


Abstract
The L1 stalk is a key mobile element of the large ribosomal subunit which interacts with tRNA during translocation. Here, we investigate the structure and mechanical properties of the rRNA H76/H75/H79 three-way junction at the base of the L1 stalk from four different prokaryotic organisms. We propose a coarse-grained elastic model and parameterize it using large-scale atomistic molecular dynamics simulations. Global properties of the junction are well described by a model in which the H76 helix is represented by a straight, isotropically flexible elastic rod, while the junction core is represented by an isotropically flexible spherical hinge. Both the core and the helix contribute substantially to the overall H76 bending fluctuations. The presence of wobble pairs in H76 does not induce any increased flexibility or anisotropy to the helix. The half-closed conformation of the L1 stalk seems to be accessible by thermal fluctuations of the junction itself, without any long-range allosteric effects. Bending fluctuations of H76 with a bulge introduced in it suggest a rationale for the precise position of the bulge in eukaryotes. Our elastic model can be generalized to other RNA junctions found in biological systems or in nanotechnology.
INTRODUCTION
The ribosome is a large and complex biomolecular machine which synthesizes proteins in all cells. It translates the genetic information written in messenger RNA (mRNA) to produce a polypeptide chain from amino acids which come attached to their transfer RNA (tRNA). In bacteria and archaea, the complete 70S, 2.4 MDa ribosome has ∼21 nm in size and consists of a small (30S) and a large (50S) subunit. The process of translation has three main phases: initiation of protein synthesis, elongation of the polypeptide chain and termination. During elongation, individual amino acids are added to the nascent polypeptide chain in an iterative manner. In the process, the individual tRNA molecules subsequently occupy three main positions in the ribosome, termed A, P and E site. One iteration, or cycle, of elongation can be divided into tRNA incorporation, peptidyl transfer and mRNA–tRNA translocation. The pretranslocation (PRE) complex is a ribosome containing a peptidyl-tRNA in the A site and deacylated tRNA in the P site. In the translocation process, the tRNA and mRNA are moved by the span of one codon (13 Å) relative to the ribosome, so that the post-translocational (POST) complex contains a peptidyl-tRNA in the P site and a deacylated tRNA in the E site. Contemporary knowledge about structural and functional dynamics of the ribosome has been recently summarized in a book (1) and several reviews (2–10).
Intense research effort has been made to characterize the structural and dynamical aspects of translocation. Studies using cryoelectron microscopy (cryo-EM) (11–18), X-ray crystallography (19–23), single-molecule FRET (smFRET) (24–33) and various molecular biological techniques (34–39), together with simulation studies focused on global changes (40–45) or on the properties of building blocks (46–51) are beginning to yield a general consensus picture. It appears that the PRE complex spontaneously interconverts between two global states, termed macrostate I (MS-I) and macrostate II (MS-II). In MS-I, the two tRNAs are in their classical A and P positions and the L1 stalk, a prominent structural feature emerging from the 50S subunit, is open away from the ribosome body. The MS-I to MS-II transition is characterized by the formation of the hybrid A/P, P/E state in which the acceptor arms of the A-site and P-site tRNAs occupy the P site and E site on the 50S subunit, respectively. Furthermore, the two subunits rotate relative to each other (the so-called ratchet-like motion) and the L1 stalk closes to interact with the tRNA in the E site. Translocation is completed by moving from the PRE to the POST complex, a process catalysed by the elongation factor G (EF-G).
A prominent role in the process of translocation is played by the L1 stalk (Figure 1). This structural protuberance on the 50S subunit consists of helices 76, 77 and 78 of 23S RNA and protein L1. H76 forms a stem on which the H77/H78/L1 complex folds into a rather compact structure resembling a hat of a mushroom. The other end (or the base) of H76 is attached to a three-way junction involving, besides H76, also helices 75 and 79. It is now widely accepted that the L1 stalk interacts with deacylated tRNA in the E site during translocation and regulates its release from the ribosome (2–4,6,8–10). The role of the L1 stalk in translocation is further corroborated by the fact that the deletion of the L1 protein destabilizes the hybrid state, thus slowing down the rate of translocation (24). Interestingly, the L1 stalk seems to be a rather new evolutionary achievement (52).
Figure 1.

(A and B) Superposition of 23S rRNA in the large ribosomal subunit of E.c. (pdb code 2AW4, red) and T.t. (1VSP, green). The H75/H76/H79 junction at the basis of the L1 stalk is highlighted. Notice the different positions of H76 in the two structures: the E.c. H76 is extended away from the ribosome body (open conformation), the T.t. H76 is tilted towards it (half-closed conformation). The central protuberance (CP) and the L7/L12 stalk are indicated, all the ribosomal proteins and the H77/H78 rRNA at the top of the L1 stalk are omitted for clarity. (A) View from the small subunit, (B) structure rotated through 90° towards the reader. (C and D) Magnified view of the T.t. L1 stalk (pdb code 1VSP) including the H75/H76/H79 junction (atomic and ribbon representation), rRNA helices H77 and H78 (magenta) and the L1 protein (cyan). The L1 protein is not complete in this structure. The orientation in (C and D) is the same as in (A and B), respectively.
Cryo-EM and X-ray studies (2–4,6,8–10) revealed at least three different positions of the L1 stalk, which were also observed in an smFRET study (27). In structures of ribosomes with a vacant E site or in isolated 50S subunits, the L1 stalk is positioned away from the ribosome body (open conformation). If a deacylated tRNA is bound in the classical E/E state of the POST complex, the L1 stalk moves towards the ribosome body to interact with the tRNA elbow (half-closed conformation). If a P/E tRNA of the hybrid PRE complex is present instead, the stalk moves still further to preserve its contacts with the tRNA (closed conformation). Particular conformational rearrangements of the L1 stalk seem to be associated with the formation of the first peptide bond, stimulated by the elongation factor EF-P. In a crystal structure of EF-P bound to the 70S ribosome together with the initiator tRNA (22), a movement of the L1 stalk larger than in the half-closed, but smaller than in the closed structure was observed.
Several recent studies have started to identify conformational intermediates between the MS-I and MS-II macrostates (17,18,23,24,30). A crystallographic study of Zhang et al. (23) yielded several structures of the ribosome in intermediate states of ratcheting. Using smFRET, Munro et al. (24) identified two distinct hybrid state intermediates: apart from the classical (A/A, P/P) state and the hybrid (A/P, P/E) state, they detected another hybrid state, (A/A, P/E) whose occupancy was enriched by a point mutation on the 23S RNA. Fu et al. (18) investigated this mutated PRE complex using cryo-EM and inferred four states. They are characterized by increasing intersubunit rotation angle and by the movement of the L1 stalk towards the ribosome body (termed inward) and in the perpendicular direction towards the 30S subunit (forward).
Although most of the studies so far have focused on the prokaryotic ribosome, structural and dynamical data for the eukaryotic ribosome are starting to emerge (53–59). The eukaryotic helix H76 contains a bulge base not present in prokaryotes, which introduces an additional hinge point into H76 and likely explains the much larger movement of the L1 stalk (53). The L1 stalk is found either close to the central protuberance (in-position) or in an open out-position. Contrary to prokaryotes where the L1 stalk in vacant ribosomes is open, the L1 stalk in vacant eukaryotic ribosomes is in the in-position (53).
Computer simulation techniques are able to provide additional useful information about functionally relevant dynamics of the ribosome. However, it is fair to admit that the ribosome presents a formidable challenge for computational studies. Unrestrained atomistic simulations are hampered not only by the enormous size of the ribosome and the timescale of its dynamics, but also by the uncertainties and absent parts due to the resolution limits of the experimental structures. While unrestrained atomistic simulations represent a ‘gold standard’ in the simulation field, they are extremely sensitive to the completeness and accuracy of the starting model (60). This means that a useful modelling of the ribosome dynamics necessarily requires the application of smart approximate steps or approaches, such as coarse-graining, the use of knowledge based potentials, applying various restraints to keep the structure in place or to impose the desired move, etc. Simulations may help in interpreting experimental data, notably my means of flexible fitting of atomic-resolution structures into the cryo-EM density maps (61) or more generally, by producing configurations that are consistent with available biochemical and biophysical measurements (45). Molecular dynamics (MD) simulations have been instrumental in elucidating mechanisms of tRNA accommodation (62,63), drug–ribosome interactions (64) or functional, ribosome-induced conformational changes in the elongation factor Tu (EF-Tu) (65).
A study combining atomistic structural data with elastic network normal mode analysis (40) found the closing and opening movement of the L1 stalk already in the first normal mode. The stalk closing was seen also in another study, employing anisotropic network model (41). Coarse-grained MD representing each protein or RNA residue as one effective bead (42,66) suggests that functionally important movements of the L1 stalk are in phase with the intersubunit rotation. A recent systematic analysis using essential dynamics coarse-graining yielded coarse-grained sites of 20–30 residues each, connected by effective harmonic springs (44). The authors found that the L1 stalk interacts very weakly with other residues, except those at its basis. Another study combined cryo-EM and X-ray data with MD simulation to identify differences in the interactions of the L1 stalk with initiator and elongator tRNA, and investigated the stalk opening in the context of the entire ribosome (43).
