


Abstract
The histone variant H2A.Z has been implicated in many biological processes, such as gene regulation and genome stability. Here, we present the identification of H2A.Z.2.2 (Z.2.2), a novel alternatively spliced variant of histone H2A.Z and provide a comprehensive characterization of its expression and chromatin incorporation properties. Z.2.2 mRNA is found in all human cell lines and tissues with highest levels in brain. We show the proper splicing and in vivo existence of this variant protein in humans. Furthermore, we demonstrate the binding of Z.2.2 to H2A.Z-specific TIP60 and SRCAP chaperone complexes and its active replication-independent deposition into chromatin. Strikingly, various independent in vivo and in vitro analyses, such as biochemical fractionation, comparative FRAP studies of GFP-tagged H2A variants, size exclusion chromatography and single molecule FRET, in combination with in silico molecular dynamics simulations, consistently demonstrate that Z.2.2 causes major structural changes and significantly destabilizes nucleosomes. Analyses of deletion mutants and chimeric proteins pinpoint this property to its unique C-terminus. Our findings enrich the list of known human variants by an unusual protein belonging to the H2A.Z family that leads to the least stable nucleosome known to date.
INTRODUCTION
In the eukaryotic nucleus, DNA is packaged into chromatin. The fundamental unit of this structure is the nucleosome consisting of a histone octamer (two of each H2A, H2B, H3 and H4) that organizes ∼147 bp of DNA (1). In order to allow or prevent nuclear regulatory proteins access to the DNA, the chromatin structure has to be flexible and dynamic. Several mechanisms ensure controlled chromatin changes, one being the incorporation of specialized histone variants (2,3).
Variants of the histone H2A family are the most diverse in sequence and exhibit distinct functions (4,5), comprising DNA damage repair, transcriptional regulation, cell cycle control and chromatin condensation, though the exact mechanisms of action are not fully understood yet. Interestingly, the highest sequence variation among H2A variants is found in the C-terminus, suggesting that differences in structure and biological function might be primarily attributed to this domain (6–9). One of the best investigated and highly conserved but also functionally enigmatic histone variant is H2A.Z. This variant is essential in most eukaryotes and possesses unique functions (10,11). H2A.Z is involved in transcriptional regulation, chromosome segregation and mitosis, acting in an organism- and differentiation-dependent manner (12,13). Furthermore, H2A.Z has been implicated in regulating epigenetic memory (14) and in inhibiting read-through antisense transcription (15). In higher eukaryotes, H2A.Z might play a role in heterochromatin organization (16), genome stability and chromosome segregation (17). Despite many efforts to elucidate the exact biological functions of H2A.Z, its roles have been and remain controversial (18). Furthermore, deregulation of H2A.Z expression or localization seems to be connected to the development of several neoplasias (19–23). Interestingly, in vertebrates two non-allelic genes coding for two highly similar H2A.Z proteins, H2A.Z.1 and H2A.Z.2, exist (24) (previously named H2A.Z-1 and H2A.Z-2, prefixes were changed due to a new histone variant nomenclature; Talbert P.B., manuscript in preparation). They have a common origin in early chordate evolution, are both acetylated on the same N-terminal lysines (25–27) and might be ubiquitinated on either one of the two C-terminal lysines (28).
Here, we report the identification and structural characterization of H2A.Z.2.2 (Z.2.2), an unusual alternative splice form of H2A.Z. We show that Z.2.2 mRNA is expressed to different degrees in all human cell lines and tissues examined, with highest levels found in brain. Cell biological and biochemical analyses consistently reveal the presence of two distinct Z.2.2 populations within the cell. The majority of Z.2.2 is freely dispersed in the nucleus, whereas only a minority is stably incorporated into chromatin, most likely through the H2A.Z-specific p400/NuA4/TIP60 (TIP60) and SRCAP chaperone complexes. In vivo and in vitro analyses, in agreement with molecular dynamic (MD) simulations, demonstrate that due to its unique docking domain Z.2.2 chromatin incorporation leads to severely unstable nucleosomes. Our data provide compelling evidence that a novel H2A.Z variant exists in humans that plays a distinct and novel role in chromatin structure regulation.
