as well as the transition time to switch from one branch to the other, 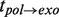 and
and 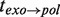 (Figure 3B).
(Figure 3B).
Figure 3.

Analysis of the pauses observed during wt T4 holoenzyme strand displacement activity. (A) An experimental trace indicating the assignment of the elongation branch (regions in green) and the degradation branch (regions in orange). Pauses during elongation and degradation are shown in dark green and brown, respectively, while the transition time from pol to exo and from exo to pol are shown in red and magenta, respectively. The bar shows the extension change corresponding to the synthesis of 100 nt. (B) Distributions of the residence pause times during the elongation (Number of events Ne = 221), and degradation (Ne = 55) branches, and the transition times for switching between pol and exo (Ne = 173 and Ne = 153) fit to either a single- or double-exponential equation. The error bars are proportional to the inverse of the square root of the number of points for each individual bin. (C) Mean residence times, 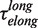 ,
, 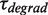 ,
, 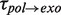 and
and 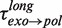 shown as a function of the applied force. Errors are estimated from the single- or double-exponential fits.
shown as a function of the applied force. Errors are estimated from the single- or double-exponential fits.
For the elongation branch, pauses with two distinctive residence times were detected: short pauses with a force-independent mean residence time 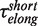 of 0.7 ± 0.2 s and long pauses with a force-dependent mean residence time
of 0.7 ± 0.2 s and long pauses with a force-dependent mean residence time 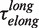 of 7 ± 1 s at 9.5 pN. The distribution of transition times from elongation to degradation followed single-exponential behaviour with a force-dependent mean residence time
of 7 ± 1 s at 9.5 pN. The distribution of transition times from elongation to degradation followed single-exponential behaviour with a force-dependent mean residence time 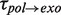 of 8 ± 1 s at 9.5 pN, which agreed with
of 8 ± 1 s at 9.5 pN, which agreed with 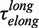 . This agreement was observed over the range of applied forces (9.5–13 pN) where the holoenzyme predominantly synthesized DNA during strand displacement activity (Figure 3C). Therefore, we associated the long pauses during the elongation branch with an on-pathway intermediate of the pol→exo transition that we called I. In contrast, the mean residence time of the short pauses observed during the elongation branch differed significantly from the pol→exo transition time; therefore, the short pauses were associated with an off-pathway inactive polymerase intermediate that we called ip. Assays performed at different holoenzyme concentrations demonstrated that only the long pauses in the elongation branch were sensitive to the holoenzyme concentration (Supplementary Figure S5). Therefore, dissociation from the DNA might be favoured or induced when the holoenzyme acquires the on-pathway, inactive I conformation.
. This agreement was observed over the range of applied forces (9.5–13 pN) where the holoenzyme predominantly synthesized DNA during strand displacement activity (Figure 3C). Therefore, we associated the long pauses during the elongation branch with an on-pathway intermediate of the pol→exo transition that we called I. In contrast, the mean residence time of the short pauses observed during the elongation branch differed significantly from the pol→exo transition time; therefore, the short pauses were associated with an off-pathway inactive polymerase intermediate that we called ip. Assays performed at different holoenzyme concentrations demonstrated that only the long pauses in the elongation branch were sensitive to the holoenzyme concentration (Supplementary Figure S5). Therefore, dissociation from the DNA might be favoured or induced when the holoenzyme acquires the on-pathway, inactive I conformation.
For the degradation branch, just one type of pause was identified with a force-dependent mean residence time 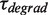 of 5 ± 1 s at 9.5 pN, which agreed with
of 5 ± 1 s at 9.5 pN, which agreed with 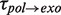 from 5 to 9.5 pN of applied force (Figure 3C and Supplementary Figure S6). Surprisingly, transitions from degradation to elongation with two distinct transition times were detected. Only ∼10% of the transitions were long and their distribution followed single-exponential behaviour with a force-dependent mean residence time
from 5 to 9.5 pN of applied force (Figure 3C and Supplementary Figure S6). Surprisingly, transitions from degradation to elongation with two distinct transition times were detected. Only ∼10% of the transitions were long and their distribution followed single-exponential behaviour with a force-dependent mean residence time 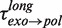 of 6 ± 2 s at 9.5 pN, similar to
of 6 ± 2 s at 9.5 pN, similar to 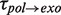 from 9.5 to 13 pN of applied force (Figure 3C and Supplementary Figure S6). Therefore, we associated the pauses during the degradation branch with an exo→pol transition that proceeded through the same on-pathway intermediate I as the pol→exo transition. In contrast, most of the exo→pol transitions (∼90%) occurred nearly instantaneously without a marked pause regardless of the applied force. Such fast transitions were not observed in the pol→exo direction, implying that the predominant pathway for the exo→pol transition was irreversible and did not involve an intermediate under our experimental conditions.
