


Abstract
The crucial role of Myc as an oncoprotein and as a key regulator of cell growth makes it essential to understand the molecular basis of Myc function. The N-terminal region of c-Myc coordinates a wealth of protein interactions involved in transformation, differentiation and apoptosis. We have characterized in detail the intrinsically disordered properties of Myc-1–88, where hierarchical phosphorylation of S62 and T58 regulates activation and destruction of the Myc protein. By nuclear magnetic resonance (NMR) chemical shift analysis, relaxation measurements and NOE analysis, we show that although Myc occupies a very heterogeneous conformational space, we find transiently structured regions in residues 22–33 and in the Myc homology box I (MBI; residues 45–65); both these regions are conserved in other members of the Myc family. Binding of Bin1 to Myc-1–88 as assayed by NMR and surface plasmon resonance (SPR) revealed primary binding to the S62 region in a dynamically disordered and multivalent complex, accompanied by population shifts leading to altered intramolecular conformational dynamics. These findings expand the increasingly recognized concept of intrinsically disordered regions mediating transient interactions to Myc, a key transcriptional regulator of major medical importance, and have important implications for further understanding its multifaceted role in gene regulation.
INTRODUCTION
The c-Myc oncoprotein (herein referred to as Myc) is a transcription factor that regulates a wealth of genes involved in a wide-variety of biological activities including apoptosis, differentiation as well as proliferation (1,2). As a critical regulator of both normal and tumor cells, Myc is highly controlled at many levels and is at the center of an extensive interactome (3–5). The Max protein heterodimerizes with the C-terminus of Myc to form a DNA-binding basic helix–loop–helix leucine-zipper (bHLHLZ) entity (Figure 1) (6). In a complex interplay with interactors governing the timing and efficiency of gene activation, Myc is able to regulate the expression of specific genes as well as entire gene networks (7–9).
Figure 1.

Domain structure of c-Myc, indicating conserved Myc homology boxes MBI, MBII, MBIIIa, MBIIIb and MBIV (1). The basic region (BR) N-terminal to the HLHLZ binds DNA in a heterodimeric complex with Max (5). The Myc fragments 1–88 studied in this work is indicated together with an expansion of MBI and phosphorylation sites T58 and S62 located therein.
The N-terminal transactivation domain of Myc (Myc TAD, comprising residues 1–143; Figure 1) is essential for Myc-mediated transformation, differentiation and apoptosis (10–13). This region serves as an interaction platform for proteins involved in chromatin and histone modification as well as ubiquitination and subsequent degradation (14), and has also been shown to independently control protein expression through mRNA translation as well as direct regulation of DNA replication (9,15). Two well-conserved and functionally critical regions among the Myc family proteins, termed Myc homology boxes, were identified primarily by the level of sequence conservation between Myc, N-Myc and L-Myc (1,9). Interactions with Myc homology box I (MBI, residues 47–63) govern the cellular stability of the protein thereby setting a time window for its activity, whereas Myc homology box II (MBII, residues 128–143) coordinates interactions with the transcriptional regulatory machinery (9,11,16) (Figure 1).
In non-transformed cells, Myc has a short protein half-life of 20–30 min, which is largely controlled through phosphorylation of T58 and S62 within MBІ. While phosphorylation at S62 by ERK or CDK kinases is critical for Myc transforming ability and transiently increases Myc cellular stability, subsequent phosphorylation of T58 by GSK3β triggers dephosphorylation of S62 by protein phosphatase 2 A (PP2A) and subsequent ubiquitin-mediated degradation through SCF-Fbwx7 (17–20). In aggressive lymphomas, mutations at or near T58, which disturb its phosphorylation, lead to accumulation and retention of Myc in its activated, S62-phosphorylated state (21,22), highlighting the importance of this region in regulating Myc activity. Additionally, regulators of MBІ have themselves been shown to be oncogenes and tumor suppressors, further emphasizing the importance of understanding the regulation and function of the MBІ regulatory hub (23).
Despite the wide range of reported Myc interactors, there is very limited per-residue information on where and how Myc-interacting proteins bind to the Myc TAD. The only structure available is that of the C-terminal SH3 domain of Bin1 (Bin1–SH3) in complex with a small c-Myc peptide within MBI comprising residues 55–68 (Myc-55–68). (24). Bin1 has been shown to act as a tumor suppressor by binding Myc, thereby controlling cell cycle transit, proliferation and apoptosis (25–29). The structural analysis showed a canonical interaction between the SH3 domain and the proline-rich region of Myc centered on two Xxx-Pro di-peptides P59–P60 and S62–P63 (24). While the affinity of unphosphorylated Myc-55–68 for Bin1–SH3 was significant (KD ∼ 4.2 µM), and unaltered by phosphorylation on T58, the same peptide phosphorylated at S62 was unable to bind Bin1–SH3 even at micromolar concentrations (24). Thus, Bin1 binding could retain Myc in its S62-unphosphorylated, inactive state, which is indirectly supported by liberated Myc cell proliferative activity when Bin1 expression is inhibited by the adenovirus E1A oncoprotein (30).
We have previously shown that, while Myc TAD is intrinsically disordered, it contains two proteolytically semi-resistant entities: Myc-1–88 and Myc-92–167, comprising MBI and MBII, respectively (31). These Myc entities were both shown to be intrinsically disordered, but at various levels of complexity (31–33). We (31) and others (34,35) have suggested that Myc may partially fold on binding to interacting partners, but to date, no structural data has been presented to support this hypothesis.
Therapeutic targeting of the Myc TAD has been proposed as a highly attractive way to achieve selective Myc inhibition (16). To support progress in this direction, more advanced molecular knowledge on the structural and biophysical behavior of the Myc TAD in free and bound states is essential. Although proteomics studies mapping Myc interactors are now emerging (3,4), and identification of Myc TAD-binding proteins have been scored using deletion and point mutants within this region (1,17,25,36–38), neither of these approaches provides information at the molecular resolution required for structural analysis or inhibitor development.
To help forward our understanding of the Myc TAD, alone and in complex with interactors, we present here a detailed characterization of the transient structure and dynamics within Myc-1–88, comprising the MBI region. We report near-complete NMR resonance assignments for Myc-1–88 which form the basis for detailed mapping of interactions and dynamics within this Myc region on a per-residue level. We used these spectroscopic probes to characterize the interaction and dynamics within Myc-1–88 upon interaction with Bin1–SH3. Unexpectedly we find a much more complex mechanism of interaction between Myc-1–88 and Bin1–SH3 compared with previous interaction studies of Bin1–SH3 with a short 13-residue Myc peptide (residues 55–67). Recent findings have provided critical examples on how transactivation domains tailor intrinsic disorder at various levels to recruit and regulate interactions involved in biologically complex events, reviewed in (32,33). We describe how the intrinsic disorder and complex dynamics in Myc together with multivalent interactions within Myc-1–88 and between Myc-1–88 and Bin1–SH3 may be critical for achieving rapid yet accurate response to cellular signals in gene regulation.