The ribosome, just as any other biomolecular machine, consists of building blocks (67). It is thus important to study properties of these blocks and how they contribute locally to the events in question. This bottom-up approach complements the global one and would enable us to understand systems composed of blocks in different contexts, such as in novel structures proposed by RNA nanotechnology (68–70).
There have been multiple studies focused on structural dynamics and flexibility of RNA building blocks (or structural motifs) using atomic-resolution, unrestrained explicit solvent MD simulations. The studied systems include the L7/L12 stalk RNA region, the flexible base of the A-site finger, the 5S rRNA three-way junction, RNA kink-turns, SSU helix 44 and others (47–51,71,72). The basic assumption of these studies is that the intrinsic structure and deformability (flexibility) of the RNA building blocks and segments are optimized to support the overall dynamics and functional thermal fluctuations of the ribosome, especially if complex structural changes within the blocks (e.g. a rupture or rearrangement of base pairing) are not involved.
Large-scale, unrestrained atomic-resolution MD simulations can be combined with a suitable coarse-grained model, defined by a small number of global coordinates and an effective energy function. Fitting the model configuration to each snapshot of an atomistic MD trajectory yields time series of the global coordinates, from which parameters of the energy function can be inferred (73–75). This method has been used to deduce sequence-dependent harmonic energy functions for DNA base pair step coordinates (76) and intra-base pair coordinates (77). The base pair step stiffness parameters (76) yield DNA mesoscopic force constants which compare favourably to experimentally observed values (78), without any adjustable parameters. Furthermore, they were found to outperform stiffness parameters from other data sources in predicting relative affinities of a benchmark protein–DNA binding system dominated by indirect readout (79). The approach has since been used in a range of problems, including the stiffness of RNA double helices (47,80,81). This indicates that unrestrained MD simulations may be able to realistically capture the magnitude of thermal structural fluctuations, at least for structures where no major conformational rearrangements take place.
In this work, we study structural and mechanical properties of the three-way junction at the base of the ribosomal L1 stalk, that is, the H76/H75/H79 rRNA junction. We propose a coarse-grained elastic model in which both the H76 helix and the junction core are represented by flexible, possibly anisotropic elements. The model is characterized by a small number of shape and stiffness parameters, which we infer from extensive atomistic, explicit-solvent MD simulations. Our results provide insights into the junction properties relevant for its biological role. The proposed elastic model should be readily applicable to other junction systems important for biological functioning or in nanotechnology.
MATERIALS AND METHODS
Starting structures
We investigated the H75/H76/H79 three-way junctions (3wj) from four different organisms (Figures 1 and 2). Starting conformations for our MD simulations were taken from the crystal structures of E.c. 50S subunit (pdb code 2AW4, resolution 3.5 Å) (82) and D.r. 50S (2ZJR, resolution 2.9 Å) (83), where the L1 stalk is in the open position, and T.t. 50S (1VSP, resolution 3.8 Å) (21), where the L1 stalk is in the half-closed position. In the crystal structure for H.m. 50S (1S72, 2.4 Å) (84), the helix H76 is not complete (there are just the first 3 bp in the stem); therefore, we modelled the missing part as a canonical A-RNA duplex using Insight II (Biosym/MSI, San Diego, CA, USA). The program allowed us to add only canonical base pairs (A–U and G = C, Figure 2) even though the native sequence of H.m. H76 has also a non-canonical A/A pair and several wobble G/U pairs [see the secondary structure of H.m. at Comparative RNA Web Site, http://www.rna.ccbb.utexas.edu, (85)]. In the X-ray structure for E.c., we changed the X-ray glycosidic syn conformation of U2092 to the anti orientation in order to allow for the formation of the conserved trans Hoogsteen/Hoogsteen (tH/H) base pair with the opposite A2227 [the conservation of the tH/H pair at this position was established using the Ribostral software (86)]. The initial syn conformation represents probably a refinement error. We also performed a test simulation using another T.t. crystal structure with higher resolution (pdb code 2J01, resolution 2.8 Å) (20). However, after 25 ns of simulation using the parm99 force field (see below for details of the simulation method), we observed unfolding of the central region. Therefore, this system was not included in the present study.
Figure 2.

2D structures of the studied three-way junctions. H75 is in grey, H76 is in green and H79 is in pink. The central UNA/GAN motif is in light yellow and the residues forming a hook-turn are in red. The original X-ray residue numbering is shown. The part of H76 in H.m. which is missing in the X-ray structure was prepared in silico (magenta). Red arrows indicate the two positions where the bulge base (adenine) was introduced.
We also studied fluctuations of the 3wj with a bulge base positioned in the middle of H76 (Figure 2) as found in the eukaryotic ribosome (see Comparative RNA Web Site). A recent eukaryotic Saccharomyces cerevisiae X-ray structure of the large subunit (56), however, shows disorder in the area of the bulge base in H76, and the eukaryotic Tetrahymena thermophila X-ray structure of the large subunit (58) contains only a lower part of H76. Hence, we prepared the system in silico using the H.m. 3wj with a modelled H76 stem as described earlier, where we manually introduced the extra base into H76 in a stacked-in conformation using Swisspdb viewer. We created two bulged systems (Figure 2). In one of them, the bulge base was introduced into the 2136–2149 strand of H76 (bulgeR), while in the other one, the bulge base was introduced into the opposite, 2226–2239 strand of H76 (bulgeL). The biologically relevant position is bulgeL, but the identity of the bulge base is not conserved (see Phylogenetic conservation map of the large ribosomal subunit for Eukaryota at Comparative RNA Web Site). In our study, we used adenine for the bulge base, as found in S. cerevisiae.
Molecular dynamics simulations
MD simulations were performed using the Amber 10 suite of programs. The RNA molecules in their starting conformations were solvated by an octahedral TIP3P water box extending 10 Å away from the solute and neutralized by sodium cations initially placed to the most negative sites around the solute. The original parm99 Na+ parameters (radius 1.868 Å and well depth 0.00277 kcal/mol) were used. After equilibration, production MD runs extending to 100 ns each were performed using the pmemd module of Amber 10 with the parm99 (87,88) and parmbsc0 (89) force fields. Standard simulation protocols described elsewhere (90) were applied. The bulged systems were simulated using the parm99 force field only.
In the simulation of E.c. 3wj with parmbsc0, we observed a disruption of H76 and a formation of a ladder-like structure, a recently reported irreversible artefact occurring in long simulations (91). Therefore, we ran a new simulation with parmbsc0 together with the parmχOL3 reparameterization for the glycosidic torsion (92,93). This force field (presently a new default RNA force field in the AMBER suite) prevents the formation of ladder-like high-anti χ structures which sooner or later occur with all the preceding force field variants. This new simulation was stable and was used in our analysis. No other simulation showed signs of unacceptable structural perturbations. The latest parmχOL3 definitely represents a substantial step forward in stabilizing long RNA simulations. However, the correction was not available before a major part of the simulation data presented here was completed. Unless the ladder-like structures form, the parm99 and parmbcs0 force fields are both considered viable alternatives for RNA simulations, although they result in somewhat different RNA structural dynamics (60,94). The main difference concerns the flips of the backbone torsions α and γ into the t/t conformation, which are present in parm99 but largely suppressed in parmbsc0 (60,94). In the absence of the ladder-like structures, parmbsc0/parmχOL3 behaves similarly to parmbsc0 (92,93). We expect that the results may be more affected by resolution limits of the starting structures than by the RNA force field.
The present results are also assumed to be insensitive to details of the ion treatment. We have used a standard Na+ net-neutral condition, which has been used in the majority of nucleic acids simulation studies published so far. It implies a biologically relevant cation concentration of ∼0.2 M. We have extensively tested different ion conditions such as excess salt simulations for a number of RNA systems and we did not observe any visible difference of the solute dynamics compared to the standard Na+ simulation protocol (51,95).
Global structure and stiffness description
We describe the global conformation of our three-way junction by specifying the configuration of H76 with respect to H75 and H79. The basic definitions are illustrated in Figure 3. We first determine the helix axis of H79, which forms the z-axis of our global coordinate system. The x-axis is defined as the projection of the helical axis of H75 onto the plane perpendicular to the z-axis. The y-axis complements the x- and z-axes to form a right-handed, orthonormal system. We then select a segment of four consecutive base pairs within H76, compute the helical axis of the segment (aH76 in Figure 3), and measure the angles ϕ and ψ as shown: ϕ is the oriented angle from the z-axis to the segment axis, ψ is the oriented angle from the x-axis to the segment axis projected onto the xy plane. Each helix axis is computed as a directional average (normalized vector sum) of the local helical axes provided by the program 3DNA (96). We describe the segment configuration by the coordinate vector (ϕ1,ϕ2) whose components are defined by:
| (1) |
Figure 3.