from 9.5 to 13 pN of applied force (Figure 3C and Supplementary Figure S6). Therefore, we associated the pauses during the degradation branch with an exo→pol transition that proceeded through the same on-pathway intermediate I as the pol→exo transition. In contrast, most of the exo→pol transitions (∼90%) occurred nearly instantaneously without a marked pause regardless of the applied force. Such fast transitions were not observed in the pol→exo direction, implying that the predominant pathway for the exo→pol transition was irreversible and did not involve an intermediate under our experimental conditions.
Figure 4A depicts a model drawn from our results illustrating the force-dependent shift in the equilibrium from the polymerization conformation to the exonuclease conformation through an inactive intermediate during strand displacement synthesis. The two main states are the active pol and exo states that presumably correspond to well-defined DNA-polymerase conformations in which the primer is accommodated in the pol and exo active sites, respectively. Two different inactive intermediates are present: an on-pathway intermediate of the pol↔exo transition, I and an off-pathway intermediate linked to the pol state, ip. An alternative irreversible pathway allows for the rapid direct transition between the exo and pol states. Dissociation of the polymerase from the DNA most likely occurs through the inactive I conformation.
Figure 4.

Kinetic scheme for the wt T4 holoenzyme primer transfer reaction in strand displacement. (A) Diagram showing the effect of applied force to shift the equilibrium from the polymerization conformation (pol) to the exonuclease conformation (exo) through an inactive intermediate (I) during strand displacement synthesis. An off-pathway intermediate (ip) and unbound polymerase state (D) are also present in the scheme. For each transition d and n correspond to the distance change in the molecular extension of the DNA in nm and the number of unwound or annealed bases, respectively, associated with the conformational transition. (B) State frequencies as defined in Equation (1) shown as a function of the applied force and fit to a single-exponential equation. The distance d and the corresponding estimation of bases unwound or annealed, n, are obtained from the fits to Equation (2).
Determination of equilibrium constants and kinetic rates
Under equilibrium conditions, the frequency of one state (i) relative to another state (j) (e.g. the ratio between the probabilities of populating the two states i and j) is given by the ratio between the transition rates from one state to the other: 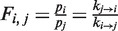 . The fact that we detected an irreversible transition from exo to pol implies that our system is not under equilibrium conditions. However, we can still assume that the system reaches several local equilibria along the reversible pathways, if the irreversible transition occurs on a much longer time scale than the other transitions. In particular, we assumed local equilibrium conditions between the inactive states (ip and I) and the active pol state at forces >9.5 pN and between the inactive I state and the active exo state at forces <8 pN. The state frequencies were computed as:
. The fact that we detected an irreversible transition from exo to pol implies that our system is not under equilibrium conditions. However, we can still assume that the system reaches several local equilibria along the reversible pathways, if the irreversible transition occurs on a much longer time scale than the other transitions. In particular, we assumed local equilibrium conditions between the inactive states (ip and I) and the active pol state at forces >9.5 pN and between the inactive I state and the active exo state at forces <8 pN. The state frequencies were computed as:
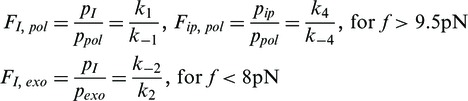 |
(1) |
where the occupancy probabilities of each state pi are computed from the distribution of strand displacement rates as described in Methods and Materials section and ki are the rates connecting the various states (Figure 4A). As shown in Figure 4B, the force clearly shifted the equilibrium between the different states with the dependence of the state frequencies Fi, j on the force f being (21):
| (2) |
where 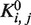 is the equilibrium constant between states i and j at zero force, kB is the Boltzmann constant, T is the temperature and
is the equilibrium constant between states i and j at zero force, kB is the Boltzmann constant, T is the temperature and 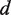 corresponds to the distance change in the molecular extension of the DNA when the polymerase switches from state i to state j at the force f. In our experimental configuration, a primer/enzyme transition involving the unwinding or reannealing of n nucleotides would result in a distance change of
corresponds to the distance change in the molecular extension of the DNA when the polymerase switches from state i to state j at the force f. In our experimental configuration, a primer/enzyme transition involving the unwinding or reannealing of n nucleotides would result in a distance change of 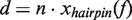 , where
, where 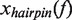 is the change in extension of the DNA substrate due to opening or closing one base pair at force f. Using Equation (2), fits to the data estimated a distance change of ∼4 nm or
is the change in extension of the DNA substrate due to opening or closing one base pair at force f. Using Equation (2), fits to the data estimated a distance change of ∼4 nm or 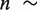 4 - 5 nt (
4 - 5 nt (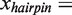 0.92 nm/bp at 11 pN) corresponding to the pol →I conformational change and ∼2 nm or
0.92 nm/bp at 11 pN) corresponding to the pol →I conformational change and ∼2 nm or 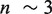 nt (
nt ( 0.7 nm/bp at 6 pN) corresponding to the I→exo conformational change (Figure 4B).