MATERIALS AND METHODS
Protein expression and purification
The plasmid pETMCSIII containing human c-Myc residues 1–88 with an N-terminal 6× His tag (31) was transformed into BL21(DE3) cells, incubated at 37°C until OD600 reached a level of 0.8, induced by 0.4 mM IPTG at 37°C for 5 h, harvested and resuspended in native lysis buffer (100 mM NaH2PO4, 10 mM Tris–HCl, 300 mM NaCl, pH 8.0), sonicated on ice 10 × 30 s and centrifuged at 10 000g for 30 min. For 15N and 13C/15N labeled media, Spectra 9 from ISOTEC or Cambridge Isotopes Laboratories was used. The pellet was resuspended in denaturing lysis buffer (100 mM NaH2PO4, 10 mM Tris–HCl, 8 M urea, pH 8.0) followed by a second sonication (10 × 30 s) and centrifugation, Ni2+-affinity chromatography under denaturing conditions using 100 mM NaH2PO4, 10 mM Tris–HCl, 8 M urea, pH 4.5 and subsequent refolding into a native buffer by dialysis against 50 mM NaH2PO4, pH 8.0, 10 mM Tris, 100 mM NaCl, 5 mM EDTA, 2 mM DDT, 5% glycerol, in four steps of 24 h each, resulted in >95% pure protein as analyzed with SDS–PAGE and MALDI–TOF–MS. The oligomeric state of Myc-1–88 was a monomer as analyzed by gel filtration (Superdex 75, GE Healthcare) using a protein concentration of 850 µM in 20 mM HEPES, pH 6.9, 100 mM NaCl, 5 mM DTT and 5% glycerol. Cloning, expression and purification of the Myc-binding Bin1–SH3 domain, comprising residues 402–482, is described in (24).
NMR samples
Buffer optimization was performed according to (39). Fifty-six buffer conditions based on HEPES, TRIS, TRICINE, MOPS, MES, phosphate and citric acid were assayed with various salt content and pH. Final conditions for Myc-1–88 were 40 mM HEPES, pH 6.8, 100 mM NaCl, 2 mM DTT, 5% glycerol; during these conditions the sample was stable for 3 weeks in the spectrometer. The same buffer was used for Bin1–SH3. Myc-1–88 and Bin1–SH3 were concentrated to 1.5 mM using Amicon Ultra concentrators with a 3.5 kDa cut-off (GE Healthcare). After concentration 10% D2O and 100 µM NaN3 were added. NMR sample concentrations were 600 µM for apo Myc-1–88 and 900 µM for the Myc-1–88:Bin1–SH3 complex. Titration experiments were performed with an initial Myc-1–88 concentration of 780 μM in a sample buffer containing 20 mM HEPES, pH 6.9, 100 mM NaCl, 5 mM DTT, 5% glycerol, 100 μM NaN3 and 10% D2O. The Bin1–SH3 protein, with the same buffer, was titrated in steps of 0.05 equivalents until the complex reached a Myc-1–88:Bin1–SH3 ratio of 1:0.8. For analysis with one equivalent or more, the Bin1–SH3 domain was added in steps of 0.25 equivalents. The 1:10 Myc-1–88:Bin1–SH3 complex was prepared separately at a final Myc-1–88 concentration of 80 μM. The chemical shift changes were monitored by 15N-HSQC during the titration and assigned by 3D experiments at the titration endpoint to resolve overlaps.
NMR spectroscopy and resonance assignments
Assignment of apo Myc-1–88 was pursued using the FAWN approach, which is a semi-automated fragment Monte Carlo approach for sequential assignment (40) using 15N-HSQC, HNCA, HNCO, HBHACONH, CBCACONH and 15N-NOESY-HSQC experiments; the assignment was manually complemented and extended to full side chains where possible by analysis of 15N- and 13C-correlated TOCSY and 13C-NOESY-HSQC experiments. Experiments on the apo state were recorded with non-uniform sampling to increase resolution as described (40). Assignment of the apo form was made at 10 and 15°C, whereas assignment and analysis of the Myc-1–88:Bin-SH3 complex was performed at 15°C. For the assignment measurements of the 1:1.5 eqivalent complex, a c-Myc concentration of 750 μM was used, whereas during interaction analysis sample concentrations down to 80 µM were used; no concentration-dependent shift changes were observed. Temperature-dependent chemical shift changes between 10 and 15°C, as well as chemical shift changes between apo and holo forms at 15°C, were small and assignments were easily transferred using Varian/Agilent BioPack pulse sequences encoding HNCA, HNCO, HNCACO, CBCACONH, HNCACB and 15N-NOESY-HSQC experiments. Bruker 600 and 800 MHz as well as Varian INOVA 600 and 800 MHz spectrometers were employed.
Relaxation experiments
15N-R1, 15N-R1ρ and {1H}-15N-NOE were measured using previously published pulse sequences (41,42) using a Varian INOVA 800 MHz spectrometer at 15°C. The experiments were performed for free Myc-1–88 at a concentration of 490 μM as well as for a 1:1.5 Myc-1–88:Bin1–SH3 complex at a final Myc concentration of 390 μM (due to addition of Bin1–SH3). For the R1 experiments 23 data points, including four duplicates, were acquired with relaxation delays ranging from 10 to 1512 ms. For the R1ρ experiments 18 data points, including eight duplicates, with delays between 2 and 100 ms were recorded. The spinlock field strengths were 2058 Hz for free Myc-1–88 and 2051 Hz for the Myc-1–88:Bin1–SH3 complex and the radio frequency carrier for the spinlock field was positioned at 119 ppm. The heteronuclear NOE was measured by recording experiments including or not including a 5 s period of 120° 1H saturation pulses. The total recovery delay was 12 s in both cases.
Data analysis
All spectra were processed with NMRpipe (43) and visualized using SPARKY (44). The compound 1H and 15N chemical shift difference, between free Myc-1–88 and Myc-1–88:Bin1–SH3, was calculated with the equation
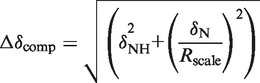 |
where Rscale is the chemical shift scaling factor, taken to be 6.5 as determined from the ratio of the average variances of the amide nitrogen and proton chemical shifts observed for the 20 common amino acid residues in proteins as deposited with the BioMagResBank (45). To distinguish significant CSPs, a cut-off of two standard deviations from the trimmed mean was calculated in an iterative procedure as described (46). For intensity analysis, the ratio of intensity of bound to free Myc-1–88 was calculated by evaluating peak intensities in corresponding HNCOs recorded at the same concentrations as in the relaxation experiment. The intensity ratio was estimated to be equal to the ratio of peak heights as derived from SPARKY with a scaling factor compensating for dilution. Secondary structure propensities were calculated using the program SSP (47) using the chemical shifts of 13Cα and 13Cβ as input.
For the relaxation experiments, peaks were integrated by fitting a combination of Lorentzian and Gaussian line-shapes using the in-house program PINT (A. Ahlner, M. Carlsson, B.-H. Jonsson and P. Lundström, available upon request). The same program was used to fit the R1 and R1ρ rates. Errors in the extracted rates were estimated using the jackknife approach (48). R2 was calculated from R1ρ and R1 experiments using the following equation
where θ is the tilt-angle, defined as arctan(B1/Ω) where B1 is the spinlock field strength and Ω is the resonance offset from the radio frequency carrier (49).