(Diagrams) Schematic representation of the coarse-grained description. The global reference system is defined by the helical axes of H79 and H75, a segment is then chosen within H76. The segment helical axis (aH76) serves to measure the bending magnitude ϕ and the bending direction ψ, from which the global coordinates ϕ1,ϕ2 are computed as shown. (Panels) Scatter plots of ϕ1,ϕ2 for some of the studied systems. Grey points: instantaneous conformations in 100 ps intervals. The coordinate fluctuations are interpreted using a harmonic, anisotropic elastic model. Red crosses: equilibrium conformations, ellipses: energy levels of kBT/2, black crosses: starting X-ray conformations. In the E.c. panel, additional X-ray structures are shown: pdb codes 2I2V (yellow), 3I1N (magenta), 3I1P (blue). The fluctuations are almost isotropic for systems without a bulge. The initial half-closed conformation (T.t., black cross) lies within the kBT/2 contour and is thus thermally accessible. The bulgeL structure observed in eukaryotes shows biologically relevant fluctuations towards the main ribosome body and towards the 30S subunit (compare with the T.t. half-closed starting conformation). Inserting the bulge at the same location but in the opposite strand (bulgeR) results in biologically less relevant movement where H76 stays away from the 30S subunit. Analogous data for the remaining systems are in Supplementary Figure S1.
The coordinates ϕ1,ϕ2 can be interpreted as bending angles in the direction of x-axis and y-axis, respectively. In our development, we assume small rotations, thus all the rotations commute in our approximation. The location of the segment within H76 is specified by the distance L from the first H76 base pair adjacent to the junction core to the middle of the segment, taken along the helix contour. The contour length L is computed as a sum of helical rises provided by 3DNA (we take half the helical rise of the step in the middle of the segment). We interpret thermal fluctuations of ϕ1,ϕ2 using a model in which the elastic energy is a general quadratic function of ϕ1,ϕ2. The model is elaborated in detail in the Supplementary Data.
The model just described cannot distinguish between the contribution to the H76 flexibility from the junction core and from the helix H76 itself. To identify these two contributions, we propose a more detailed model in which the helix H76 is represented by a homogeneous, intrinsically straight, flexible elastic rod, attached to a flexible spherical hinge representing the junction core (Figure 4). Both the helix and the core are in general assumed anisotropic. Consider a segment whose position within H76 is given by the contour length L. We express the segment coordinates ϕ1,ϕ2 as a sum of two terms,
| (2) |
where ϕJ1,ϕJ2 are contributions to ϕ1,ϕ2 from the junction core and ϕH1,ϕH2 are contributions to ϕ1,ϕ2 from the helix (Figure 4). Just as for ϕ1,ϕ2, the values of ϕH1,ϕH2 depend on L. The values of ϕJ1,ϕJ2 describe the orientation of the core and do not depend on L. The elastic energy is assumed to have the form:
| (3) |
where EJ is a general quadratic function of ϕJ1,ϕJ2 and EH is a general quadratic function of ϕH1,ϕH2. Thus, the total elastic energy is a sum of the energies associated with the junction core and with the helix. The coordinates ϕJ1,ϕJ2 and ϕH1,ϕH2 together describe the configuration of the model, but are not directly measurable. This is because our H76 segment has a finite length and therefore L cannot be chosen arbitrarily small. The measurable coordinates are only ϕ1,ϕ2 as functions of L, from which the properties of the model must be deduced.
Figure 4.

(Left) Schematic illustration of the flexible core, flexible helix model. The coordinates ϕ1,ϕ2 (Figure 3) are decomposed into the contribution from the flexible junction core (ϕJ1,ϕJ2) and from the flexible helix (ϕH1,ϕH2): ϕ1 = ϕJ1 + ϕH1, ϕ2 = ϕJ2 + ϕH2. (Panels) Fitting of the isotropic flexible core, flexible helix model. The contour length is the distance along H76 from its base pair closest to the core to the middle of the segment chosen in H76. The bending angle variation  is defined by Equation (6). The model predicts a linear relation between the contour length and the bending angle variation [Equation (5)]. The simulated data (blue crosses) satisfy the linear relation very well. The model stiffness parameters are inferred from the fitted linear functions (red lines). The intersection of the line with the y-axis determines the stiffness of the junction core aJ, the line slope determines the stiffness of the helix expressed by persistence length Lp [Equation (5) and (7)]. The stiffness parameters inferred from the data are shown. The complete list of the inferred parameters is in Table 1, analogous plots for the remaining systems are in Supplementary Figure S3.
is defined by Equation (6). The model predicts a linear relation between the contour length and the bending angle variation [Equation (5)]. The simulated data (blue crosses) satisfy the linear relation very well. The model stiffness parameters are inferred from the fitted linear functions (red lines). The intersection of the line with the y-axis determines the stiffness of the junction core aJ, the line slope determines the stiffness of the helix expressed by persistence length Lp [Equation (5) and (7)]. The stiffness parameters inferred from the data are shown. The complete list of the inferred parameters is in Table 1, analogous plots for the remaining systems are in Supplementary Figure S3.
The model is described in detail in Supplementary Data. Since the helix is assumed intrinsically straight, its equilibrium direction is given by the equilibrium values 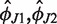 of the junction core coordinates ϕJ1,ϕJ2. These are equal to the means of ϕ1,ϕ2,
of the junction core coordinates ϕJ1,ϕJ2. These are equal to the means of ϕ1,ϕ2,
| (4) |
The stiffness parameters of the junction core and the helix are related to the second moments of the coordinates—see Supplementary Data for the general, anisotropic case. If both the core and the helix are assumed isotropic, one obtains,
| (5) |
where aJ is the stiffness constant of the junction core, aH is the stiffness constant of the helix and 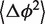 is the bending angle variation defined by:
is the bending angle variation defined by:
| (6) |
Thus, for the isotropic model, the bending angle variation 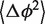 is a linear function of the contour length L, where the absolute term determines the junction core stiffness and the linear term determines the helix stiffness. The bending angle variation
is a linear function of the contour length L, where the absolute term determines the junction core stiffness and the linear term determines the helix stiffness. The bending angle variation  can be understood as the mean square angular deviation between the instantaneous direction of H76 and its equilibrium direction. Notice that we do not assume H76 to be rigid. Rather, it is assumed to be flexible, so that its instantaneous direction depends on the position within H76, that is, on the contour length L. It is convenient to express the helix stiffness constant in the form of a persistence length Lp using the formula (97),
can be understood as the mean square angular deviation between the instantaneous direction of H76 and its equilibrium direction. Notice that we do not assume H76 to be rigid. Rather, it is assumed to be flexible, so that its instantaneous direction depends on the position within H76, that is, on the contour length L. It is convenient to express the helix stiffness constant in the form of a persistence length Lp using the formula (97),
| (7) |
It can be shown (see Supplementary Data) that the absolute term in Equation (7) is equal to the bending angle variation 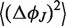 associated with the core, while the linear term is equal to the bending angle variation
associated with the core, while the linear term is equal to the bending angle variation 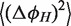 associated with the helix. Thus, the absolute term represents the contribution to
associated with the helix. Thus, the absolute term represents the contribution to 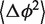 coming from the junction core.
coming from the junction core.
We split the MD trajectories into individual snapshots (taken in 10 ps intervals) and analyse the RNA structure in each snapshot using the 3DNA program to obtain the local helical axes and H76 helical rises. From these, we compute the coordinates ϕ1,ϕ2 and their moments, replacing the canonical ensemble averages by averages over the trajectories. The model fitting to the simulated data is described below.
RESULTS AND DISCUSSION
Global fluctuations of H76
The coordinates ϕ1,ϕ2 for selected systems are plotted in Figure 3, analogous plots for the remaining systems are in Supplementary Figure S1. The coordinate ϕ1 can be interpreted as the bending angle of H76 towards H75, that is, towards the main body of the ribosome, ϕ2 can be understood as the bending angle in the perpendicular direction, away from the 30S subunit. The plots show the orientation of a 4-bp segment within H76 whose last bp is the third bp from the end (the two last bp are excluded to avoid end-effects). The red crosses in Figure 3 indicate the equilibrium values of ϕ1,ϕ2. The numerical values are listed in Supplementary Table S1. The equilibrium values for the parm99 and parmbsc0 simulations differ by one to several degrees (9° in one case), which may be related to the different backbone dynamics characteristic for the two force fields, as discussed in ‘Methods’ section. These differences do not qualitatively affect the equilibrium conformation (which is clearly open except D.r., see below) and can be considered rather minor. The red ellipses are isoenergetic contours corresponding to the elastic energy of kBT/2. We define the anisotropy of the system as the ratio (greater or equal to 1) of the ellipse semiaxes. All of our simulated systems except the bulged ones exhibit anisotropy close to 1 (Supplementary Table S1). Thus, our results indicate that the bending fluctuations of bacterial and archeal H76 are essentially isotropic.
The black crosses in Figure 3 represent the starting structures used in our MD simulations. In the E.c., H.m. and D.r. starting structures, the L1 stalk is in the open conformation. Figure 3 and Supplementary Figure S1 show that the equilibrium MD structures are close to the starting structures, except for D.r. where the deviation is probably caused by the instability at the base of the helix (see Supplementary Data). In our T.t. starting structure, the L1 stalk is in the half-closed conformation. The simulated structure undergoes a conformational change to the open form (Figure 3 and Supplementary Figure S1) within several nanoseconds. This is expected, since the half-closed L1 stalk in the ribosome is stabilized by the interactions with the E-site tRNA, which is absent in our system.
Comparing the T.t. starting conformation and the kBT/2 energy contours (Figure 3 and Supplementary Figure S1), we see that the energy cost of the half-closed conformation does not exceed kBT. This suggests that the half-closed conformation of the L1 stalk could be reached by local thermal fluctuations of the L1 stalk itself, without any long-range allosteric rearrangements.