0.7 nm/bp at 6 pN) corresponding to the I→exo conformational change (Figure 4B).
The overall kinetics of the primer transfer reaction in the strand displacement configuration can be determined by combining the measurements of the state frequencies and the mean lifetimes of the different states [Supplementary Materials Equation (S11)]. The kinetic rates obtained from this analysis are shown in Figure 5A. Like the state frequencies, the kinetic rates depended exponentially on the applied force as (21):
| (3) |
where 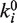 is the rate at zero force and
is the rate at zero force and 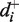 is the distance to the transition state. In Supplementary Tables S2 and S3 we present the various rates, equilibrium constants and distances between states obtained from fitting the force-dependent extracted rates and state frequencies to Equations (2) and (3).
is the distance to the transition state. In Supplementary Tables S2 and S3 we present the various rates, equilibrium constants and distances between states obtained from fitting the force-dependent extracted rates and state frequencies to Equations (2) and (3).
Figure 5.

Predictions from the kinetic model. (A) Kinetic rates (as shown in Figure 4A) estimated from the measurements of the state frequencies and mean lifetime of each state using Equation (S11). Error bars are obtained by propagating the errors associated with the mean lifetime and state frequencies. (B) A comparison of the measured mean strand displacement rate for the wt T4 holoenzyme (purple squares) to the predicted rates for the elongation (green line) and degradation (orange line) branches as estimated from Equations (S7) and (S9) and for the whole kinetic scheme including the two branches (magenta line) as estimated from Equation (S4) shown as a function of the applied force. Error bars are the s.e.m.
Validation of the model
The extracted rates can be used to predict the mean strand displacement rate of the holoenzyme as described by Equation (S4) in the Supplementary Materials. As shown in Figure 5B the predicted mean strand displacement rate agreed well with the experimental results obtained for the wt T4 holoenzyme over the entire range of forces we investigated, suggesting that the minimal model we proposed satisfactorily describes the main steps involved in the primer transfer reaction during the strand displacement mode. Interestingly, the experimental results were also approximated very well by the prediction obtained from the separate elongation branch at high forces (Equation (S7)) and the degradation branch at low forces (Equation (S9)). These results validate the local equilibrium condition assumed in Equation (1).
We also analyzed the strand displacement activity of the holoenzyme made with the exonuclease deficient polymerase gp43exo (Supplementary Figure S7). The mean strand displacement rate measured with the exo- and wt T4 holoenzymes agreed only at high forces. At low forces the exo T4 holoenzyme performed strand displacement DNA synthesis at a very slow rate of ∼1 nt/s, in contrast to the wt T4 holoenzyme that exhibited exonuclease activity. Interestingly, the mean strand displacement rate of the exo T4 holoenzyme as a function of the applied force was reproduced using the same reaction scheme as for the wt T4 holoenzyme where the exo state was considered as an extra inactive state [Supplementary Materials Equation (S5)]. These results strongly suggest that the exo T4 holoenzyme visited the same DNA–protein conformations as the wt T4 holoenzyme during the primer transfer reaction in the strand displacement mode.