MATERIALS AND METHODS
See Supplementary Materials and Methods section for detailed protocols.
Cell culture, transfection, FACS and cloning
Cell lines were grown in DMEM medium (PAA) supplemented with 10% FCS (Sigma) and 1% penicillin/streptomycin at 37°C and 5% CO2. Cells were transfected using FuGene HD (Roche Applied Science) according to the manufacturer’s instructions. For details on cell selection, FACS and cloning of expression plasmids see Supplementary Materials and Methods section.
RNA expression analysis
RNA isolation and cDNA generation were performed as previously described (29). Data were analyzed with the advanced relative quantification tool of the Lightcycler 480 (Roche) software including normalization to HPRT1 and HMBS levels. Statistical evaluation was done using t-test (two-tailed distribution, heteroscedastic). Total RNA from different human tissues was commercially acquired from: Applied Biosystems: normal lung, breast and tumor breast, lung and ovary; Biochain: tumor lung, breast, thyroid and bone, normal testis, cerebellum, cerebral cortex, hippocampus, thalamus and total fetal brain; amsbio: frontal lobe.
Histone extraction, RP–HPLC purification, sucrose gradient, cellular fractionation and salt stability experiments
Acid extraction of histones was done as previously described (30). Histones were separated by RP–HPLC as previously described (29). Fractions were dried under vacuum and stored at −20°C.
Details on MNase digest and sucrose gradient fractionation can be found in Supplementary Materials and Methods section.
Fractionation and salt stability experiments were carried out as described previously (31–33) with minor changes. For details on these methods see Supplementary Materials and Methods section.
Antibodies
For the generation of a Z.2.2-specific antibody (αZ.2.2), a peptide spanning the last C-terminal amino acids GGEKRRCS of Z.2.2 was synthesized (Peptide Specialty Laboratories GmbH) and coupled to BSA and OVA, respectively. Development of Z.2.2-specific monoclonal antibodies in rats was done as previously described (29). The αZ.2.2 clone 1H11-11 of rat IgG1 subclass was applied in this study. Rabbit αZ.2.2 antibody (rabbit 2, bleed 3) was generated by the Pineda-Antikörper-Service company using the identical peptide epitope followed by affinity purification. Following other primary antibodies were used: αGAPDH (sc-25778, Santa Cruz), αGFP (Roche Applied Science), αH2A (ab 13923, abcam), αH3 (ab1791, abcam) and αH2A.Z (C-terminus: ab4174, abcam; N-terminus: ab18263, abcam). Following secondary antibodies and detection kits were used in immunoblots: GFP-Z.2.2 and GFP-Bbd histones (αGFP) and endogenous Z.2.2 (αZ.2.2) were detected using HRP-conjugated secondary antibodies (Amersham) with ECL advance (Amersham), all other proteins were detected using ECL (Amersham). Detection of recombinant proteins to evaluate histone stoichiometry of in vitro assembled nucleosomes was carried out using IRDye-labeled secondary antibodies (LI-COR).
Fluorescence microscopy of cells and chromosomes
Preparation of cells and chromosome spreads for fluorescence microscopy was done as previously reported (34). Wide-field fluorescence imaging was performed on a PersonalDV microscope system (Applied Precision) equipped with a 60×/1.42 PlanApo oil objective (Olympus), CoolSNAP ES2 interline CCD camera (Photometrics), Xenon illumination and appropriate filtersets. Iterative 3D deconvolution of image z-stacks was performed with the SoftWoRx 3.7 imaging software package (Applied Precision).
FRAP and exponential fitting
For details see Supplementary Materials and Methods section.