The bulged systems (Figures 2 and 3) exhibit larger and more anisotropic fluctuations compared to those where no bulge is present. We introduced the bulge in a stacked-in conformation either at the position where it is found in eukaryotic ribosomes (bulgeL) or at the same location but in the opposite strand (bulgeR). Figure 3 shows that the H76 global dynamics is very different in the two cases (see Supplementary Data for a more detailed description of the local bulge dynamics). While in bulgeR the H76 fluctuations are moderately anisotropic (anisotropy 1.3) and essentially stay away from the 30S subunit, the H76 movements in the biologically relevant bulgeL system are more anisotropic (anisotropy 1.5) and directed towards the ribosome body and towards the 30S subunit, as expected from the L1 stalk functional role. The equilibrium structure of bulgeL (red cross in Figure 3) is shifted towards the main ribosome body and towards the 30S subunit, in the same direction as the half-closed conformation of the T.t. structure (black cross in the T.t. panel of Figure 3). This is consistent with the in-position of the L1 stalk observed in vacant eukaryotic ribosomes (53). In contrast, the equilibrium conformation of the non-native bulgeR points away from the 30S subunit. The very different equilibrium and dynamics of H76 with bulges at the same location but in different strands could explain why nature has chosen one particular strand of H76 to introduce the bulge.
Our global conformational description can be compared with other published approaches. Fu et al. (18) introduced rotation angles of H76 in two perpendicular directions around a hinge point at the junction core. One of them (‘inward’) captures the rotation towards the main body of the ribosome, the other (‘forward’) refers to the rotation in the perpendicular direction, towards the 30S subunit. To clarify the relationship between our coordinates ϕ1,ϕ2 and the rotation angles of Fu et al., we measured global conformations of some of the E.c. X-ray structures they studied. They are shown as the additional colour crosses in the E.c. panel of Figure 3. Although our coordinate values are higher than those of Fu et al., the trends are identical (Supplementary Table S1). Taking the structure 2I2V (resolution 3.2 Å, yellow cross) (98) as a reference, we see that the structure 3I1N (3.2 Å, magenta cross) (23) is bent away from the ribosome body and towards the 30S subunit, while the opposite bending direction is observed for the 3I1P structure (3.2 Å, blue cross) (23). These bending deviations are entirely consistent with those described by Fu et al. Thus, our coordinates capture the same type of movement as theirs: ϕ1 corresponds to the ‘inward’ rotation of Fu et al., ϕ2 corresponds to their negative ‘forward’ rotation. In the study by Trabuco et al. (43), the global reference system and the H76 orientation were based on the principal axes of inertia for suitably chosen parts of the large subunit. The change between the open and the half-closed conformation in terms of their angular coordinates roughly corresponds to the changes in ϕ1,ϕ2 observed here.
A flexible core, flexible helix model of the three-way junction
The analysis presented so far suggests isotropic H76 fluctuations, but does not identify their origin. Are they predominantly due to the flexible junction core, due to the flexible H76 helix, or both? To clarify the issue, we propose a model in which both the H76 helix and the junction core are modelled as harmonic, possibly anisotropic flexible elements (see ‘Methods’ section and Figure 4).
We first test the validity of the model against the simulated data. The means of ϕ1,ϕ2 are nearly independent of L (SD 1–3°), suggesting that H76 in its equilibrium conformation is indeed close to straight, as the model requires. The diagonal entries of the covariance matrices of ϕ1,ϕ2 are well described by linear functions in L, while the off-diagonal entries are very small (Supplementary Figure S2). This implies an essentially homogeneous helix with very small stiffness variations. Furthermore, the two diagonal entries in each matrix have similar values (Supplementary Figure S2), implying isotropic stiffness of both the helix and the core. Thus, the simulation data are consistent with a model of an intrinsically straight, isotropically flexible H76 helix attached to an isotropically flexible junction core.
The isotropic model can now be parameterized. For each system, we first compute the means 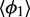 and
and 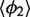 for different values of L and then calculate their averages over L. This gives estimates of the parameters
for different values of L and then calculate their averages over L. This gives estimates of the parameters 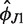 and
and  describing the H76 equilibrium configuration with respect to H75 and H79 [Equation (4)]. To estimate the stiffness parameters, we notice that the model predicts the bending angle variance
describing the H76 equilibrium configuration with respect to H75 and H79 [Equation (4)]. To estimate the stiffness parameters, we notice that the model predicts the bending angle variance 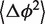 defined by Equation (6) to be a linear function of L [Equation (5)]. The absolute term in Equation (5) is related to the core stiffness constant aJ, the linear term is related to the helix stiffness constant aH, or equivalently to its persistence length Lp [Equation (7)]. Thus, by plotting
defined by Equation (6) to be a linear function of L [Equation (5)]. The absolute term in Equation (5) is related to the core stiffness constant aJ, the linear term is related to the helix stiffness constant aH, or equivalently to its persistence length Lp [Equation (7)]. Thus, by plotting 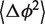 as a function of L and fitting a linear function, we obtain estimates for both the core and the helix stiffness parameters (Figure 4). The intersection of the line with the y-axis determines the stiffness of the core, the line slope determines the stiffness of the helix. Furthermore, the y-axis intersection gives the contribution to
as a function of L and fitting a linear function, we obtain estimates for both the core and the helix stiffness parameters (Figure 4). The intersection of the line with the y-axis determines the stiffness of the core, the line slope determines the stiffness of the helix. Furthermore, the y-axis intersection gives the contribution to 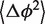 coming from the junction core (see ‘Methods’ section).
coming from the junction core (see ‘Methods’ section).
We performed this analysis for the E.c., H.m. and T.t. systems (Figure 4 and Supplementary Figure S3). The initial movement of the T.t. H76 from the half-closed to the open conformation took no more than several nanoseconds and its influence was neglected. The D.r. system was not analysed due to instabilities at the base of H76 (see Supplementary Data). For E.c. and T.t., the local helical structure of H76 was not well defined in several locations (each containing wobble pairs), where the helical rise showed large fluctuations and its value could not be reliably estimated (areas in Figure 4, Supplementary Figures S2 and S3 with no crosses). The contour length over that area was computed as the distance between the base pair centres at the sides, each projected onto the local helical axis of the adjacent helical domain.
The inferred model parameters are summarized in Table 1. The equilibrium configuration of H76 lies roughly in the yz plane perpendicular to H75 (small absolute values of 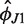 ) and points away from the 30S subunit (large positive
) and points away from the 30S subunit (large positive 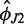 ), as expected for the open conformation. The force field-dependent differences are quite small except
), as expected for the open conformation. The force field-dependent differences are quite small except 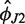 for T.t. where the difference is 11°, but the qualitative features just described remain unaffected. The deduced stiffness parameters (Table 1) indicate that the junction core is rather flexible. Indeed, the values of aJ in Table 1 are comparable to the stiffness constants for the most flexible intra-base pair or base pair step angular coordinates of DNA (76,77). Our estimated H76 persistence lengths Lp are similar to those of simulated A-RNA duplexes (80,81,99) and can be compared with the experimental RNA persistence length of 70–80 nm (100). Notice that the parmbsc0 values of the persistence length are consistently higher than the parm99 ones and fit well within the experimental range. Given that the experimental value also includes the effect of static disorder and was obtained in the presence of Mg2+, which soften the helix (100), it is possible that our persistence length is still somewhat underestimated.
for T.t. where the difference is 11°, but the qualitative features just described remain unaffected. The deduced stiffness parameters (Table 1) indicate that the junction core is rather flexible. Indeed, the values of aJ in Table 1 are comparable to the stiffness constants for the most flexible intra-base pair or base pair step angular coordinates of DNA (76,77). Our estimated H76 persistence lengths Lp are similar to those of simulated A-RNA duplexes (80,81,99) and can be compared with the experimental RNA persistence length of 70–80 nm (100). Notice that the parmbsc0 values of the persistence length are consistently higher than the parm99 ones and fit well within the experimental range. Given that the experimental value also includes the effect of static disorder and was obtained in the presence of Mg2+, which soften the helix (100), it is possible that our persistence length is still somewhat underestimated.
Table 1.
Parameters of the isotropic model
| Simulation | Helix direction 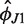 (deg) (deg) |
Helix direction 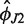 (deg) (deg) |
Core stiffness aJ (kcal/mol.deg2) | Helix persistence length Lp (nm) | Core bending angle variationa (%) |
|---|---|---|---|---|---|
| E.c. parm99 | −7 | 13 | 0.0048 | 68 | 44 |
| H.m. parm99 | −4 | 21 | 0.0089 | 64 | 27 |
| T.t. parm99 | 5 | 21 | 0.0091 | 57 | 25 |
| E.c. bsc0b | −6 | 17 | 0.0060 | 74 | 41 |
| H.m. bsc0 | −3 | 23 | 0.0079 | 79 | 35 |
| T.t. bsc0 | 3 | 32 | 0.0100 | 75 | 29 |
aFraction of the bending angle variation due to the junction core.
bWith the parmχOL3 correction.