Holoenzymes from different families display similar strand displacement behaviour
While the T7 holoenzyme presented a faster polymerization rate (∼500 nt/s at high forces) and a faster exonuclease rate (∼-200 nt/s) during the strand displacement mode than the T4 holoenzyme, the strand displacement activity at various forces demonstrated qualitatively similar behaviour to that observed for the T4 holoenzyme. Like the T4 holoenzyme, the T7 holoenzyme exhibited a force-controlled switch between polymerization and degradation activity during strand displacement DNA synthesis (Supplementary Figure S8). An analysis of the pauses during the elongation and degradation branches and the transition times 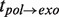 and
and 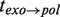 also presented similarities to the T4 case (Supplementary Figure S9). In particular, two different pathways were detected for the exo→pol transition: the first through an on-pathway intermediate I and the second without intermediates. These results demonstrate that the strand displacement activity of the T4 and T7 holoenzymes, from two different polymerase families, followed similar kinetic pathways; therefore, the scheme presented in Figure 4A likely represents a general kinetic scheme for replicative polymerases.
also presented similarities to the T4 case (Supplementary Figure S9). In particular, two different pathways were detected for the exo→pol transition: the first through an on-pathway intermediate I and the second without intermediates. These results demonstrate that the strand displacement activity of the T4 and T7 holoenzymes, from two different polymerase families, followed similar kinetic pathways; therefore, the scheme presented in Figure 4A likely represents a general kinetic scheme for replicative polymerases.
Cyclic polymerase assay and single-molecule Sanger sequencing application
We have taken advantage of the fact that the equilibrium between the polymerization and exonuclease activities of the T4 holoenzyme can be shifted and modulated through the applied force to develop a cyclic polymerase assay (CPA). The polymerization activity of the T4 holoenzyme may be studied by monitoring the molecular extension of a hairpin substrate as the enzyme synthesizes DNA at a fixed applied force Ftest; by reducing the assisting force (Fexo = 5 pN), the exonuclease activity can then be switched on inducing the wt T4 holoenzyme to degrade the newly synthesized strand while the hairpin reforms. The exonuclease activity, induced by the fork regression pressure, stops when the holoenzyme reaches the primer tail (Figure 1A) allowing for the complete regeneration of the hairpin substrate. Many cycles of synthesis and degradation can be repeated, as long as the enzyme does not pass the apex of the loop in the hairpin substrate (Figure 6A). If the enzyme passed the apex, the disappearance of duplex DNA ahead of the complex providing regression pressure prevented induction of the exonuclease activity and degradation of the newly synthesized strand upon lowering the force. The inclusion of modified nucleotides, such as LNA bases, placed in the template strand before the apex act as a roadblock in the progression of the holoenzyme preventing it from reaching the apex. On a short (∼100 bp) hairpin (SH) containing a three-nucleotide LNA roadblock ahead of the apex, the T4 holoenzyme synthesized DNA until it reached the LNA roadblock. Polymerization was stopped and exonuclease activity could be induced by reducing the force, thus allowing for full recovery of the original hairpin substrate (Figure 6B). The advantage of the CPA is that many cycles can be repeated quickly and easily, generating sufficient amounts of data for statistical analysis in a short time.
Figure 6.

Cyclic polymerase assay and application for single-molecule Sanger sequencing. Schematics (left panels) and experimental traces (right panels) of the CPA on the 1.2 Kbp long DNA hairpin (LH) substrate (A) and 100 bp short DNA hairpin (SH) substrate with LNA block (B). Synthesis (green) took place at higher applied forces (Ftest) until the LNA block (purple) was reached or the force was decreased to Fexo inducing the exonuclease activity (orange) and recovering the initial hairpin. (C) Utilizing the CPA for single-molecule Sanger sequencing, a trace recorded during a single cycle displayed transient pauses due to the low dATP concentration, as well as a permanent arrest due to the incorporation of ddATP (left panel). Distribution of the frequency of enzyme arrests corresponded with the expected position of adenosine nucleotides in the known DNA test sequence (right panel, N = 53). Bars show the extension change corresponding to the synthesis or degradation of 100 nt and 10 nt for the LH and SH results, respectively.
We have applied the CPA to demonstrate the possibility of single-molecule DNA sequencing by the Sanger method (13). In our single-molecule experiments, the incorporation of ddATP by the T4 holoenzyme from a pool of nucleotides containing dNTPs and ddATP produced a clear arrest in the molecular extension trace. The position of the arrest was correlated to the position of the complementary thymine base in the template strand of the hairpin substrate (Figure 6C). When the applied force was decreased, this chain-terminating nucleotide could be removed by the exonuclease activity allowing repeated cycles to sample all of the thymidine nucleotide positions along the sequence. Repeating the process separately with ddGTP, ddCTP and ddTTP in the nucleotide pool could provide the complete sequence of the DNA hairpin.