Stable isotope labeling with amino acids in cell culture (SILAC) and mass spectrometric identification of H2A.Z-specific chaperone complexes
HeLa cells expressing GFP-Z.2.1 or GFP-Z.2.2 were SILAC labeled and nuclear extracts were prepared as described before (35,36). High-resolution LC MS/MS analysis was performed on an Orbitrap platform: details on the experimental procedure are found in Supplementary Materials and Methods section. Mass spectrometric (MS) operation and raw data analysis (37) are described in Supplementary Materials and Methods section. A complete list of all proteins identified is found in Supplementary Table S1.
Immunoflurescence microscopy of cell cycle-dependent GFP-Z.2.1 and GFP-Z.2.2 chromatin incorporation
Details on the experimental labeling (38) and microscopy procedures are found in Supplementary Materials and Methods section.
Expression of recombinant human histone proteins in Escherichia coli, in vitro octamer and nucleosome reconstitution
Histones were expressed, purified and assembled into octamers as described (39) and mononucleosomes were assembled on DNA containing the 601-positioning sequence (40) according to (39,41). For details on in vitro octamer and nucleosome reconstitution, see Supplementary Materials and Methods section.
Single molecule Förster resonance energy transfer
Single molecule Förster resonance energy transfer (smFRET) single molecule burst analysis followed by the removal of multi-molecular events (42–45) are described in detail in the Supplementary Materials and Methods section.
Molecular modeling and MD simulations
The molecular modeling suite YASARA-structure version 9.10.29 was employed, utilizing the AMBER03 force field (46) for the protein and the general amber force field (GAFF) (47) throughout this study. The partial charges were computed using the AM1/BCC procedure (48) as implemented in YASARA structure (49). The starting point for molecular modeling was the crystal structure of a nucleosome core particle containing the histone variant H2A.Z (PDB 1F66) (50). Missing side chain atoms were added (Glu E 634). The missing N-terminal and C-terminal residues were not modeled, although they might interact with the neighboring DNA, e.g. in the case of missing C-terminal residues in H2A.Z (119–128; GKKGQQKTV). All structures were solvated in a water box with 0.9% NaCl and neutralized (51). The structures were initially minimized using steepest descent and simulating annealing procedures. All deletions and mutations were introduced sequentially using YASARA structure. MD simulations were carried out at 300 K over 2.5 ns in an NPT ensemble using PME. All simulations were performed four times using various starting geometries. The 2.5 ns MD trajectories were sampled every 25 ps, resulting in 100 simulation frames per run, which were evaluated after an equilibration phase of 500 ps to derive statistical averages and properties of the corresponding variant. Finally, the interaction energy of H2A and H3 was calculated from a simulation of the solvated octamer and the isolated (H3–H4)2 tetramer or the isolated respective H2A.Z–H2B dimer. The interaction energy is calculated as energy difference of the solvated octamer minus the solvated (H3–H4)2 tetramer and H2A.Z–H2B dimer.
RESULTS
Alternative splicing of H2A.Z.2 occurs in vivo
Two non-allelic intron-containing genes with divergent promoter sequences that code for H2A.Z variants exist in vertebrates (24,27). In humans, the H2A.Z.2 (H2AFV) primary transcript is predicted to be alternatively spliced thereby generating five different gene products (Supplementary Figure S1A). Using PCR and confirmed by sequencing we detected not only H2A.Z.2.1 (Z.2.1) but also H2A.Z.2.2 (Z.2.2) mRNA, though none of the other splice variants in human cells (Supplementary Figure S1B) showing that the H2A.Z.2 primary transcript is indeed alternatively spliced in vivo. Interestingly, database searches found Z.2.2 mRNA to be predicted in chimpanzee (Pan troglodytes) and Northern white-cheeked gibbon (Nomascus leucogenys) as well. In addition, the coding sequence of the unique exon 6 was present downstream of the H2AFV locus of several other primate genomes, such as gorilla (Gorilla gorilla gorilla), macaque (Macaca mulatta), orangutan (Pongo abelii) and white-tufted-ear marmoset (Callithrix jacchus) (data not shown). In all of these primates, with the exception of marmoset, the resulting protein sequence, if translated, is 100% identical to the unique human Z.2.2 peptide. Further searches revealed that the genomes of horse, and to a certain extent also rabbit and panda bear, contain sequences downstream of their H2AFV loci that could, if translated, lead to proteins with some similarities to human Z.2.2, although they are much more divergent and even longer (rabbit, panda bear). Due to these differences, it is highly likely that those species do not express a Z.2.2 protein homolog. Surprisingly, we could not detect Z.2.2-specific sequences in mouse, rat or other eukaryotic genomes, suggesting that Z.2.2 might be primate specific.