GU wobble pairs do not substantially affect the H76 flexibility
It has been proposed that the movement of the L1 stalk can be facilitated by the cluster of wobble pairs in H76, present in nearly all species (2,19). This view was partially based on an NMR study of the tRNAAla microhelix by Ramos and Varani (101), where the authors identified local conformational averaging between multiple conformers and a static distortion at the G3/U70 wobble pair. However, they also noticed that the overall structure of the microhelix was close to the A-form RNA and that base stacking was similar to what would be expected for a G-C containing helix. Our results are in fact consistent with the findings of Ramos and Varani. Indeed, although the helical structure of our H76 locally fluctuates at various positions containing wobble pairs, the helix as a whole remains straight and exhibits uniform, isotropic flexibility comparable to that of a regular A-RNA duplex (Table 1). In conclusion, the GU wobble pairs do not modulate the flexibility of H76 in any extent that would be detectable by the simulations.
The role of the L1 stalk junction core
The junction core has also been suggested as a putative hinge region for movement of the L1 stalk (2,30) The model proposed here a priori considers both the core and the helix to be flexible, which enables us to quantify their contributions to flexibility by fitting the model to the simulated fluctuations. Our results indicate that both the core and the helix contribute substantially. The contribution of the core to the H76 bending angle variation is found to be 25–45% (Table 1). In conclusion, there is a hinge at the core of the L1 stalk three-way junction, but the hinge has a nonzero stiffness and is isotropic. Another, somewhat larger contribution to the overall flexibility comes from the basic flexibility of the long A-RNA H76 helix.
Influence of the ribosomal environment
In this work we study the L1 stalk three-way junction in isolation. In the ribosome, however, the junction makes contacts with its structural environment. The question thus arises as to how these contacts modulate the dynamics and flexibility of the junction. As described below, the H75 and H79 helices make numerous tertiary contacts with the surrounding rRNA and ribosomal proteins. In contrast, H76 makes no contact with its surroundings, except perhaps for the first two of its base pairs. Thus, H75 and H79 can be considered attached to the ribosome, while H76 is essentially free. In our description, we mimic this situation by fixing our global coordinate system onto H75 and H79, as detailed in ‘Methods’ section. The helical axes of H75 and H79 defining the coordinate system are computed as directional averages of the local helical axes, which largely eliminates the effect of local structural fluctuations within the helices. There may be an additional effect due to the relative motion between the effectively rigid helices, but we checked that these movements are small. In conclusion, our approach should yield a rather realistic description of the L1 stalk junction with H75 and H79 fixed, corresponding to the junction built into the ribosome.
The intrinsic flexibility described by our model reflects the most basic RNA flexibility mode which does not involve any changes of the RNA secondary or tertiary structures. It defines the region of easily accessible positions of the L1 stalk RNA, spontaneously sampled by thermal fluctuations. We suggest that other ribosomal elements may utilize this intrinsic L1 stalk rRNA flexibility when imposing, or making use of, different L1 stalk functional states. Within the range of its intrinsic flexibility as identified by our model, the RNA can passively adapt to structural changes of the ribosome on the time scale of <100 ns.
Comparison with the L7/L12 stalk three-way junction and other flexible RNA systems
Different RNA building blocks exhibit different thermally driven structural fluctuations as a result of differences in their topology and intra-molecular interactions. MD simulations represent an established method to capture these fluctuations. Structural fluctuations seen in simulations can be analysed using various approaches, such as visual inspection, clustering, distance and angle measurements, principal component analysis, etc. Upon defining a suitable set of global coordinates, one can deduce the intrinsic stiffness associated with these coordinates, as done in this work. Each of the approaches represents a different viewpoint from which the molecular fluctuations are assessed and which together compose the overall picture of the RNA structural dynamics. The precise quantitative meaning of flexibility and anisotropy, however, may differ between the approaches.
Recently, Besseova et al. (50) carried out extensive simulation analysis of three ribosomal RNA three-way junctions that occur in dynamical or potentially dynamical, functionally important regions of the large ribosomal subunit. All of these junctions (the H90–H92, H42–H44 and 5S rRNA junctions) were of type C (102), so that their topology is different from the type A junction of the L1 stalk studied here. In type C junctions, two of the helices (P1 and P3) are parallel to each other and usually make interhelical contacts, while the third one (P2) is coaxially stacked on P1. Thus, globally there are just two interconnected stems, the P1/P3 domain and the P2 helix. The global deformation of the junctions was quantified using an inter-stem angle defined by the centres of mass of 3 bp, one in each of the two stems and the third one in the middle of the junction. The character of the global motion was further assessed by visually inspecting structures of maximal and minimal inter-stem angle, for which atoms of one of the stems were superimposed. In a related study, Reblova et al. (103) investigated the elbow-shaped kink-turn Kt-38 at the base of A-site finger (H38) from the H.m. ribosome and equivalently positioned molecular elbows in eubacteria using simulations and Cryo-EM data. The global motion was again described using an interhelical angle and structure superposition. Another very flexible RNA element studied recently is the reverse K-turn (51). The global conformational descriptors used include the atomic root mean square deviation, radius of gyration and end-to-end distance. In all of these three studies, the analysis techniques employed there suggest that the structural motifs investigated possess anisotropic (or directional) flexibility. In contrast, our analysis indicates that the prokaryotic and archaeal L1 stalk three-way junction is globally isotropic, as detailed earlier. A precise quantitative comparison, however, would require a description of all the systems from a common viewpoint, which is beyond the scope of the present work.
Similarly, a simulation and elasticity study of intrinsic flexibility of the rRNA helix h44 from the small ribosomal subunit and a canonical A-RNA duplex (47) used Essential dynamics analysis to suggest that this particular system is isotropic, similar to the H76 portion of the L1 stalk rRNA. Again, since the global geometry description of this system was different from ours, a rigorous, quantitative comparison of flexibility and anisotropy cannot be accomplished. The basic analysis of the simulations should nevertheless provide a relevant qualitative assessment of fluctuations of the different rRNA elements.
As noticed already by Hagerman (100), a proper description of the conformational variation of non-helical elements should also take into account the finite stiffness of the flanking helices. His analysis shows that, for instance, the core of yeast tRNAPhe is not substantially more flexible than an equivalent length of pure helix. An MD study discussed earlier (103) suggests that the flexible helical arms attached to Kt-38 and the equivalent elbow elements substantially enhance the range of their angular fluctuations. In our case, we observe an analogous phenomenon: our data are best explained by a model where both the junction core and the helix are flexible. While rigid helix models are fully justified if the junction core is very flexible and the helices are short, finite stiffness of both the core and the helix must be considered for the systems studied here. In other words, the flexibility in our system is smoothly distributed over multiple nucleotides, which may be easier to achieve than attempting to construct a highly localized RNA hinge and, in addition, it is probably a more robust evolutionary solution.
Our approach can be contrasted with studies aimed at identifying RNA conformations constrained by secondary structure. Sim and Levitt (104) proposed a model of rigid helices, each characterized by its orientation with respect to a reference helix. Al-Hashimi and co-workers (105–108) describe the geometry of a two-way junction by specifying the general rotation of one rigid helix with respect to the other one. Notice, however, that the aim of these works is different from ours. Those studies typically generate a rather narrow ensemble of topologically admissible conformations, from which tertiary contacts and intermolecular interactions select and stabilize specific conformers. In contrast, our aim is to understand functionally relevant mechanical properties of these conformers which contribute to the stochastic dynamics of the whole molecular machine.
Interactions within the junction and their stability
The helical stems are predominantly composed of canonical base pairs and wobble pairs while the junction core, which is specifically shaped, contains three conserved non-Watson–Crick (non-WC) pairs, namely a trans Hoogsteen/Watson–Crick (tH/W) A/U base pair, a trans Hoogsteen/Sugar-Edge (tH/S) A/G ‘sheared’ base pair and a tH/H base pair (U/A in E.c., H.m. and T.t. and U/C in D.r.) (Figure 2). The interface between H76/H79 and H75 exhibits two significant bends of the sugar–phosphate backbone. The first one, positioned in the strand connecting H79 and H75, is a hook turn motif found at various places of 16S and 23S rRNAs (109), while the second one connecting H75 and H76 has not been classified and resembles the sharp bend seen in kink-turn structures (110) (Figure 2 and Supplementary Figure S4). These bends are interconnected by the tH/H pair which involves a single hydrogen bond and has a relatively long C1′–C1′distance (over 12 Å for U/A and U/C). We carried out a search for the tH/H pairs in ribosomal RNAs using the FR3D program (111) and noticed that they often occur in irregular ribosomal RNA segments. The tH/W and tH/S pairs together with the two adjacent bulge bases form an UNA/GAN motif which is also found in the core of the H20/H21/H22 junction of 16S rRNA (112). The UNA/GAN motif is closely related to the UAA/GAN internal loop, a structural motif with a consensus sequence localized at various places of 23S rRNA and also in other RNAs (112). Sugar–phosphate and base–base contacts form a stabilizing network of H-bonds in the central part of the junction (for more details see Supplementary Data and Supplementary Table S2). The simulated junction structures sampled conformations around their starting structures and did not unfold at the 100-ns time scale. Details about the structure stability are in Supplementary Data and Supplementary Figure S5.