DISCUSSION
We have utilized a detailed force analysis to investigate the strand displacement synthesis activity of replicative polymerases. Mechanical tension applied to a DNA–enzyme complex is a useful variable to gain insight into biochemical steps that are coupled to movement (21). For example, tension applied to a ssDNA template has been used to study the polymerization catalytic cycle of the T7 holoenzyme and Klenow fragment (11,12) and the primer transfer pathway between polymerase active sites as part of the proof-reading ability of the Φ29 polymerase (22). Mechanical tension applied as an assisting force to open dsDNA is a useful tool to investigate the mechanism of motor proteins that unwind duplex DNA. This approach has been used to study the unwinding activity of numerous helicases (23–27) and the robust strand displacement synthesis activity DNA polymerases (28).
Mechanical force affects the holoenzyme primer extension and strand displacement activities differently
In this work we investigated the T4 and T7 holoenzyme strand displacement and primer extension reactions on a DNA hairpin manipulated with magnetic tweezers. Our results revealed that the progression of the holoenzyme was strongly affected by the presence of upstream duplex DNA. The primer extension synthesis was moderately hindered by the applied force, revealing a polymerase step size of one nucleotide for both the T4 and T7 holoenzymes; whereas the strand displacement activity was strongly stimulated by forces assisting the opening of the hairpin. In a simplified view, the T4 and T7 holoenzymes carried out efficient strand displacement synthesis on a primed DNA hairpin when assisted by a large force (a force close to the tension required to unzip the hairpin) and switched to a processive exonuclease activity when the force was reduced. Finding a single nucleotide step size for the polymerase primer extension is natural, the larger values reported in previous single-molecule findings (11,12) were probably caused by lower experimental sensitivity which made distinguishing between activity and pausing a real challenge.
Force-induced polymerase/exonuclease switch led to cyclic polymerase assay with potential applications for DNA sequencing
The observed force-induced polymerase/exonuclease switch allowed us to develop a CPA, as illustrated in Figure 6, in which the polymerase reaction can be cycled between synthesis and exonuclease activities by modulating the applied force. This assay is very useful in order to characterize the polymerase activity as it overcomes one of the main drawbacks of single-molecule polymerase assays, which are usually one-shot (i.e. assays that cannot be repeated).
The CPA could also be relevant for single-molecule DNA sequencing. Due to the hairpin design of our substrate, the incorporation of one nucleotide resulted in an increase in molecular extension of ∼0.8 nm. This implies that single nucleotide resolution should be easier to achieve with that set-up than with the approach based on sequencing by RNA-polymerase (29) where single nucleotide progression led to ∼0.3 nm extension change. A further advantage of the CPA over previous single-molecule sequencing assays is that many cycles can be repeated quickly and easily generating sufficient amounts of data for statistical analysis on the same molecule in a short time. Moreover, since many molecules can be monitored at once in a magnetic tweezers apparatus (∼60 in the present implementation), simultaneous sequencing of individual molecules with different sequences would be possible.
Model of primer transfer in strand displacement DNA synthesis
From the quantitative measurement of the strand displacement DNA synthesis activity of the bacteriophage T4 replicative holoenzyme obtained by directly measuring the effect of external forces, we propose a force-induced primer transfer pathway between the enzyme’s polymerization and exonuclease sites (Figure 4A). The extracted kinetic rates, equilibrium constants and conformational changes for each state along the force-induced primer transfer pathway are compatible with those detected in kinetic and structural studies. Note that the main states proposed (the active pol and exo states and the inactive on-pathway I state) also agree with those proposed previously for the primer transfer reaction of Φ29 in the primer extension configuration (22).
Structural data for RB69 polymerase (a structural and functional T4 homolog) with a primed DNA molecule bound in the pol conformation show the 5′ end of the template strand exiting the pol active site with a sharp almost 90° bend in the phosphate backbone in order to expose the first unpaired base of the template strand towards the 3′ end of the primer strand for extension (30,31). This sharp bending of the template strand, together with the mechanical tension applied along the primer/template dsDNA and the 5′ ssDNA tail of the DNA hairpin, may generate a destabilizing stress at the fork junction promoting the unwinding of n1 nucleotides of the upstream DNA duplex ensuring proper alignment of the template strand in the pol active site. This explains why polymerization activity is favoured as the applied force is increased in the strand displacement mode, but also explains the inhibition of polymerization at very large forces in the primer extension mode due the inability of the protein to generate enough counter force for proper template-polymerase alignment.