Binding affinity measurements using SPR
Surface plasmon resonance (SPR) measurements were performed using a Biacore 3000 instrument at 25°C together with sensor chip CM5 research grade (GE Healthcare). Both ligand and reference channels were activated by the freshly mixed EDC/NHS solution (Amine coupling kit, GE Healthcare) (injection time 7 min, flow; 10 µl/min). Myc1–88 was diluted in acetate buffer pH 4.5 to final concentration 3 µM and immobilized on the ligand channel to 3400 RU by amine binding (flow 10 µl/min) and subsequent ethanolamine (GE Healthcare) deactivation (injection time 7 min, flow; 10 µl/min). Optimal regeneration conditions were carefully selected, choosing the condition with the most stable baseline. Assisted by the BIACORE 3000 wizard, several injections of Bin1–SH3 were made at a range of flow rates and highest Bin1–SH3 concentration to check for limitations by mass transport; no such effects were found. Bin1–SH3 was prepared for the kinetic study by dialysis against running buffer (40 mM HEPES, 100 mM NaCl, 2 mM DTT, 0.005% Tween-20, pH 7.0), followed by stepwise dilution into the same buffer, in order to generate a series of concentrations (111.0–6.9 µM). The kinetic measurements were performed with a 5-min injection time followed by 6 min dissociation time and 2.5 min regeneration, using 5 mM NaOH (flow 30 µl/min). Kinetic data was evaluated using the BIAEvaluation 4.1 software (GE Healthcare). All sensorgrams were double reference subtracted. Fits were analyzed using both the Langmuir 1:1 and the parallel reaction model, and were further evaluated using visual inspection, χ2 values and T-values. A fit was accepted if χ2 < 10 and T > 10 (50). To check for Bin1–Bin1 binding, a control experiment was performed using Bin1–SH3 both as a ligand and analyte. Except for the immobilization of Bin1–SH3, which was slightly revised (acetate buffer pH4, 7 µM, 2330 RU), the surface and buffer were prepared the same way as in the myc-1–88–Bin1–SH3 experiment. Bin1–SH3 was injected over the surface at a concentration of 140 µM. Injection time was set to 10 min (flow 30 µl/min). The injection of Bin1–SH3 was repeated twice. No binding between free and immobilized Bin1–SH3 was detected.
Myc-1–88 sequence analysis and disorder prediction
The conservation score is based on a multiple sequence alignment calculated with Clustalw2. Sequences in the multiple alignment were obtained from a Blast search of the Myc-1–88 sequence in the Swissprot database. The conservation score for each position is calculated by extracting the vector in the BLOSUM62 substitution matrix for the amino acid residue at that position in human Myc and then taking the Euclidean distance between that vector and the centroid vector of all residues in that position, similar to the c-score in (51). The values are further normalized between zero and one. Disorder predictions were made using a set of current disorder predictors through available web interfaces as reviewed (52) and as described in (53–56).
RESULTS
NMR assignment of Myc1–88
We chose NMR to obtain a detailed per residue characterization of its structural and dynamic properties, since Myc-1–88 had already showed biophysical characteristics of disorder (31). After extensive buffer optimization, the resolution in the HSQC spectrum of Myc-1–88 (Figure 2) is characteristic of an intrinsically disordered protein, with all amide proton shifts confined to 7.7–8.6 ppm. However, while intrinsically disordered proteins typically have uniformly sharp resonances, the Myc-1–88 spectra demonstrated heterogeneity of peak intensities and peak shapes likely reflecting an interconverting and transiently interacting heterogeneous ensemble of states. Despite a high level of overlap in the HSQC (Figure 2), indirect dimensions in the 3D spectra were highly resolved through use of non-uniform sampling (35) (Supplementary Figure S1). Near-complete backbone assignments were obtained at 10°C using the semi-automated amide-anchored assignment strategy FAWN (40), manually confirmed and extended to side chains based on 13C-HSQC-NOESY and -TOCSY experiments. Out of a total of 88 residues, M1, P2, L3, P45, S46, E47, P59, S67 and C70 could not be assigned due to low NH intensity and/or extensive overlap. In addition, assignments of residues D17, E29, Q34, Q35 and L40 were lost at 15°C due to increased amide exchange and/or unresolved overlap. All assigned proline residues (7 out of 11) were found to be in the trans conformation based on chemical shift analysis as well as on the presence of strong NOEs between Hδ and the preceding Hα as identified from the 13C-HSQC-NOESY. Less than 10 minor peaks were observed in the HSQC which might result from cis–trans isomerization at one of the multiple prolines; however, none of these resonances could be unambiguously assigned due to their low intensity and the absence of cross peaks in 3D experiments. Therefore, with the current Myc-1–88 construct and optimized sample conditions, no cis contributions could be identified or assigned.
Figure 2.

15N-HSQC spectrum of Myc-1–88 recorded at 15°C with overlaid assignments. The glycine region (δ 15N < 112 ppm) has been omitted for clarity but contains peaks corresponding to G84 and G68.
Myc-1–88 contains multiple regions of transient secondary structure
The resonance assignments of Myc-1–88 were used to probe for the presence of transient secondary structure by evaluating secondary structure propensity (SSP) (47). In two regions, SSPs ranging from −0.2 to 0.35 were observed (Figure 3A). These values indicate the presence of transient structure, given that values of +1 and −1 are expected for fully formed helices and extended regions, respectively, and values around zero for no preferential sampling of secondary structure. Residues 26–34 and 47–52 show dNN(i,i + 2) and dαN(i,i + 3) NOEs typical for helices in the 15N-HSQC-NOESY spectra (Figure 3D) although the intensities were weaker than those expected for a fully formed structured helix thus supporting transient structure formation. Furthermore, weak NOEs indicative of a fluctuating β-turn with a proline in position +2 were observed at residues 20-QPYF (Figure 3D); this motif may be stabilized by the Y22-F23-Y24 hydrophobic cluster and is in agreement with negative SSPs (Figure 3A). A tentative stretch of transient extended structure at residues 55–63 is suggested by negative SSPs for non-proline residues; the presence of significant trans proline content (see above) is also likely to be stabilizing for polyproline II (PPII) extended structure. A short continuous stretch of sequential NOEs may support transient extended structure at 74–78. Residues 1–20 and 79–88 were disordered as judged by both NOE and SSP analysis, and residues 35–41 were not possible to evaluate at the NOE level due to the close overlap in 1HN and 15N shifts. The regions with transient structure are summarized in Figure 3C.
Figure 3.

Identification of transient structure in Myc-1–88 by NMR. (A) SSP of Myc-1–88 based on experimental data. A region with values close to one indicates a fully formed α-helix whereas values close to minus one and zero are indicative of fully formed β-strand or no preferential secondary structure, respectively (47). (B) Sequence conservation in Myc-1–88 described on a normalized c-score scale as described in ‘Materials and methods’ section: high (0.8–1.0, star) intermediate (0.6–0.8, dot) and low (0–0.6, unlabeled). (C) Graphical representation of transient structures based on combined SSP and NOE evaluation. (D) Schematic representation of characteristic secondary structure NOEs as identified in 15N-NOESY-HSQC spectra. The dNN(i, i + 1) NOEs are classified as strong, intermediate or weak by the intensity of the bar, while other NOEs are indicated as present irrespective of magnitude. Light gray indicates partial overlap whereas completely overlapping NOEs are excluded.
Importantly, the regions with transiently ordered regions are well conserved in Myc proteins (Figure 3B). In particular, residues 45–65 correspond to the previously identified MBI region, which directly interacts with a large number of proteins involved in phosphorylation-directed Myc regulatory events (1,9,18,20). The more N-terminal transiently structured region includes Myc residues 24–31, which were previously identified by mutational analysis to interact with TRRAP, a functionally important Myc partner involved in regulating histone acetylation (57). Furthermore, Myc residues 1–46 appear required for p300 binding together with the MBI region (58) and Myc-Δ1-40 no longer binds JPO2 (59). Notably, Myc residues 15–35 contain a highly conserved sequence pattern also present in N-, L- and v-Myc (Figure 3B), suggesting that this region could be involved in interactions throughout the transforming members of the Myc protein family.
The disordered and transiently ordered regions of Myc were difficult to distinguish by current disorder predictors (52). DisEMBL (53) and PONDR-FIT (54) correctly recognized both transiently ordered regions but predicted the disordered segment 10–20 to be ordered, but only OnD-CRF (55) could correctly identify both disordered and transiently ordered regions in Myc-1–88. Myc residues 71–81, predicted as transiently extended, as well as residues 12–28, which comprise the N-terminal β-turn-helix motif, were identified by ANCHOR to have propensity for disorder-to-order transition on binding a globular partner (56).