Ribosomal contacts of the 3wj
The H75 and H79 helices make a number of tertiary contacts with the surrounding RNA and ribosomal proteins. In contrast, H76 makes no contact with its surroundings, except for 2–3 H-bonds between the first two base pairs of T.t. H76 (2093G = C2196 and 2094G = C2195) and the ribosomal protein L9 (Supplementary Figure S6 and description in Supplementary Data). The structural element closest to H76 is H68. The shortest distance between H76 and H68 is 7.3 Å in D.r., 6.1 Å in H.m. and 5.6 Å in E.c. In the T.t. ribosome, which is in the half-closed conformation and thus has the L1 stalk tilted towards the body of the 50S subunit, the shortest distance between H76 and H68 is only 4.0 Å (Supplementary Figure S6). Specific interactions of H75 and H79 with the surrounding ribosomal structures in all four systems are described in Supplementary Data [the base-backbone interactions are classified according to Zirbel et al. (113)].
CONCLUSION
In this work, we study the global structure and flexibility of the H76/H75/H79 three-way junction found at the basis of the L1 stalk, a biologically important mobile element of the large ribosomal subunit. We propose a new coarse-grained model in which both the junction core and the H76 helix are modelled as harmonic, possibly anisotropic flexible elements. The elements are characterized quantitatively, by means of their equilibrium shape and stiffness parameters. The parameters are inferred from a set of large-scale, unrestrained atomic-resolution molecular dynamics simulations.
The approach proposed here is rather general and can be extended to other types of RNA junctions important in biological systems or in nanotechnology. The quantitative characterization of RNA building blocks in terms of their mechanical properties would make it possible to understand the behaviour of larger systems or to combine the blocks into nanostructures with desired mechanical features.
Our results indicate that both the junction core and the helix H76 are isotropically flexible, and both contribute substantially to the overall H76 movement. The H76 thermal fluctuations in the isolated junction can spontaneously reach the half-closed conformation observed in the translocating ribosome. Introducing a bulge base into H76 as found in eukaryotes induces a very flexible, anisotropic hinge within the helix. The observed bulge dynamics helps to explain the in-position of the L1 stalk found in vacant eukaryotic ribosomes (53) and may rationalize the precise positioning of the H76 bulge in eukaryotes.
We conclude that the prokaryotic L1 stalk rRNA three-way junction is an intrinsically flexible RNA region, whose flexibility stems from the common isotropic flexibility of the long A-RNA H76 helix complemented by the isotropic-hinge flexibility of the junction core. It thus seems to differ from other recently studied flexible RNA systems such as the L7/L12 stalk junction, kink-turns and elbow segment at the base of the A-site finger, which appear to be anisotropically flexible (50,51,103). The eukaryotic H76 enhances its flexibility in an anisotropic manner using the single bulge in its upper part. The L1 stalk rRNA junction allows the stalk to spontaneously sample various conformations at minimal energy cost, at the time scale of ∼100 ns. This suggests that the junction may act as a passive flexible element, allowing the L1 stalk to easily adapt to the surrounding structures and optimize its interactions at various stages of translocation. The local flexibility thus enhances the global movement of the L1 stalk during translocation which itself takes place at much longer time scales of 100 ms to several seconds (27,30) and involves concerted motions of large ribosomal domains.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables S1 and S2 and Supplementary Figures S1–S6.
FUNDING
Czech Science Foundation (203/09/1476, P208/11/1822, P208/12/1878, P305/12/G03 to K.R. and J.S., P208/12/G016 to F.L.); Grant Agency of the Academy of Sciences of the Czech Republic (KJB400040901 to K.R. and J.S.); Academy of Sciences of the Czech Republic (J.E. Purkyně Fellowship and Z40550506 to F.L.) and ‘CEITEC—Central European Institute of Technology’ from European Regional Development Fund (CZ.1.05/1.1.00/02.0068 to K.R. and J.S.). Funding for open access charge: Czech Science Foundation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Joachim Frank for useful discussions and Jie Fu for providing information about their choice of global coordinates.
REFERENCES
- 1.Rodnina MV, Wintermeyer W, Green R. Ribosomes. Structure, Function, and Dynamics. Springer, Wien, New York. 2011 [Google Scholar]
- 2.Korostelev A, Ermolenko DN, Noller HF. Structural dynamics of the ribosome. Curr. Opin. Chem. Biol. 2008;12:674–683. doi: 10.1016/j.cbpa.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–1242. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- 4.Blanchard SC. Single-molecule observations of ribosome function. Curr. Opin. Struct. Biol. 2009;19:103–109. doi: 10.1016/j.sbi.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agirrezabala X, Frank J. Elongation in translation as a dynamic interaction among the ribosome, tRNA, and elongation factors EF-G and EF-Tu. Q. Rev. Biophys. 2009;42:159–200. doi: 10.1017/S0033583509990060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunkle JA, Cate JHD. Ribosome structure and dynamics during translocation and termination. Annu. Rev. Biophys. 2010;39:227–244. doi: 10.1146/annurev.biophys.37.032807.125954. [DOI] [PubMed] [Google Scholar]
- 7.Agirrezabala X, Frank J. From DNA to proteins via the ribosome: structural insights into the workings of the translation machinery. Hum. Genomics. 2010;4:226–237. doi: 10.1186/1479-7364-4-4-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aitken CE, Petrov A, Puglisi JD. Single ribosome dynamics and the mechanism of translation. Annu. Rev. Biophys. 2010;39:491–513. doi: 10.1146/annurev.biophys.093008.131427. [DOI] [PubMed] [Google Scholar]
- 9.Frank J, Gonzalez RL., Jr Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu. Rev. Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrov A, Kornberg G, O’Leary S, Tsai A, Uemura S, Puglisi JD. Dynamics of the translational machinery. Curr. Opin. Struct. Biol. 2011;21:137–145. doi: 10.1016/j.sbi.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 12.Gao H, Sengupta J, Valle M, Korostelev A, Eswar N, Stagg SM, Van Roey P, Agrawal RK, Harvey SC, Sali A, et al. Study of the structural dynamics of the E. coli 70S ribosome using real-space refinement. Cell. 2003;113:789–801. doi: 10.1016/s0092-8674(03)00427-6. [DOI] [PubMed] [Google Scholar]
- 13.Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J. Locking and unlocking of ribosomal motions. Cell. 2003;114:123–134. doi: 10.1016/s0092-8674(03)00476-8. [DOI] [PubMed] [Google Scholar]
- 14.Frank J, Gao H, Sengupta J, Gao N, Taylor DJ. The process of mRNA-tRNA translocation. Proc. Natl. Acad. Sci. USA. 2007;104:19671–19678. doi: 10.1073/pnas.0708517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agirrezabala X, Lei J, Brunelle JL, Ortiz-Meoz RF, Green R, Frank J. Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol. Cell. 2008;32:190–197. doi: 10.1016/j.molcel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Julián P, Konevega AL, Scheres SH, Lázaro M, Gil D, Wintermeyer W, Rodnina MV, Valle M. Structure of ratcheted ribosomes with tRNAs in hybrid states. Proc. Natl Acad. Sci. USA. 2008;105:16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- 18.Fu J, Munro JB, Blanchard SC, Frank J. Cryoelectron microscopy structures of the ribosome complex in intermediate states during tRNA translocation. Proc. Natl Acad. Sci. USA. 2011;108:4817–4821. doi: 10.1073/pnas.1101503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 2006;126:1065–1077. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Selmer M, Dunham CM, Murphy FV, IV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 21.Korostelev A, Trakhanov S, Asahara H, Laurberg M, Lancaster L, Noller HF. Interactions and dynamics of the Shine Dalgarno helix in the 70S ribosome. Proc. Natl Acad. Sci. USA. 2007;104:16840–16843. doi: 10.1073/pnas.0707850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blaha G, Stanley RE, Steitz TA. Formation of the first peptide bond: the structure of EF-P bound to the 70S ribosome. Science. 2009;325:966–970. doi: 10.1126/science.1175800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Dunkle JA, Cate JHD. Structures of the ribosome in intermediate states of ratcheting. Science. 2009;325:1014–1017. doi: 10.1126/science.1175275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munro JB, Altman RB, O’Connor N, Blanchard SC. Identification of two distinct hybrid state intermediates on the ribosome. Mol. Cell. 2007;25:505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fei J, Kosuri P, MacDougall DD, Gonzalez RL., Jr Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol. Cell. 2008;30:348–359. doi: 10.1016/j.molcel.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Cornish PV, Ermolenko DN, Noller HF, Ha T. Spontaneous intersubunit rotation in single ribosomes. Mol. Cell. 2008;30:578–588. doi: 10.1016/j.molcel.