Without sufficient destabilizing stress generated by the polymerase or an assisting force to keep the fork junction open, reannealing will occur creating a regression pressure on the polymerase that may lead to polymerase-template destabilization and loss of the template in the pol active site. In turn this favours the polymerase stalling and kinetic partitioning to the I state. We propose that the DNA-polymerase conformation in which the polymerase loses the template strand while the DNA fork regresses n1 nucleotides corresponds to the inactive I intermediate detected in our experiments. The measured conformational change between pol and I suggested that this transition involves the first n1 ∼4–5 bp of the DNA fork. In agreement with these results, the analysis presented in the companion article (6) shows that the T4 holoenzyme in the pol conformation is able to destabilize the first few base pairs of the upstream DNA duplex. This ability to destabilize the DNA duplex, thereby preventing polymerase stalling, is essential for the efficient coupling between helicase and polymerase.
We found that the polymerase can also be exchanged with polymerase in solution during strand displacement activity. Our concentration analysis revealed that such exchange might be mainly through the I intermediate. Therefore, the polymerase might frequently dissociate when going from the pol to the exo active conformations. This is consistent with assays demonstrating that the primer transfer reaction in T4 occurs intermolecularly (32). On the other hand, the T4 polymerase in this case is present as the holoenzyme, i.e. complexed with the clamp protein, which would favour intramolecular switching between the pol and exo sites.
Persistent regression pressure from the fork junction felt by the polymerase may lead to the unwinding of the first n2 base pairs of the primer/template duplex allowing the primer to be transferred from the pol to the exo site of the polymerase. Our measurements imply a DNA-polymerase conformational change of 2 nm consistent with the opening of ∼3 bp of the primer/template duplex required for accommodating the primer in the exo site, in agreement with several structural studies (31,33). Note that the measured exonuclease rate of 100 nt/s coincides with the measured rate for the hydrolysis of the terminal phosphodiester bond by wt T4 polymerase (100 s−1) (19) and is independent of the applied force implying that the rate limiting step of the exonuclease reaction is not coupled to polymerase movement (21).
We detected that the majority of the exo→pol transitions occurred through a fast irreversible transition rather than returning slowly through the I intermediate. Such a fast transition has been measured in primer extension bulk assays (32). This is a desired characteristic for the proofreading ability of replicative polymerases; once the erroneous base has been removed, a quick exit from the exo site prevents removing more bases than necessary. The equilibrium constant between the pol and exo states in the primer extension mode estimated from strand displacement measurements (Supplementary Materials), 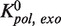 =
= 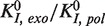 ∼10−4, agreed reasonably well with previous bulk measurements (19).
∼10−4, agreed reasonably well with previous bulk measurements (19).
Explanation for poor strand displacement synthesis in replicative polymerases
Many replicative holoenzymes exhibit rapid and processive primer extension DNA synthesis, but inefficient strand displacement DNA synthesis. However, little is known about what causes this inefficiency. Our investigation of the T4 and T7 holoenzyme strand displacement activities demonstrates that the origin of this inefficiency is the stalling of the holoenzyme induced by the fork regression pressure that leads to a force-induced shift between the active pol and exo polymerase conformations. The fork regression pressure acts to destabilize the polymerase-template interaction leading to loss of the template strand in the pol active site, polymerization stalling and kinetic partitioning to the exo conformation.
Normally, the role of the exonuclease activity in a replicative polymerase is to increase the fidelity of DNA replication. When an incorporation error occurs, the poor hybridization of the mismatch with the template strand triggers the transfer of the primer to the exonuclease domain for removal of the nucleotide. Such proofreading activity does not require processive exonuclease activity, yet recent studies have shown the ability of the T4 and RB69 DNA polymerases to catalyze the processive excision of two consecutive nucleotides (32). Here, we found that the processivity of the exonuclease reaction by the T4 and T7 holoenzymes can be much larger, reaching thousands of nucleotides. The biological purpose of this processive exonuclease activity may be in replisome disassembly to reset a stalled replication fork to a symmetrical situation. For instance, if the helicase prematurely dissociates from the replisome, the regression pressure exerted by the fork junction would trigger the processive exonuclease activity of the polymerase degrading the newly synthesized leading strand until the last Okazaki fragment on the lagging strand. However, single-stranded DNA-binding protein gp32 will modulate this activity in vivo, since gp32 markedly blocks processive exonuclease activity (6). The poor strand displacement capability of replicative polymerases might also play a biological role in the correct processing of the ends of Okazaki fragments to avoid generating long 5′-flaps during the maturation of Okazaki fragments.