Multivalent interactions of Myc with Bin1–SH3
To better understand the roles of disorder and transient order in the interaction of Myc TAD with target proteins, we investigated the interaction between Myc-1–88 and the SH3 domain of Bin1 by SPR technology. Both kinetic and steady-state measurements show saturable Myc binding at lower concentrations of Bin1–SH3, but at higher concentrations of analyte a second binding event is indicated from the increased responses obtained (Figure 4). Since Bin1–SH3 is monomeric under our experimental conditions [up to 1 mM concentrations, (24)] and no SPR response is obtained in a Bin1-to-Bin1 control experiment (see ‘Materials and methods’ section), we can exclude oligomerization of Bin1–SH3 as a potential cause of this higher response. Alternatively, Myc-1–88 may harbor two binding sites for Bin1–SH3 with different affinities. In agreement with this, while the kinetic data could not be fit to a 1:1 Langmuir model (Figure 4A), the data fits well to a parallel reaction model, which assumes that the immobilized ligand (Myc-1–88) has two binding sites to which the analyte (Bin1–SH3) can bind independently (Figure 4A). This model fits two binding sites with KDs of 33 and 200 µM, with kon/koff of 590 M−1s−1/0.019 s−1 and 6.7 M−1s−1/0.0013 s−1, respectively. It should be noted that since the lower-affinity binding does not reach saturation during the measurements, the stoichiometry and affinity of this site (or sites) are therefore poorly estimated. Previous chemical shift perturbation (CSP) analysis showed no binding outside the canonical Bin1–SH3 binding site on extending Myc-55–68 to Myc-1–153 in a titration with labeled Bin1–SH3 (24), excluding a second binding site on Bin1. Thus, our SPR results together with previous CSP mapping suggests that the Bin1–SH3 canonical SH3 binding site can bind to two (or more) sites on Myc.
Figure 4.

SPR measurements suggest multivalent binding of Myc-1–88 to Bin1–SH3. Myc-1–88 was immobilized and Bin1–SH3 injected over the surface. (A) Kinetic experiments. Overlaid sensorgrams show experimental data (solid lines) and simultaneously fitted functions using a parallel reaction model with two binding sites (dashed lines). A 1:1 Langmuir model fits very poorly as indicated to the middle response (gray line); no Langmuir fit could be made to all curves simultaneously. The concentration series includes 111.0, 55.5, 27.7, 13.9 and 6.9 µM of Bin1–SH3. (B) Steady-state binding experiments of Bin1–SH3 to Myc-1–88. Based on the data points at lower concentrations a plateau in binding would be expected at higher concentration, but instead the response increases further in both kinetic and steady-state experiments, thus indicating that Myc-1–88 may display one or more additional binding sites for Bin1–SH3 with lower affinity.
Per-residue NMR mapping of effects of target protein binding to Myc
To investigate how disordered Myc-1–88 binds to a folded target protein on a per residue basis, the NMR assignment of Myc-1–88 was used as a basis for joint evaluation of amide CSPs and changes in HNCO peak intensities. While the chemical shift reflects the immediate chemical environment and can be a sensitive probe for localizing protein interactions, conclusions based entirely on chemical shift data can be misleading in cases of pronounced conformational flexibility, with extensive sampling of a wide and heterogeneous conformational space (60). We therefore also analyzed the ratios of the HNCO peak intensities of Bin1-bound Myc-1–88 versus free Myc-1–88. Since NMR peak intensities are affected by the linewidth, which is narrow in the free state and broadened by interactions, intensities are lowered on interaction with a slowly tumbling folded domain, leading to a reduced peak intensity ratio relative to the free state (60).
The complex was analyzed at Myc-1–88:Bin1–SH3 ratios of 1:1.5, where the high-affinity site should be up to ∼88% saturated (Figures 5 and 6). Titration of unlabeled Bin1–SH3 into labeled Myc-1–88 resulted in complete disappearance of residue S62 (Figure 5) already at 10% saturation of Bin1. This is consistent with chemical exchange on the millisecond time scale, in agreement with the kinetic parameters of association determined by SPR. The early disappearance of S62 from the spectra suggests that this residue experiences a large chemical shift change for 15N and/or 1HN upon binding of Bin1–SH3 giving rise to intermediate exchange broadening; this provides strong evidence that S62 is indeed involved in Bin1–Myc recognition. Smaller but significant CSPs are observed for residues 56, 58, 61, 64, 66, 68 and 69 in fast exchange on the NMR time scale (Figures 5 and 6A). These Myc-1–88 CSP values are maintained in a 1:10 excess of Bin1–SH3, suggesting high saturation of this site already in the 1:1.5 complex (Supplementary Figure S2). Concomitantly reduced peak ratios for residues 55, 58, 61, 64, 65 and 66 are observed on 1:1.5 complex formation (Figure 6B), but the reduction is smaller than would be expected for tight binding (60) to residues 55–66 as suggested previously (24). A tendency toward reduced intensity ratios without significant CSPs is also observed for residues in the region 37–48, although this part of the plot has more limited information due to overlap and missing assignments. This region contains a proline-rich segment (P42–P43–A44–P45) not previously identified as a Bin1-binding element but that shares sequence characteristics of SH3 domain targets. An additional region of broadening upon binding is observed at the extreme C-terminus; while no canonical SH3-binding sequence is apparent, binding may be to non-canonical sequences or the broadening may be due to transient contacts with other segments that do bind Bin1. Taken together, CSP and intensity ratios are consistent with Myc anchoring into the binding groove of Bin1–SH3 at S62–P63 (24), but in agreement with SPR measurements also suggest concomitant and multivalent binding both to the adjacent P59–P60 di-peptide recognition element and to the proline-rich P42–P43–A44–P45 segment. The presence of reduced intensity ratios already at a Myc-Bin ratio of 1:1.5 supports the relevance of multivalent Bin1 binding.
Figure 5.
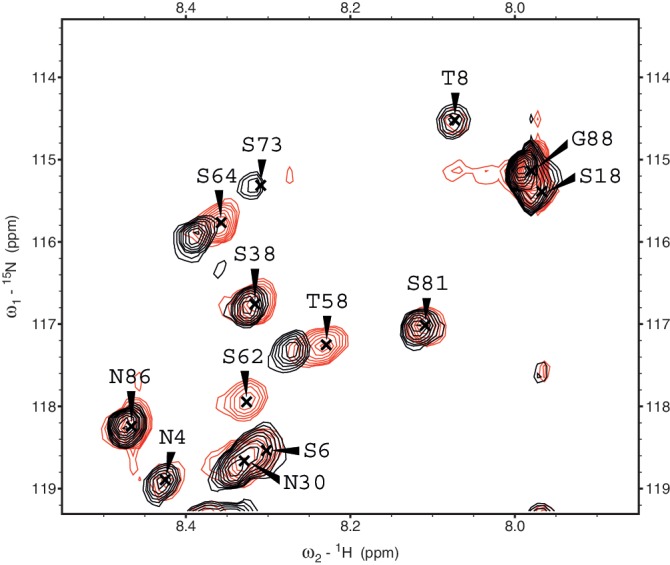
Selected HSQC region showing superimposed spectra of Myc-1–88 in the free state (red, labeled) and in a 1:1.5 complex with Bin1–SH3 (black, unlabeled). In the bound state, the resonance for S62 is broadened beyond detection, thus no peak for S62 is observed in the bound state in the selected region or elsewhere.
Figure 6.