Next, we wanted to determine to what degree all three H2A.Z mRNAs are expressed in different human cell lines and tissues and performed quantitative PCR (qPCR). Z.2.2 mRNA was present to different degrees in all human cell lines and tissues tested, though less abundant than Z.1 and Z.2.1 mRNAs that are expressed in similar amounts (Supplementary Figure S1C and D). Z.2.2 constituted between 5% and 15% of total Z.2 transcripts in all cell lines and tissues, with the exception of brain, where it was statistically significant upregulated (p = 1.7 × 10−4; Figure 1A). In some regions of this particular organ Z.2.2 accounted for up to 50% of all Z.2 transcripts pointing toward an exciting brain-specific function of this novel variant.
Figure 1.

Identification of Z.2.2. (A) qPCR with cDNA from different human cell lines and tissues using primers specific for Z.2.1 and Z.2.2. Data were normalized to HPRT1 and HMBS expression levels. Controls generated without reverse transcriptase (no RT) were used to assess amplification threshold. Shown are the levels of Z.2.2 mRNA as percentages of total Z.2 transcripts (Z.2.1 + Z.2.2). For an evaluation of absolute expression levels see Supplementary Figure S1C and D. (B) Amino acid alignment of human Z.1, Z.2.1 and Z.2.2 proteins using ClustalW Alignment (MacVector 10.0.2). Identical amino acids are highlighted in dark gray, similar amino acids in light gray and changes are set apart on white background. Known acetylation sites are depicted with stars and ubiquitination sites with circles. A schematic representation of the secondary structure of Z.1 and Z.2.1 is shown below the alignment, including depiction of the H3/H4 docking domain (50). M6 and M7 boxes indicate regions important for H2A.Z-specific biological functions in D. melanogaster (60). (C) IB analyses of RP–HPLC purified fractions from different human (HEK293, HeLa, HeLa Kyoto and U2OS) and mouse (NIH3T3) cell lines using a polyclonal rabbit αZ.2.2 and αH2A.Z (αZ, C-terminal) antibodies. Recombinant Z.2.2 protein (rZ.2.2) was loaded in the first lane as positive control for αZ.2.2 antibody. Similar results were obtained when using a monoclonal rat αZ.2.2 antibody (data not shown).
Encouraged by our findings we next investigated whether the endogenous protein is present in vivo. The distinctive feature of Z.2.2 is its C-terminus that is 14 amino acids shorter and contains six amino acids differences compared to Z.2.1 (Figure 1B). Due to this shortened C-terminal sequence, ubiquitination sites at positions K120 and K121 (28) and part of the H3/H4 docking domain (50) are lost in Z.2.2. We generated antibodies against Z.2.2’s unique C-terminal amino acids (αZ.2.2) in rats and rabbits and confirmed their specificity in immunoblots (IB) with recombinant Z.2.1 and Z.2.2 proteins (Supplementary Figure S1E and data not shown). We extracted histones from several human and mouse cell lines, purified them by reversed phase–high performance liquid chromatography (RP–HPLC) and analyzed obtained fractions by IB (Figure 1C). Using αZ.2.2 (polyclonal rabbit), we observed a signal of the calculated weight of Z.2.2 that elutes shortly before Z.1- and Z.2.1-containing fractions in all human samples. Similar results were obtained with a monoclonal αZ.2.2 rat antibody (data not shown). In agreement with the finding that Z.2.2-specific exon 6 sequences are mainly restricted to primate genomes, we could detect Z.2.2 protein in human but not in mouse cells (Figure 1C). In summary, our data show that Z.2.2 protein indeed exists in vivo, albeit at a low expression level.