Generality of this finding
Hairpin-induced switches in DNA polymerase activity have been observed for several polymerases, including DNA polymerase α (3,34), E. coli Pol II (1) and Pol III (2), and bacteriophage T4 (4), suggesting that this may be a general phenomenon, but each polymerase shows a different degree of sensitivity when encountering template secondary structures or fork junctions. All of these polymerases function in the context of a replisome with helicases whose primary job is to unwind duplex DNA. The companion article (6) shows that the presence of the helicase at the fork is essential to relieve the fork regression pressure on the holoenzyme preventing holoenzyme stalling.
The present findings explain why replicative polymerases are inefficient in performing strand displacement synthesis without assistance. The control of the partitioning between polymerization and exonuclease activities by the force applied to the replication fork appears as a new feature that must be taken into account in the context of a replisome; specifically in the coupling between the polymerase and the helicase during leading-strand synthesis. The detailed analysis done here can be repeated for other polymerases. Furthermore, the force-controlled polymerase/exonuclease switch offers interesting applications for single-molecule DNA sequencing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary Figures 1–9, Supplementary Text and Supplementary Reference [35].
FUNDING
Human Frontier Science Program [RGP0003/2007-C] to V.C. and S.J.B.; the National Institutes of Health [GM013306] to S.J.B.; and the European Research Council [MAGREPS 267 862] to V.C.. Funding for open access charge: European Research Council [MAGREPS 267 862].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank B. Ibarra for discussions on the work and reading of the manuscript and M. Baaden for stimulating discussions.
REFERENCES
- 1.Sherman LA, Gefter ML. Studies on the mechanism of enzymatic DNA elongation by Escherichia coli DNA polymerase II. J. Mol. Biol. 1976;103:61–76. doi: 10.1016/0022-2836(76)90052-8. [DOI] [PubMed] [Google Scholar]
- 2.LaDuca RJ, Fay PJ, Chuang C, McHenry CS, Bambara RA. Site-specific pausing of deoxyribonucleic acid synthesis catalyzed by four forms of Escherichia coli DNA polymerase III. Biochemistry. 1983;22:5177–5188. doi: 10.1021/bi00291a018. [DOI] [PubMed] [Google Scholar]
- 3.Weaver DT, DePamphilis ML. The role of palindromic and non-palindromic sequences in arresting DNA synthesis in vitro and in vivo. J. Mol. Biol. 1984;180:961–986. doi: 10.1016/0022-2836(84)90266-3. [DOI] [PubMed] [Google Scholar]
- 4.Hacker KJ, Alberts BM. The rapid dissociation of the T4 DNA polymerase holoenzyme when stopped by a DNA hairpin helix. A model for polymerase release following the termination of each Okazaki fragment. J. Biol. Chem. 1994;269:24221–24228. [PubMed] [Google Scholar]
- 5.Benkovic SJ, Valentine AM, Salinas F. Replisome-mediated DNA replication. Ann. Rev. Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- 6.Manosas M, Spiering MM, Ding F, Croquette V, Benkovic SJ. Collaborative coupling between polymerase and helicase for leading-strand synthesis in bacteriophage T4. Nucleic Acids Res. 2012;40:6187–6198. doi: 10.1093/nar/gks254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 8.Reddy MK, Weitzel SE, von Hippel PH. Assembly of a functional replication complex without ATP hydrolysis: a direct interaction of bacteriophage T4 gp45 with T4 DNA polymerase. Proc. Natl Acad. Sci. USA. 1993;90:3211–3215. doi: 10.1073/pnas.90.8.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaboord BF, Benkovic SJ. Accessory proteins function as matchmakers in the assembly of the T4 DNA polymerase holoenzyme. Curr. Biol. 1995;5:149–157. doi: 10.1016/s0960-9822(95)00036-4. [DOI] [PubMed] [Google Scholar]
- 10.Mark DF, Richardson CC. Escherichia coli thioredoxin: a subunit of bacteriophage T7 DNA polymerase. Proc. Natl. Acad. Sci. USA. 1976;73:780–784. doi: 10.1073/pnas.73.3.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wuite GJ, Smith SB, Young M, Keller D, Bustamante C. Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature. 2000;404:103–106. doi: 10.1038/35003614. [DOI] [PubMed] [Google Scholar]