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cornish PV, Ermolenko DN, Staple DW, Hoang L, Hickerson RP, Noller HF, Ha T. Following movement of the L1 stalk between three functional states in single ribosomes. Proc. Natl Acad. Sci. USA. 2009;106:2571–2576. doi: 10.1073/pnas.0813180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sternberg SH, Fei J, Prywes N, McGrath KA, Gonzalez RL., Jr Translation factors direct intrinsic ribosome dynamics during translation termination and ribosome recycling. Nat. Struct. Mol. Biol. 2009;16:861–868. doi: 10.1038/nsmb.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fei J, Bronson JE, Hofman JM, Srinivas RL, Wiggins CH, Gonzalez RL., Jr Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc. Natl Acad. Sci. USA. 2009;106:15702–15707. doi: 10.1073/pnas.0908077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munro JB, Altman RB, Tung CS, Cate JH, Sanbonmatsu KY, Blanchard SC. Spontaneous formation of the unlocked state on the ribosome is a multistep process. Proc. Natl Acad. Sci. USA. 2010;107:709–714. doi: 10.1073/pnas.0908597107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aitken CE, Puglisi JD. Following the intersubunit conformation of the ribosome during translation in real time. Nat. Struct. Mol. Biol. 2010;17:793–800. doi: 10.1038/nsmb.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munro JB, Wasserman MR, Altman RB, Wang L, Blanchard SC. Correlated conformational events in EF-G and the ribosome regulate translocation. Nat. Struct. Mol. Biol. 2010;17:1470–1477. doi: 10.1038/nsmb.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fei J, Richard AC, Bronson JE, Gonzalez RL., Jr Transfer RNA-mediated regulation of ribosome dynamics during protein synthesis. Nat. Struct. Mol. Biol. 2011;18:1043–1051. doi: 10.1038/nsmb.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rakauskaite R, Dinman JD. An arc of unpaired ‘hinge bases’ facilitates information exchange among functional centers of the ribosome. Mol. Cell. Biol. 2006;26:8992–9002. doi: 10.1128/MCB.01311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komoda T, Sato NS, Phelps SS, Namba N, Joseph S, Suzuki T. The A-site finger in 23 S rRNA acts as a functional attenuator for translocation. J. Biol. Chem. 2006;281:32303–32309. doi: 10.1074/jbc.M607058200. [DOI] [PubMed] [Google Scholar]
- 36.Sergiev PV, Kiparisov SV, Burakovsky DE, Lesnyak DV, Leonov AA, Bogdanov AA, Dontsova OA. The conserved A-site finger of the 23 S rRNA: just one of the intersubunit bridges or a part of the allosteric communication pathway? J. Mol. Biol. 2005;353:116–123. doi: 10.1016/j.jmb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Spirin AS. The ribosome as a conveying thermal ratchet machine. J. Biol. Chem. 2009;284:21103–21119. doi: 10.1074/jbc.X109.001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhodin MH, Dinman JD. An extensive network of information flow through the B1b/c intersubunit bridge of the yeast ribosome. PLoS One. 2011;6:e20048. doi: 10.1371/journal.pone.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ninio J. Multiple stages in codon-anticodon recognition: double-trigger mechanisms and geometric constraints. Biochimie. 2006;88:963–992. doi: 10.1016/j.biochi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Tama F, Valle M, Frank J, Brooks CL., III Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc. Natl Acad. Sci. USA. 2003;100:9319–9323. doi: 10.1073/pnas.1632476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Rader AJ, Bahar I, Jernigan RL. Global ribosome motions revealed with elastic network model. J. Struct. Biol. 2004;147:302–314. doi: 10.1016/j.jsb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Trylska J, Tozzini V, McCammon JA. Exploring global motions and correlations in the ribosome. Biophys. J. 2005;89:1455–1463. doi: 10.1529/biophysj.104.058495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trabuco LG, Schreiner E, Eargle J, Cornish P, Ha T, Luthey-Schulten Z, Schulten K. The role of L1 stalk-tRNA interaction in the ribosome elongation cycle. J. Mol. Biol. 2010;402:741–760. doi: 10.1016/j.jmb.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z, Sanbonmatsu KY, Voth GA. Key intermolecular interactions in the E. coli 70S ribosome revealed by coarse-grained analysis. J. Am. Chem. Soc. 2011;133:16828–16838. doi: 10.1021/ja2028487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitford PC, Ahmed A, Yu Y, Hennelly SP, Tama F, Spahn CM, Onuchic JN, Sanbonmatsu KY. Excited states of ribosome translocation revealed through integrative molecular modeling. Proc. Natl Acad. Sci. USA. 2011;108:18943–18948. doi: 10.1073/pnas.1108363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rázga F, Koca J, Sponer J, Leontis NB. Hinge-like motions in RNA kink-turns: the role of the second A-minor motif and nominally unpaired bases. Biophys. J. 2005;88:3466–3485. doi: 10.1529/biophysj.104.054916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Réblová K, Lankas F, Rázga F, Krasovska MV, Koca J, Sponer J. Structure, dynamics, and elasticity of free 16S rRNA helix 44 studied by molecular dynamics simulations. Biopolymers. 2006;82:504–520. doi: 10.1002/bip.20503. [DOI] [PubMed] [Google Scholar]
- 48.Reblova K, Razga F, Li W, Gao H, Frank J, Sponer J. Dynamics of the base of ribosomal A-site finger revealed by molecular dynamics simulations and cryo-EM. Nucleic Acids Res. 2009;38:1325–1340. doi: 10.1093/nar/gkp1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reblova K, Strelcova Z, Kulhanek P, Besseova I, Mathews DH, Van Nostrand K, Yildirim I, Turner DH, Sponer J. An RNA molecular switch: intrinsic flexibility of 23S rRNA helices 40 and 68 5′-UAA/5′-GAN internal loops studied by molecular dynamics methods. J. Chem. Theor. Comput. 2010;6:910–929. [PMC free article] [PubMed] [Google Scholar]
- 50.Besseová I, Réblova K, Leontis NB, Sponer J. Molecular dynamics simulations suggest that RNA three-way junctions can act as flexible RNA structural elements in the ribosome. Nucleic Acids Res. 2010;38:6247–6264. doi: 10.1093/nar/gkq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sklenovsky P, Florova P, Banas P, Reblova K, Lankas F, Otyepka M, Sponer J. Understanding RNA flexibility using explicit solvent simulations: the ribosomal and group I intron reverse kink-turn motifs. J. Chem. Theor. Comput. 2011;7:2963–2980. doi: 10.1021/ct200204t. [DOI] [PubMed] [Google Scholar]
- 52.Bokov K, Steinberg SV. A hierarchical model for evolution of 23S ribosomal RNA. Nature. 2009;457:977–980. doi: 10.1038/nature07749. [DOI] [PubMed] [Google Scholar]
- 53.Spahn CM, Gømez-Lorenzo MG, Grassucci RA, Jorgensen R, Andersen GR, Beckmann R, Penczek PA, Ballesta JPG, Frank J. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 2004;23:1008–1019. doi: 10.1038/sj.emboj.7600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor DJ, Devkota B, Huang AD, Topf M, Narayanan E, Sali A, Harvey SC, Frank J. Comprehensive molecular structure of the eukaryotic ribosome. Structure. 2009;17:1591–1604. doi: 10.1016/j.str.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armache JP, Jarasch A, Anger AM, Villa E, Becker T, Bhushan S, Jossinet F, Habeck M, Dindar G, Franckenberg S, et al. Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-A resolution. Proc. Natl Acad. Sci. USA. 2010;107:19748–19753. doi: 10.1073/pnas.1009999107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Crystal structure of the eukaryotic ribosome. Science. 2010;330:1203–1209. doi: 10.1126/science.1194294. [DOI] [PubMed] [Google Scholar]
- 57.Budkevich T, Giesebrecht J, Altman RB, Munro JB, Mielke T, Nierhaus KH, Blanchard SC, Spahn CMT. Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol. Cell. 2011;44:214–224. doi: 10.1016/j.molcel.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science. 2011;334:941–948. doi: 10.1126/science.1211204. [DOI] [PubMed] [Google Scholar]
- 59.Ramakrishnan V. The eukaryotic ribosome. Science. 2011;331:681–682. doi: 10.1126/science.1202093. [DOI] [PubMed] [Google Scholar]
- 60.Ditzler MA, Otyepka M, Sponer J, Walter NG. Molecular dynamics and quantum mechanics of RNA: conformational and chemical change we can believe in. Acc. Chem. Res. 2010;43:40–47. doi: 10.1021/ar900093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitford PC, Geggier P, Altman RB, Blanchard SC, Onuchic JN, Sanbonmatsu KY. Accommodation of aminoacyl-tRNA into the ribosome involves reversible excursions along multiple pathways. RNA. 2010;16:1196–1204. doi: 10.1261/rna.2035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanbonmatsu KY, Joseph S, Tung C-S. Simulating movement of tRNA into the ribosome during decoding. Proc. Natl Acad. Sci. USA. 2005;102:15854–15859. doi: 10.1073/pnas.0503456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vaiana AC, Sanbonmatsu KY. Stochastic gating and drug-ribosome interactions. J. Mol. Biol. 2009;386:648–661. doi: 10.1016/j.jmb.2008.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Villa E, Sengupta J, Trabuco LG, LeBarron J, Baxter WT, Shaikh TR, Grassucci RA, Nissen P, Ehrenberg M, Schulten K, et al. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc. Natl Acad. Sci. USA. 2009;106:1063–1068. doi: 10.1073/pnas.0811370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trylska J. Coarse-grained models to study dynamics of nanoscale biomolecules and their applications to the ribosome. J. Phys. Condens. Matter. 2010;22:453101. doi: 10.1088/0953-8984/22/45/453101. [DOI] [PubMed] [Google Scholar]