NMR analysis of Myc-1–88 in the absence and presence of Bin1–SH3. Omitted histogram bars correspond to missing or overlapped residues and prolines. (A) Myc-1–88 CSPs at a Myc-1–88:Bin1–SH3 ratio of 1:1.5. The cut-off value for significant CSPs is shown as a dashed line and calculated as described in ‘Materials and methods’ section. (B) Ratios of peak intensities with and without Bin1, derived from HNCO experiments at a Myc-1–88:Bin1–SH3 ratio of 1:1.5. In the absence of interactions, or if interactions are the same in free and bound forms, the intensity ratio would be 1 (gray line). Gain/loss of interactions in the bound state lead to decreased/increased peak intensity ratio, respectively.
Smaller but significant shift changes outside the Bin1–SH3 target sites described above are observed for residues 23–28 in the transiently structured region, residues 48–52 within the MBI region and in residues 73–75 and 80 C-terminal to MBI (Figure 6A); these CSPs increase slightly in 10-fold excess of Bin1 (Supplementary Figure S2). However, sequences in these regions do not have typical SH3 binding motifs and, in contrast to the binding sites described above, show significantly increased ratios on Bin1 binding already at a Myc-Bin ratio of 1:1.5. This suggests that in the free state, resonances in regions comprising residues 4–25, 49–54 and 73–77 are broadened by interactions with other parts of Myc-1–88, whereas in the complex, such interactions are sampled much less frequently, presumably due to steric hindrance by multivalent Bin1–SH3 binding (56). The effects observed on chemical shifts and intensity ratios in these regions is thus likely not due to direct contacts with Bin1–SH3.
Changes in motional properties of Myc-1–88 upon Bin-1-SH3 binding
To investigate whether Bin1–SH3 binding results in reduced intrinsic disorder within the Myc binding site(s), the dynamic properties of Myc-1–88 on a rapid time scale (ps–ns) were assayed in the absence and presence of ligand binding. Backbone 15N relaxation experiments (R1, R1ρ, NOE) were performed on the apo and complex forms of Myc-1–88 (Figure 7). Measurements were made at 1.5 equivalents of Bin1–SH3 relative to Myc in order to achieve high saturation of the primary site while limiting binding to second, lower affinity site(s). Peaks with overlap leading to difficulties in fitting the data were not included in the evaluation.
Figure 7.

15N Relaxation parameters of free Myc-1–88 (open circles) and Myc-1–88 bound to Bin1–SH3 in a 1:1.5 ratio (filled circles). (A) {1H}-15N-NOE. (B) R1 relaxation rate constants. (C) R2 relaxation rate constants.
Relaxation parameters for interpretable residues show that in the unbound state, the entire Myc-1–88 behaves as an intrinsically disordered protein with intermediate mobility (Figure 7; empty symbols). The magnitude of the heteronuclear NOEs are consistent with a disordered region, but are higher than expected for single-domain intrinsically disordered proteins. Instead, their magnitude resembles those of longer intrinsically disordered regions linked to well-folded globular domains (61,62). Residues 49–54, with transient helical structure as suggested from analysis of chemical shifts and NOESY cross peaks, also have consistently higher values of the heteronuclear NOE and R2 than flanking residues. NOEs of similar magnitude are also observed for residues 26, 30, 31 and 32 in the second transiently structured region comprising residues 22–33. R1 values are fairly consistent throughout the sequence and the order of magnitude of R1 agrees with Myc-1–88 being monomeric under NMR conditions. The surprisingly high R2 values in the free state in segments 4–32, 51–56 and 73–80 resemble R2 values observed in collapsed states of lysozyme presenting long-range intramolecular interactions (63). This agrees with HNCO intensity ratios higher than 1, suggesting loss of interactions between transient structures in the free state on Bin1 binding (Figure 6C). The C-terminus of Myc-1–88 as well as the middle region (residues 36–45) appears more flexible than the N-terminus, with lower and even negative NOEs and R2 values below 10 s−1.
Upon Bin1–SH3 binding, the Myc-1–88 relaxation data (Figure 7, filled symbols) shows that dynamics are consistent with continued disorder within Myc-1–88 upon binding. The absence of intermolecular NOEs in Myc-1–88 NOESY spectra (data not shown) is consistent with distance averaging in a heterogeneous and dynamic ensemble (64). Still, significant changes in relaxation do occur. In the bound state, residues 56 and 58 in the canonical Bin1–SH3 binding site show significantly lower R1 than surrounding residues, suggesting that these residues sample dynamic properties of a larger protein complex. S62 could not be studied in the bound state due to its extensive broadening by millisecond dynamics, but L61 shows a significantly higher NOE in the bound state suggesting decreased dynamics in the bound state as expected. Furthermore, residues 27, 30 and 31, in, or adjacent to, the N-terminal transiently ordered region also show similarly small but significant changes in NOE and/or R1 values. Residues in the more flexible middle and C-terminal region show very minor or insignificant differences between bound and free forms. The lower R2 values observed for residues 1–30, 49–56 and 61–82 in the transiently ordered segments of the bound form compared to the free state suggest that fewer long-range interactions are sampled by these regions in the conformational ensemble of the disordered protein domain (63). Thus, Bin1–SH3 binding to Myc-1–88 induces subtle, residue-specific alterations in dynamics, primarily within Myc regions that are also identified by CSPs, responding to both Bin-1-SH3 interactions and altered long-range interactions involving transiently ordered segments and/or hydrophobic clusters. Importantly, however, binding maintains overall intrinsic disorder in Myc-1–88 with no large-scale evidence for stable structure in the complex.
DISCUSSION
Understanding the molecular basis of Myc regulation and function has been a cornerstone of the fields of cancer biology and signal transduction for over 20 years. As Myc is downstream of many signaling cascades, it has been referred to as an intracellular sentinel of the extracellular milieu that functions as a central hub for rapidly responding to stimuli and regulating a number of cellular functions (1,16,65). However, despite the wide range of identified Myc-interacting proteins, to this date, there is very little molecular information on where and how these proteins bind to the Myc transactivation domain. Therefore, this study focuses on advancing our understanding of how Myc coordinates its multiple functions by analyzing in detail the biophysical properties of the Myc TAD region, in particular the environment surrounding the highly conserved MBI.
In this study we show that although Myc-1–88 as a whole is intrinsically disordered as judged by NMR chemical shift dispersion and relaxation, there are two distinct regions that appear to transiently sample secondary structure based on their NOE and SSP signatures. In residues 22–33, a fluctuating beta turn is followed by a fractionally populated short helical segment. Residues 48–55 also sample helical properties, and are followed by residues 56–65 which have a fluctuating extended character, likely stabilized in part by the proline-rich nature of the sequence. Relaxation measurements support the transient population of secondary structure with slightly higher values of NOE and R2 in these regions. Importantly, both of these transiently ordered regions actively participate in interactions conveying and reacting to post-translational modifications in the regulation of Myc activity (Figure 8) (1,9,17,18,20,36,38,57–59,66).
Figure 8.

Biophysical and biological functionalities in Myc-1–88. (A) Myc-1–88 is intrinsically disordered (green), with transient secondary propensity in residues 23–33 (yellow) and 48-65 (red); phosphorylation sites T58 and S62 are indicated. (B) Identified interactors to Myc-1–88 can be classified in three groups as indicated by double arrows: those mapped by deletion mutants covering entire Myc1–88, those identified to interact with MBI by deletion mapping or where interaction is regulated by phosphorylation to T58 and/or S62, and those that bind the N-terminal region including the first transiently structured region. (C) In the free state, Myc-1–88 shows transient and fluctuating interactions (dashed arrows) involving secondary structure elements and hydrophobic clusters, suggesting the presence of condensed disordered states in the conformational ensemble (only one of many such possible states is shown). By multivalent binding to Bin1 binding sites at P59–P63 and P42–P45 (solid arrows) and possible wobbling, or sliding, to the less optimal P57–P60 site (curved arrow), Bin1 sterically hinders interactions present in the free state.