GFP-Z.2.2 is partially incorporated into chromatin
Having demonstrated the existence of this novel variant in vivo, we next sought to clarify whether Z.2.2 constitutes a bona fide histone by being part of the chromatin structure. Due to high background of all our αZ.2.2 antibodies in IB with cell extracts (data not shown), we generated HeLa Kyoto cell lines stably expressing GFP-tagged H2A variants (HK-GFP cells) for subsequent analyses. Expression levels of GFP-tagged histone variants were determined by FACS (Supplementary Figure S2A) and by comparing expression levels of GFP-tagged variants with endogenous H2A.Z proteins in IB (Supplementary Figure S2B). GFP-Z.1 and -Z.2.1 were expressed in similar amounts as the endogenous H2A.Z protein, and GFP-Z.2.2 expression levels were considerably lower than those of other GFP-tagged H2A variants, with the exception of GFP-H2A.Bbd (Barr body deficient; Bbd). These data show that all GFP-H2A variants were not expressed in abnormal amounts in cell clones used for further analyses.
In fluorescence microscopy, GFP-Z.2.2 exhibited a sole but rather diffuse nuclear distribution similar to GFP-Bbd, suggesting that both variants might have similar properties (Figure 2A). Additionally, GFP-Z.2.2 was detected in condensed mitotic chromosomes, with a faint residual staining in the surrounding area (Figure 2B), suggesting that it is incorporated into chromatin, although to a lesser extent than other GFP-H2A variants. To discriminate between a potential non-specific DNA binding and nucleosomal incorporation of Z.2.2 we purified mononucleosomes by sucrose gradient centrifugation. GFP-Z.2.2 was detected by IB in fractions containing mononucleosomes (Figure 2C), suggesting that Z.2.2 is indeed a nucleosomal constituent.
Figure 2.
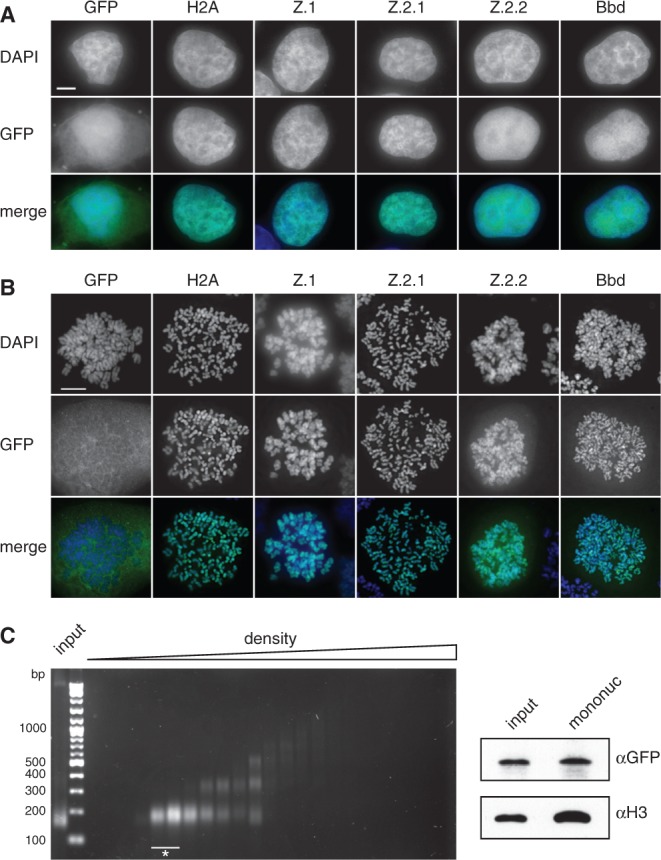
Z.2.2 localizes to the nucleus and is partially incorporated into chromatin. (A) Fluorescence imaging of stably transfected HeLa Kyoto cells shows nuclear localization of all GFP-H2A variants (middle). DNA was counterstained with DAPI (top). Overlay of both channels in color is shown at the bottom (Merge; GFP: green, DAPI: blue). Scale bar = 5 µm. (B) Deconvolved images of metaphase spreads of HeLa Kyoto cells stably expressing GFP-H2A variants (middle). Merged images in color are shown below (GFP: green; DAPI: blue). Scale bar = 10 µm. (C) Chromatin from HeLa Kyoto cells stably expressing GFP-Z.2.2 was digested with MNase followed by a purification of mononucleosomes using sucrose gradient centrifugation. Isolated DNA from subsequent sucrose gradient fractions was analyzed by agarose gel electrophoresis (left). Fractions containing pure mononucleosomes (marked with asterisk) were combined and analyzed by IB (right) using αGFP antibody for the presence of GFP-Z.2.2 (top), and αH3 (bottom).