- 12.Maier B, Bensimon D, Croquette V. Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl Acad. Sci. USA. 2000;97:12002–12007. doi: 10.1073/pnas.97.22.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frey MW, Nossal NG, Capson TL, Benkovic SJ. Construction and characterization of a bacteriophage T4 DNA polymerase deficient in 3'–>5' exonuclease activity. Proc. Natl Acad. Sci. USA. 1993;90:2579–2583. doi: 10.1073/pnas.90.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nossal NG. DNA replication with bacteriophage T4 proteins. Purification of the proteins encoded by T4 genes 41, 45, 44, and 62 using a complementation assay. J. Biol. Chem. 1979;254:6026–6031. [PubMed] [Google Scholar]
- 16.Rush J, Lin TC, Quinones M, Spicer EK, Douglas I, Williams KR, Konigsberg WH. The 44P subunit of the T4 DNA polymerase accessory protein complex catalyzes ATP hydrolysis. J. Biol. Chem. 1989;264:10943–10953. [PubMed] [Google Scholar]
- 17.Manosas M, Spiering MM, Zhuang Z, Benkovic SJ, Croquette V. Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat. Chem. Biol. 2009;5:904–912. doi: 10.1038/nchembio.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gosse C, Croquette V. Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys. J. 2002;82:3314–3329. doi: 10.1016/S0006-3495(02)75672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 20.Patel SS, Wong I, Johnson KA. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 21.Bustamante C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Ann. Rev. Biochem. 2004;73:705–748. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 22.Ibarra B, Chemla YR, Plyasunov S, Smith SB, Lázaro JM, Salas M, Bustamante C. Proofreading dynamics of a processive DNA polymerase. EMBO J. 2009;28:2794–2802. doi: 10.1038/emboj.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dessinges MN, Lionnet T, Xi XG, Bensimon D, Croquette V. Single-molecule assay reveals strand switching and enhanced processivity of UvrD. Proc. Natl Acad. Sci. USA. 2004;101:6439–6444. doi: 10.1073/pnas.0306713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumont S, Cheng W, Serebrov V, Beran RK, Tinoco I, Jr, Pyle AM, Bustamante C. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature. 2006;439:105–108. doi: 10.1038/nature04331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson DS, Bai L, Smith BY, Patel SS, Wang MD. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell. 2007;129:1299–1309. doi: 10.1016/j.cell.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lionnet T, Spiering MM, Benkovic SJ, Bensimon D, Croquette V. Real-time observation of bacteriophage T4 gp41 helicase reveals an unwinding mechanism. Proc. Natl. Acad. Sci. USA. 2007;104:19790–19795. doi: 10.1073/pnas.0709793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ribeck N, Kaplan DL, Bruck I, Saleh OA. DnaB helicase activity is modulated by DNA geometry and force. Biophys. J. 2010;99:2170–2179. doi: 10.1016/j.bpj.2010.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim S, Schroeder CM, Xie XS. Single-molecule study of DNA polymerization activity of HIV-1 reverse transcriptase on DNA templates. J. Mol. Biol. 2010;395:995–1006. doi: 10.1016/j.jmb.2009.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenleaf WJ, Block SM. Single-molecule, motion-based DNA sequencing using RNA polymerase. Science. 2006;313:801. doi: 10.1126/science.1130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 31.Hogg M, Wallace SS, Doublie S. Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J. 2004;23:1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fidalgo da Silva E, Reha-Krantz LJ. DNA polymerase proofreading: active site switching catalyzed by the bacteriophage T4 DNA polymerase. Nucleic Acids Res. 2007;35:5452–5463. doi: 10.1093/nar/gkm591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shamoo Y, Steitz TA. Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell. 1999;99:155–166. doi: 10.1016/s0092-8674(00)81647-5. [DOI] [PubMed] [Google Scholar]
- 34.Villani G, Fay PJ, Bambara RA, Lehman IR. Elongation of RNA-primed DNA templates by DNA polymerase alpha from Drosophila melanogaster embryos. J. Biol. Chem. 1981;256:8202–8207. [PubMed] [Google Scholar]
- 35.Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.