The identification of transiently structured segments in Myc TAD and their correlation with sites of protein interaction resembles p53, where such segments nucleate folding-on-binding to regulatory partner proteins (67–71). The dynamic nature of the p53 N-terminal transactivation domain can allow interactions with multiple, often competing partners (70–73) and allows it to be readily modified by post-translational modifications that also modulate interactions (72,74). Molecular understanding of disordered p53 TAD behavior in its free and bound states has been crucial in understanding its specificity and interactions, and has helped in the design of efficient inhibitors leading to p53 activation, cell cycle arrest and apoptosis of tumor cells (75–77). Given the central role of Myc in cancer, it is therefore critical to understand the details of intrinsic structure and dynamics of the Myc TAD and how these effect partner interactions.
Surprisingly, no folding-on-binding occurs in Myc-1–88 when it binds Bin1–SH3; instead disorder is maintained throughout Myc-1–88, also within transiently structured regions. In fact, dynamics on the millisecond level is even increased for Myc-1–88 as a whole in complex with Bin1–SH3. Binding to the previously identified Bin1–SH3 site, centered on P59–P60–L61–S62–P63, is indeed observed by small CSPs and localized small changes in dynamics, but these effects are not limited to the Bin1–SH3 binding site, and do not result in stabilized secondary structure or conformational restriction at the binding site or anywhere else in Myc-1–88. This observation differs significantly from the highly ordered complex described for the Myc peptide complex with Bin1–SH3 (24).
Critical to understanding this behavior is our observation of the effects of multivalent binding of Bin1–SH3. Although different methods were used to measure affinities, it appears that Myc-1–88 binds Bin1–SH3 with lower affinity to at least two sites (KD = 33 and 200 µM, SPR) compared to the higher-affinity single-site binding displayed by the Myc-55–68 peptide (4.2 µM, fluorescence). Our NMR data suggest that extending the target sequence to Myc-1–88 allows for Bin1–SH3 binding not only to the S62-centered P59–P60–L61–S62–P63 site, but also to another proline-rich segment P42–P43–A44–P45, and possibly also ‘wobbling’ or sliding, to an electrostatically slightly less favorable P57–T58–P59–P60 site, centered on T58 (Figure 8). NMR data report binding of Bin1–SH3 to the previously identified binding site comprising Myc residues 58–66 as characterized in the Myc-55–68:Bin1–SH3 complex (24), however, strong binding to this site alone would have given rise to much larger effects in CSPs, intensity ratios and NMR relaxation for these residues than observed here (Figures 6 and 7). In agreement with SPR data suggesting multivalent binding, reduced intensity ratios in the region around Q37-D48, of similar magnitude as in the primary site, suggest that the P42–P45 proline-rich sequence might bind Bin1–SH3 at lower affinity although assignments in this region are sparse due to extensive overlap. CSPs N-terminal to S62 could suggest a secondary or ‘shifted’ primary site around T58, in the context of a proline-rich sequence that could be consistent with SH3 domain binding (L56–P57–T58–P59–P60). Compared to the primary site, centered on S62 and electrostatically anchored to the Bin1–SH3 binding cleft by conserved arginine residues (24), the P57–T58–P59–P60 site, centered on T58 is electrostatically less attractive but still favorable. Although the same primary site is targeted by Bin1–SH3 binding to Myc-1–88 as previously described for a shorter peptide, binding to additional sites in the longer Myc thus appears to result in altered dynamical properties of these two complexes. A biological role for multivalent binding to both these sites is supported by the finding of large numbers of mutations in lymphoma tumors and tumor cell lines at both proposed Bin1–SH3 target sites, in Myc residues 57–63 and 36–44 (21,22,78).
The underlying role of the dynamics of Myc-1–88 both in isolation and in its multivalent complex with Bin1–SH3 could be to prevent stable binding to its critical regulatory interaction sites. From a biological point of view, lower affinity and less than fully populated binding to the unphosphorylated P59–P63 site would be most beneficial. If Bin-1 binding to Myc led to complete inaccessibility of the MBI, Myc would not be able to be timely modified in a cellular situation where phosphorylation of S62 is required for proper growth response. Biophysically, the limited effects on dynamics on Myc–Bin1–SH3 binding suggests that Bin1–SH3 may probe the phosphorylation state by capturing Myc in a highly dynamic complex centered on S62, but simultaneously sampling an ensemble of adjacent interactions in a dynamic manner, which leads to a rapid exchange between the primary site and weaker adjacent binding sites. Although this exchange appears to lead to a lower overall affinity, the benefits of dynamically sampling other interactions, including the linker region between the two transiently ordered regions, and the accessibility to T58, may be critical for biological sampling of the status of Myc post-translational modifications by Bin1 as well as by other interacting regulators.
In addition, multivalent Bin1 binding appears to affect significantly larger regions of Myc than those directly targeted by Bin1–SH3 binding. Sizeable regions outside the Bin1 binding sites in Myc-1–88 show small but significant CSPs, but lack the reduced peak intensities that signify binding by disordered proteins (33,60,79). Rather, in complex with Bin1–SH3, these regions in Myc-1–88 experience increased intensity ratios (Figure 6B), which in agreement with decreased values of R2 (Figure 7C) suggest that Bin1–SH3 binding disrupts transient interactions involving these segments. Similar effects on R2 was observed for mutations in lysozyme hindering the formation of collapsed states bridged by long-range interactions between hydrophobic clusters and/or transient structure elements (63). Thus, our data suggest that in the free state, fluctuating secondary structure elements in Myc residues 22–33 and 48–68, as well as adjacent conserved hydrophobic clusters at residues 71–81 and 5–15 loosely interact, thus shifting the population in the conformational ensemble toward more compactly disordered condensed states (Figure 8) (63), which is in agreement with the identification of Myc-1–88 as a stable proteolytic fragment (31). This condensed ensemble is likely stabilized by electrostatic interactions (33,80): the Myc-1–49 region is predominantly negatively charged with a pI of 3.5, while the pI of Myc-50–88 is 9.2. Multivalent Bin1–SH3 binding to Myc-1–88 hinders the formation of such intramolecular interactions (Figure 8) resulting in a shift in the conformational ensemble toward less compact states, but with retained local dynamics on the ps-µs time scale as sampled by NOE and R1 measurements.
From this growing perspective of Myc biophysics being ruled by interacting transient structures, it is not surprising that interaction patterns to Myc have been difficult to address using deletion mapping or mutation screens. While mutations would typically be useful for confirming binding residues, in the case of disordered proteins that have transient structural contacts such as Myc, these mutations may have confounding structural effects that are difficult to predict leading to results that are not clearly interpretable. Deleting a transiently structured segment may result in dynamic and transient long-range effects which may affect both affinity and specificity, and which cannot be predicted based on sequence information alone. In particular, a commonly used deletion mutant where the entire MBI region is deleted completely changes the electrostatic properties of the Myc N-terminal region, which is highly likely to affect the distribution of condensed ensemble states involving interactions between transiently ordered regions of Myc-1–88 as described here, since electrostatic interactions have been shown to highly contribute to the sampling of compactly disordered states in other intrinsically disordered proteins (33,80). In an extended perspective, it is not unlikely that transient tertiary interactions would involve other parts of Myc as well. In fact, a weak interaction between MBI and MBII was already identified earlier in our lab (31) and the involvement of both MBII and interaction sites within Myc-1–88 has been shown for TRRAP (57) and TBP (31,81). Further analysis of longer Myc fragments including both Myc boxes will be required to address these issues.