To analyze the extent of Z.2.2 chromatin incorporation in more detail, we isolated soluble (sol) and chromatin (chr) fractions from HK-GFP cells. IB analyses revealed, as expected, that similar to GFP-Bbd, GFP-Z.2.2 is predominantly nuclear soluble, with only minor amounts present in chromatin (Figure 3A). Based on fractionation and fluorescence imaging results, we hypothesized that this novel variant behaves in a different manner as compared to other H2A variants with regard to chromatin exchange mobility in vivo. To test this prediction, we performed fluorescence recovery after photobleaching (FRAP) experiments with HK-GFP cells. Using spinning disk confocal microscopy we monitored the kinetic behavior of H2A variants with variable intervals over 2 min (short-term) up to several hours (long-term) after bleaching a 5 µm × 5 µm square nuclear region (Figure 3B and Supplementary Figure S3). As expected, GFP alone showed the highest mobility. In contrast, GFP-H2A, -Z.1 and -Z.2.1 showed a slow recovery, which is in agreement with a previous report (52). GFP-Bbd has been described to exhibit low nucleosomal stability and a fast FRAP kinetic (53), which we also observed in our experiment. Interestingly, GFP-Z.2.2 showed an even faster recovery than GFP-Bbd, with ∼80% of initial fluorescence reached after 1 min. Careful assessment and bi-exponential fitting of FRAP data allowed us to also calculate ratios of fractions with fast, intermediate and slow recovery and their respective half-time of recovery (t1/2) as an indication of exchange rate thereby revealing quantitative differences between Z.2.2 and other H2A variants (Figure 3D, Supplementary Figure S3C and E). For Z.2.2 as well as for Bbd, we identified a fast fraction of unbound or very transiently interacting molecules (78%, t1/2 ∼ 1.1 s and 52%, t1/2 ∼ 2.5 s, respectively; for comparison GFP t1/2 ∼ 0.4 s) and a substantially slower fraction with a t1/2 in the range of 7–9 min. In contrast, GFP-H2A, -Z.1 and -Z.2.1 showed no fast mobile fraction but intermediate slow fractions with t1/2 in the range of 8–17 min and a second even slower class exchanging with a t1/2 of a few hours. For comparison, we measured the linker histone H1.0 (54–57) and the histone binding protein HP1α (58,59), both DNA-associated proteins, and found that HP1α shows an overall much faster recovery than all H2A variants. In contrast to Z.2.2 and Bbd, no unbound fraction of H1.0 was detected. More importantly, with regards to the bound Z.2.2 and Bbd fractions overall H1.0 showed a faster recovery, arguing against an unspecific DNA-association of Z.2.2 and Bbd. In agreement with cell biological and biochemical analyses, these data clearly demonstrate that a large fraction of the splice variant Z.2.2 is very rapidly exchanged or chromatin unbound, and a minor population is incorporated into chromatin.