- 67.Leontis NB, Lescoute A, Westhof E. The building blocks and motifs of RNA architecture. Curr. Opin. Struct. Biol. 2006;16:279–287. doi: 10.1016/j.sbi.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jaeger L, Westhof E, Leontis NB. TectoRNA: modular assembly units for the construction of RNA nano-objects. Nucleic Acids Res. 2001;29:455–463. doi: 10.1093/nar/29.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaeger L, Chworos A. The architectonics of programmable RNA and DNA nanostructures. Curr. Opin. Struct. Biol. 2006;16:531–543. doi: 10.1016/j.sbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 70.Novikova IV, Hassan BH, Mirzoyan MG, Leontis NB. Engineering cooperative tecto-RNA complexes having programmable stoichiometries. Nucleic Acids Res. 2010;39:2903–2917. doi: 10.1093/nar/gkq1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rázga F, Koca J, Mokdad A, Sponer J. Elastic properties of ribosomal RNA building blocks: molecular dynamics of the GTPase-associated center rRNA. Nucleic Acids Res. 2007;35:4007–4017. doi: 10.1093/nar/gkm245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Curuksu J, Sponer J, Zacharias M. Elbow flexibility of the kt38 RNA kink-turn motif investigated by free-energy molecular dynamics simulations. Biophys. J. 2009;97:2004–2013. doi: 10.1016/j.bpj.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lankas F, Sponer J, Hobza P, Langowski J. Sequence-dependent elastic properties of DNA. J. Mol. Biol. 2000;299:695–709. doi: 10.1006/jmbi.2000.3781. [DOI] [PubMed] [Google Scholar]
- 74.Gonzalez O, Maddocks JH. Extracting parameters for base-pair level models of DNA from molecular dynamics simulations. Theor. Chem. Acc. 2001;106:76–82. [Google Scholar]
- 75.Lankas F, Gonzalez O, Heffler LM, Stoll G, Moakher M, Maddocks JH. On the parameterization of rigid base and basepair models of DNA from molecular dynamics simulations. Phys. Chem. Chem. Phys. 2009;11:10565–10588. doi: 10.1039/b919565n. [DOI] [PubMed] [Google Scholar]
- 76.Lankas F, Sponer J, Langowski J, Cheatham TE., III DNA basepair step deformability inferred from molecular dynamics simulations. Biophys. J. 2003;85:2872–2883. doi: 10.1016/S0006-3495(03)74710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lankas F, Sponer J, Langowski J, Cheatham TE., III DNA deformability at the base pair level. J. Am. Chem. Soc. 2004;126:4124–4125. doi: 10.1021/ja0390449. [DOI] [PubMed] [Google Scholar]
- 78.Becker N, Everaers R. From rigid base pairs to semiflexible polymers: coarse-graining DNA. Phys. Rev. E, Stat. Nonlin, Soft. Mather. Phys. 2007;76:021923. doi: 10.1103/PhysRevE.76.021923. [DOI] [PubMed] [Google Scholar]
- 79.Becker NB, Wolff L, Everaers R. Indirect readout: detection of optimized subsequences and calculation of relative binding affinities using different DNA elastic potentials. Nucleic Acids Res. 2006;34:5638–5649. doi: 10.1093/nar/gkl683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Noy A, Pérez A, Lankas F, Luque F, Orozco M. Relative flexibility of DNA and RNA: a molecular dynamics study. J. Mol. Biol. 2004;343:627–638. doi: 10.1016/j.jmb.2004.07.048. [DOI] [PubMed] [Google Scholar]
- 81.Faustino I, Pérez A, Orozco M. Toward a consensus view of duplex RNA flexibility. Biophys. J. 2010;99:1876–1885. doi: 10.1016/j.bpj.2010.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JHD. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 83.Harms JM, Wilson DN, Schluenzen F, Connell SR, Stachelhaus T, Zaborowska Z, Spahn CMT, Fucini P. Translational regulation via L11: molecular switches on the ribosome turned on and off by thiostrepton and micrococcin. Mol. Cell. 2008;30:26–38. doi: 10.1016/j.molcel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 84.Klein DJ, Moore PB, Steitz TA. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 2004;340:141–177. doi: 10.1016/j.jmb.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 85.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Müller KM, et al. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mokdad A, Leontis NB. Ribostral: an RNA 3D alignment analyzer and viewer based on basepair isostericities. Bioinformatics. 2006;22:2168–2170. doi: 10.1093/bioinformatics/btl360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 88.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 89.Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Réblová K, Fadrná E, Sarzynska J, Kulinski T, Kulhánek P, Ennifar E, Koca J, Sponer J. Conformations of flanking bases in HIV-1 RNA DIS kissing complexes studied by molecular dynamics. Biophys. J. 2007;93:3932–3949. doi: 10.1529/biophysj.107.110056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mlýnsky V, Banás P, Hollas D, Réblova K, Walter NG, Sponer J, Otyepka M. Extensive molecular dynamics simulations showing that canonical G8 and protonated A38H+ forms are most consistent with crystal structures of hairpin ribozyme. J. Phys. Chem. B. 2010;114:6642–6652. doi: 10.1021/jp1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Banas P, Hollas D, Zgarbova M, Jurecka P, Orozco M, Cheatham TE, III, Sponer J, Otyepka M. Performance of molecular mechanics force fields for RNA simulations: stability of UUCG and GNRA hairpins. J. Chem. Theory Comput. 2010;6:3836–3849. doi: 10.1021/ct100481h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zgarbova M, Otyepka M, Sponer J, Mladek A, Banas P, Cheatham TE, III, Jurecka P. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011;7:2886–2902. doi: 10.1021/ct200162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Besseova I, Otyepka M, Reblova K, Sponer J. Dependence of A-RNA simulations on the choice of the force field and salt strength. Phys. Chem. Chem. Phys. 2009;11:10701–10711. doi: 10.1039/b911169g. [DOI] [PubMed] [Google Scholar]
- 95.Réblová K, Šponer JE, Špacková N, Beššeová I, Šponer J. A-minor tertiary interactions in RNA kink-turns. Molecular dynamics and quantum chemical analysis. J. Phys. Chem. B. 2011;115:13897–13910. doi: 10.1021/jp2065584. [DOI] [PubMed] [Google Scholar]
- 96.Lu X-J, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grosberg,A.Y. and Khokhlov,A.R. (1994) Statistical Physics of Macromolecules. AIP Press, New York.
- 98.Berk V, Zhang W, Pai RD, Cate JHD. Structural basis for mRNA and tRNA positioning on the ribosome. Proc. Natl Acad. Sci. USA. 2006;103:15830–15834. doi: 10.1073/pnas.0607541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pérez A, Noy A, Lankas F, Luque FJ, Orozco M. The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 2004;32:6144–6151. doi: 10.1093/nar/gkh954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hagerman PJ. Flexibility of RNA. Annu. Rev. Biophys. Biomol. Struct. 1997;26:139–156. doi: 10.1146/annurev.biophys.26.1.139. [DOI] [PubMed] [Google Scholar]
- 101.Ramos A, Varani G. Structure of the acceptor stem of Escherichia coli tRNAAla: role of the G3.U70 base pair in synthetase recognition. Nucleic Acids Res. 1997;25:2083–2090. doi: 10.1093/nar/25.11.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lescoute A, Westhof E. Topology of three-way junctions in folded RNAs. RNA. 2006;12:83–93. doi: 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Réblová K, Rázga F, Li W, Gao H, Frank J, Sponer J. Dynamics of the base of ribosomal A-site finger revealed by molecular dynamics simulations and Cryo-EM. Nucleic Acids Res. 2010;38:1325–1340. doi: 10.1093/nar/gkp1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sim AY, Levitt M. Clustering to identify RNA conformations constrained by secondary structure. Proc. Natl Acad. Sci. USA. 2011;108:3590–3595. doi: 10.1073/pnas.1018653108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature. 2007;450:1263–1267. doi: 10.1038/nature06389. [DOI] [PubMed] [Google Scholar]
- 106.Bailor MH, Musselman C, Hansen AL, Gulati K, Patel DJ, Al-Hashimi HM. Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. Nat. Protoc. 2007;2:1536–1546. doi: 10.1038/nprot.2007.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Frank AT, Stelzer AC, Al-Hashimi HM, Andricioaei I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: new insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 2009;37:3670–3679. doi: 10.1093/nar/gkp156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science. 2010;327:202–206. doi: 10.1126/science.1181085. [DOI] [PubMed] [Google Scholar]
- 109.Szép S, Wang J, Moore PB. The crystal structure of a 26-nucleotide RNA containing a hook-turn. RNA. 2003;9:44–51. doi: 10.1261/rna.2107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl Acad. Sci. USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008;56:215–252. doi: 10.1007/s00285-007-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee JC, Gutell RR, Russell R. The UAA/GAN internal loop motif: a new RNA structural element that forms a cross-strand AAA stack and long-range tertiary interactions. J. Mol. Biol. 2006;360:978–988. doi: 10.1016/j.jmb.2006.05.066. [DOI] [PubMed] [Google Scholar]
- 113.Zirbel CL, Sponer JE, Sponer J, Stombaugh J, Leontis NB. Classification and energetics of the base-phosphate interactions in RNA. Nucleic Acids Res. 2009;37:4898–4918. doi: 10.1093/nar/gkp468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.