The current analysis of disordered properties and transient structure in Myc-1–88 has general implications for the study of Myc interactions. Our work has shown that Myc-1–88 behaves as an intricate intrinsically disordered but yet transiently structured entity where interactions are governed by dynamics at several time scales. Thus, rather than perceiving an array of interaction sequences which can be occupied independently, interactions to the Myc N-terminal region are likely to be multivalent and transient as an intrinsic feature of the dynamic response required by the biological environment in which Myc must function. Taken together, our findings expand the increasingly recognized concept of intrinsically disordered regions mediating transient interactions to Myc, a key transcriptional regulator of major medical importance, and have important implications for further understanding its multifaceted roles in gene regulation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1 and 2.
FUNDING
VINNOVA (Visiting Professor fellowship to M.S.); the CIHR Strategic Training Program in Protein Folding and Interaction Dynamics (to V.C.); the Swedish Research Council (to M.S., P.L.); the Swedish Cancer Foundation (to M.S.); the Swedish Child Cancer Foundation (to M.S.); the Canadian Cancer Society (to J.D.F.-K., L.Z.P., C.H.A.); Ontario Research Fund (GL2-01-030; to L.Z.P.); the NIH Protein Structure Initiative grant U54 GM094597 (to C.H.A.); the Canada Research Chairs Program (to C.H.A., L.Z.P.); the Swedish NMR Centre; the Knut and Alice Wallenberg Foundation and Linköping University. Funding for open access charge: Linköping University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Vivian Morad and Shili Duan are acknowledged for protein preparation, Erik Martinsson for graphics assistance. Prof. Lars-Gunnar Larsson, Amanda Wasylishen, Sam Kim and Manpreet Kalkat are acknowledged for critical reading of the article.
REFERENCES
- 1.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat. Rev. Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 2.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agrawal P, Yu K, Salomon AR, Sedivy JM. Proteomic profiling of Myc-associated proteins. Cell Cycle. 2010;9:4908–4921. doi: 10.4161/cc.9.24.14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalkat M, Wasylishen AR, Kim SS, Penn L. More than MAX: discovering the Myc interactome. Cell Cycle. 2011;10:374–379. doi: 10.4161/cc.10.3.14645. [DOI] [PubMed] [Google Scholar]
- 6.Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 7.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin. Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 8.Dang CV. Enigmatic MYC conducts an unfolding systems biology symphony. Genes Cancer. 2010;1:526–531. doi: 10.1177/1947601910378742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole MD, Cowling VH. Transcription-independent functions of MYC: regulation of translation and DNA replication. Nat. Rev. Mol. Cell Biol. 2008;9:810–815. doi: 10.1038/nrm2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato GJ, Barrett J, Villa-Garcia M, Dang CV. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol. Cell. Biol. 1990;10:5914–5920. doi: 10.1128/mcb.10.11.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stone J, de Lange T, Ramsay G, Jakobovits E, Bishop JM, Varmus H, Lee W. Definition of regions in human c-myc that are involved in transformation and nuclear localization. Mol. Cell. Biol. 1987;7:1697–1709. doi: 10.1128/mcb.7.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freytag S, Dang C, Lee W. Definition of the activities and properties of c-myc required to inhibit cell differentiation. Cell Growth Differ. 1990;1:339–343. [PubMed] [Google Scholar]
- 13.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 14.Varlakhanova NV, Knoepfler PS. Acting locally and globally: Myc's ever-expanding roles on chromatin. Cancer Res. 2009;69:7487–7490. doi: 10.1158/0008-5472.CAN-08-4832. [DOI] [PubMed] [Google Scholar]
- 15.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer. 2010;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 16.Cowling VH, Cole MD. Mechanism of transcriptional activation by the Myc oncoproteins. Semin. Cancer Biol. 2006;16:242–252. doi: 10.1016/j.semcancer.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 17.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 18.Hann SR. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006;16:288–302. doi: 10.1016/j.semcancer.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl Acad. Sci. USA. 2010;107:58–63. doi: 10.1073/pnas.0900121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vervoorts J, Lüscher-Firzlaff J, Lüscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006;281:34725–34729. doi: 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- 21.Bahram F, von der Lehr N, Cetinkaya C, Larsson L-G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–2110. [PubMed] [Google Scholar]
- 22.Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC. Phosphorylation regulates c-Myc's oncogenic activity in the mammary gland. Cancer Res. 2011;71:925–936. doi: 10.1158/0008-5472.CAN-10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 24.Pineda-Lucena A, Ho CSW, Mao DYL, Sheng Y, Laister RC, Muhandiram R, Lu Y, Seet BT, Katz S, Szyperski T, et al. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J. Mol. Biol. 2005;351:182–194. doi: 10.1016/j.jmb.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 25.Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat. Genet. 1996;14:69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 26.Ge K, Minhas F, Duhadaway J, Mao N-C, Wilson D, Buccafusca R, Sakamuro D, Nelson P, Malkowicz SB, Tomaszewski J, et al. Loss of heterozygosity and tumor suppressor activity of Bin1 in prostate carcinoma. Int. J. Cancer. 2000;86:155–161. doi: 10.1002/(sici)1097-0215(20000415)86:2<155::aid-ijc2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 27.Elliott K, Sakamuro D, Basu A, Du W, Wunner W, Staller P, Gaubatz S, Zhang H, Prochownik E, Eilers M, et al. Bin1 functionally interacts with Myc and inhibits cell proliferation via multiple mechanisms. Oncogene. 1999;18:3564–3573. doi: 10.1038/sj.onc.1202670. [DOI] [PubMed] [Google Scholar]
- 28.DuHadaway JB, Sakamuro D, Ewert DL, Prendergast GC. Bin1 mediates apoptosis by c-Myc in transformed primary cells. Cancer Res. 2001;61:3151–3156. [PubMed] [Google Scholar]
- 29.Telfer JF, Urquhart J, Crouch DH. Suppression of MEK/ERK signalling by Myc: role of Bin-1. Cell. Signalling. 2005;17:701–708. doi: 10.1016/j.cellsig.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Kinney EL, Tanida S, Rodrigue AA, Johnson JK, Tompkins VS, Sakamuro D. Adenovirus E1A oncoprotein liberates c-Myc activity to promote cell proliferation through abating Bin1 expression via an Rb/E2F1-dependent mechanism. J. Cell. Physiol. 2008;216:621–631. doi: 10.1002/jcp.21437. [DOI] [PubMed] [Google Scholar]
- 31.Fladvad M, Zhou KS, Moshref A, Pursglove S, Safsten P, Sunnerhagen M. N and C-terminal sub-regions in the c-myc transactivation region and their joint role in creating versatility in folding and binding. J. Mol. Biol. 2005;346:175–189. doi: 10.1016/j.jmb.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 32.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 33.Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010;23:105–116. doi: 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- 34.McEwan IJ, Dahlman-Wright K, Ford J, Wright APH. Functional interaction of the c-Myc transactivation domain with the TATA binding protein: evidence for an induced fit model of transactivation domain folding. Biochemistry. 1996;35:9584–9593. doi: 10.1021/bi960793v. [DOI] [PubMed] [Google Scholar]
- 35.Burton RA, Mattila S, Taparowsky EJ, Post CB. B-Myc: N-terminal recognition of Myc binding proteins. Biochemistry. 2006;45:9857–9865. doi: 10.1021/bi060379n. [DOI] [PubMed] [Google Scholar]