Figure 3.
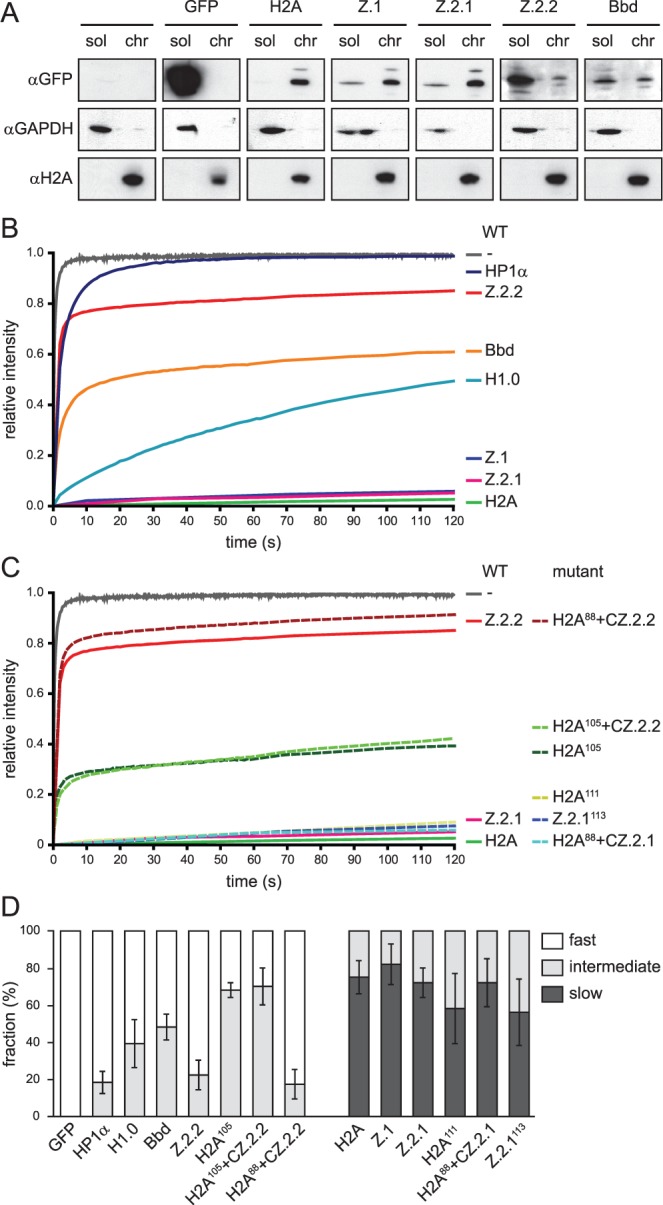
The majority of Z.2.2 protein is nuclear soluble and highly mobile in a sequence-dependent manner. (A) HK-GFP cells were subjected to biochemical fractionation. Fractions sol and chr of identical cell equivalents were probed in IB with αGFP (top), αH2A (middle) and αGAPDH (bottom). (B) FRAP quantification curves of average GFP signal relative to fluorescence intensity prior to bleaching are depicted for GFP, GFP-tagged wild-type H2A variants, linker histone H1.0 and heterochromatin protein 1α (HP1α). Mean curves of 10–29 cells are shown for each construct. Error bars are omitted for clarity. (C) FRAP quantification curves similar to (B) are depicted for GFP, GFP-tagged wild-type H2A, Z.2.1, Z.2.2 and mutant constructs. (D) Quantitative evaluation of FRAP curves. Plot shows calculated mobility fraction sizes of different wild-type and mutant H2A variant constructs, as well as H1.0 and HP1α, based on bi-exponential fitting of FRAP data. Error bars indicate SD (see Supplementary Figure S3 for long-term FRAP and for numerical values).