- 36.Ponzielli R, Katz S, Barsyte-Lovejoy D, Penn LZ. Cancer therapeutics: targeting the dark side of Myc. Eur. J. Cancer. 2005;41:2485–2501. doi: 10.1016/j.ejca.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arnold HK, Zhang X, Daniel CJ, Tibbitts D, Escamilla-Powers J, Farrell A, Tokarz S, Morgan C, Sears RC. The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. EMBO J. 2009;28:500–512. doi: 10.1038/emboj.2008.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanelis V, Chong PA, Forman-Kay JD. NMR spectroscopy to study the dynamics and interactions of CFTR. Methods Mol Biol. 2011;741:377–403. doi: 10.1007/978-1-61779-117-8_25. [DOI] [PubMed] [Google Scholar]
- 40.Lemak A, Gutmanas A, Chitayat S, Karra M, Farès C, Sunnerhagen M, Arrowsmith C. A novel strategy for NMR resonance assignment and protein structure determination. J. Biomol. NMR. 2011;49:27–38. doi: 10.1007/s10858-010-9458-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 42.Korzhnev DM, Skrynnikov NR, Millet O, Torchia DA, Kay LE. An NMR experiment for the accurate measurement of heteronuclear spin-lock relaxation rates. J. Am. Chem. Soc. 2002;124:10743–10753. doi: 10.1021/ja0204776. [DOI] [PubMed] [Google Scholar]
- 43.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 44.Goddard TG, Kneller DG. SPARKY 3, University of California, San Francisco. [Google Scholar]
- 45.Mulder FAA, Schipper D, Bott R, Boelens R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 1999;292:111–123. doi: 10.1006/jmbi.1999.3034. [DOI] [PubMed] [Google Scholar]
- 46.Schumann F, Riepl H, Maurer T, Gronwald W, Neidig K-P, Kalbitzer H. Combined chemical shift changes and amino acid specific chemical shift mapping of protein–protein interactions. J. Biomol. NMR. 2007;39:275–289. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- 47.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mosteller F, Tukey JW. Data Analysis and Regression - A Second Course in Statistics. Philippines: Addison-Wesley Publishing Company Inc.; 1977. [Google Scholar]
- 49.Palmer AG, Massi F. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem. Rev. 2006;106:1700–1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- 50.Biacore. BIAevaluation 4.0 Software Handbook. 2004. Biacore AB: Uppsala, Sweden. [Google Scholar]
- 51.Hedlund J, Johansson J, Persson B. BRICHOS - a superfamily of multidomain proteins with diverse functions. BMC Res. Notes. 2009;2:180. doi: 10.1186/1756-0500-2-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009;19:929–949. doi: 10.1038/cr.2009.87. [DOI] [PubMed] [Google Scholar]
- 53.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 54.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. BBA-Proteins Proteom. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Sauer UH. OnD-CRF: predicting order and disorder in proteins conditional random fields. Bioinformatics. 2008;24:1401–1402. doi: 10.1093/bioinformatics/btn132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dosztányi Z, Mészáros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 58.Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, Farina A, Martinez E. Dual Regulation of c-Myc by p300 via Acetylation-Dependent Control of Myc Protein Turnover and Coactivation of Myc-Induced Transcription. Mol. Cell. Biol. 2005;25:10220–10234. doi: 10.1128/MCB.25.23.10220-10234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang A, Ho CSW, Ponzielli R, Barsyte-Lovejoy D, Bouffet E, Picard D, Hawkins CE, Penn LZ. Identification of a novel c-Myc protein interactor, JPO(2), with transforming activity in medulloblastoma cells. Cancer Res. 2005;65:5607–5619. doi: 10.1158/0008-5472.CAN-05-0500. [DOI] [PubMed] [Google Scholar]
- 60.Baker JM, Hudson RP, Kanelis V, Choy WY, Thibodeau PH, Thomas PJ, Forman-Kay JD. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 2007;14:738–745. doi: 10.1038/nsmb1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinheiro AS, Marsh JA, Forman-Kay JD, Peti W. Structural signature of the MYPT1-PP1 interaction. J. Am. Chem. Soc. 2011;133:73–80. doi: 10.1021/ja107810r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelker MS, Page R, Peti W. Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: a novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J. Mol. Biol. 2009;385:11–21. doi: 10.1016/j.jmb.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klein-Seetharaman J, Oikawa M, Grimshaw SB, Wirmer J, Duchardt E, Ueda T, Imoto T, Smith LJ, Dobson CM, Schwalbe H. Long-range interactions within a nonnative protein. Science. 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]
- 64.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 65.Oster SK, Ho CSW, Soucie EL, Penn LZ. Adv. Cancer Res. 2002;84:81–154. doi: 10.1016/s0065-230x(02)84004-0. [DOI] [PubMed] [Google Scholar]
- 66.Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999;18:2942–2954. doi: 10.1038/sj.onc.1202725. [DOI] [PubMed] [Google Scholar]
- 67.Kaustov L, Yi GS, Ayed A, Bochkareva E, Bochkarev A, Arrowsmith CH. p53 transcriptional activation domain: a molecular chameleon? Cell Cycle. 2006;5:489–494. doi: 10.4161/cc.5.5.2489. [DOI] [PubMed] [Google Scholar]
- 68.Botuyan MV, Momand J, Chen Y. Solution conformation of an essential region of the p53 transactivation domain. Fold. Des. 1997;2:331–342. doi: 10.1016/S1359-0278(97)00047-3. [DOI] [PubMed] [Google Scholar]
- 69.Lee H, Mok KH, Muhandiram R, Park K-H, Suk J-E, Kim D-H, Chang J, Sung YC, Choi KY, Han K-H. Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem. 2000;275:29426–29432. doi: 10.1074/jbc.M003107200. [DOI] [PubMed] [Google Scholar]
- 70.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 71.Di Lello P, Jenkins LMM, Jones TN, Nguyen BD, Hara T, Yamaguchi H, Dikeakos JD, Appella E, Legault P, Omichinski JG. Structure of the Tfb1/p53 complex: insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Molecular Cell. 2006;22:731–740. doi: 10.1016/j.molcel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 72.Feng H, Jenkins LMM, Durell SR, Hayashi R, Mazur SJ, Cherry S, Tropea JE, Miller M, Wlodawer A, Appella E, et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure. 2009;17:202–210. doi: 10.1016/j.str.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bochkareva E, Kaustov L, Ayed A, Yi G-S, Lu Y, Pineda-Lucena A, Liao JCC, Okorokov AL, Milner J, Arrowsmith CH, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl Acad. Sci. USA. 2005;102:15412–15417. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee CW, Martinez-Yamout MA, Dyson HJ, Wright PE. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry. 2010;49:9964–9971. doi: 10.1021/bi1012996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu B, Gilkes DM, Chen J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res. 2007;67:8810–8817. doi: 10.1158/0008-5472.CAN-07-1140. [DOI] [PubMed] [Google Scholar]
- 76.Pazgier M, Liu M, Zou G, Yuan W, Li C, Li C, Li J, Monbo J, Zella D, Tarasov SG, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl Acad. Sci. USA. 2009;106:4665–4670. doi: 10.1073/pnas.0900947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y, Gu B, Wei G, Yuan W, Zhan C, et al. D-peptide inhibitors of the p53–MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc. Natl Acad. Sci. USA. 2010;107:14321–14326. doi: 10.1073/pnas.1008930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt's lymphoma and mouse plasmacytomas. Nat Genet. 1993;5:56–61. doi: 10.1038/ng0993-56. [DOI] [PubMed] [Google Scholar]
- 79.Mittag T, Orlicky S, Choy WY, Tang X, Lin H, Sicheri F, Kay LE, Tyers M, Forman-Kay JD. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl Acad. Sci. USA. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl Acad. Sci. USA. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hateboer G, Timmers HTM, Rustgi AK, Billaud M, Vantveer LJ, Bernards R. TATA-binding protein and the retinoblastoma gene-product bind to overlapping epitopes on c-Myc and adenovirus E1A protein. Proc. Natl Acad. Sci. USA. 1993;90:8489–8493. doi: 10.1073/pnas.90.18.8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.