Z.2.2’s unique docking domain, but not its shortened length, weakens chromatin association
The functional importance of specific C-terminal domains of H2A.Z has previously been demonstrated by nucleosomal structure analyses (7,50) and in rescue experiments in flies (60). Since the C-terminus of Z.2.2 is shorter and has a distinct sequence when compared to Z.1 and Z.2.1, it is not clear which of these features determines Z.2.2’s unusual chromatin-association.
Therefore, we generated deletion and domain-swap constructs (Supplementary Figure S3D) for FRAP experiments (short-term: Figure 3C and long-term: Supplementary Figure S3B). Surprisingly, C-terminal deletions of GFP-H2A (H2A111) and GFP-Z.2.1 (Z.2.1113) to mimic the shortened length of Z.2.2 did not affect their original mobility in short-term and only modestly in long-term FRAP. Hence, the mere shortening of the C-terminus is not sufficient to weaken stable chromatin association.
To investigate whether the unique six C-terminal amino acids of Z.2.2 are sufficient to generate highly mobile proteins, we created a further C-terminally truncated GFP-H2A construct (H2A105) and added the Z.2.2 specific C-terminal six amino acids (H2A105 + CZ.2.2). Although both mutant constructs are slightly more mobile than H2A111, their indistinguishable recovery kinetics demonstrate that the unique six C-terminal amino acids of Z.2.2 alone are not sufficient to cause its extreme mobility in vivo.
To explore whether the complete Z.2.2 docking domain is able to induce high-protein mobility, we transferred the respective domain of either Z.2.1 (amino acids 91–127) or Z.2.2 (amino acids 91–113) onto a C-terminally truncated H2A (H2A88 + CZ.2.1 and H2A88 + CZ.2.2, respectively). Interestingly, only the docking domain of Z.2.2, but not the one of Z.2.1, confers high mobility. In conclusion, the six unique C-terminal amino acids of Z.2.2 prevent chromatin-association of a large proportion of this protein, but only when present in the context of the preceding H2A.Z-specific docking domain sequence.
Z.2.2 interacts with H2A.Z-specific TIP60 and SRCAP chaperone complexes and is deposited into chromatin outside of S-phase
Our so far obtained data strongly imply that at least a minor amount of the cellular Z.2.2 protein is incorporated into nucleosomes. Since previous studies have shown that evolutionary conserved Swr1-related ATP-dependent chromatin remodelers specifically exchange canonical H2A–H2B with H2A.Z–H2B dimers within nucleosomes (10,61), we wondered if such complexes are also able to actively deposit Z.2.2 into chromatin. HK cells and HK cells stably expressing GFP-Z.2.1 or -Z.2.2 were SILAC labeled, soluble nuclear proteins isolated, GFP-tagged Z.2.1 and Z.2.2-associated proteins precipitated using GFP nanotrap beads and identified by quantitative mass spectrometry (Figure 4 and Supplementary Table S1 for a complete list of all identified proteins). Whereas the majority of proteins are background binders clustering around 0, specific interactors can be found on the right side having a high ratio H/L or ratio L/H for Z.2.1 and Z.2.2, respectively. In accordance with previous studies (62–65), we found GFP-Z.2.1 to be part of two major complexes, the SRCAP and the p400/NuA4/TIP60 (TIP60) complexes (Figure 4A), as we were able to detect all of their thus far identified members, with the exception of actin, as significant outliers. Interestingly, GFP-Z.2.2 also associated with both SRCAP and TIP60 complexes (Figure 4B), showing an almost identical binding composition as GFP-Z.2.1 (Figure 4C). These results strongly imply that Z.2.2 is, similar to other H2A.Z variants, actively deposited into chromatin through specific chaperone complexes.
Figure 4.
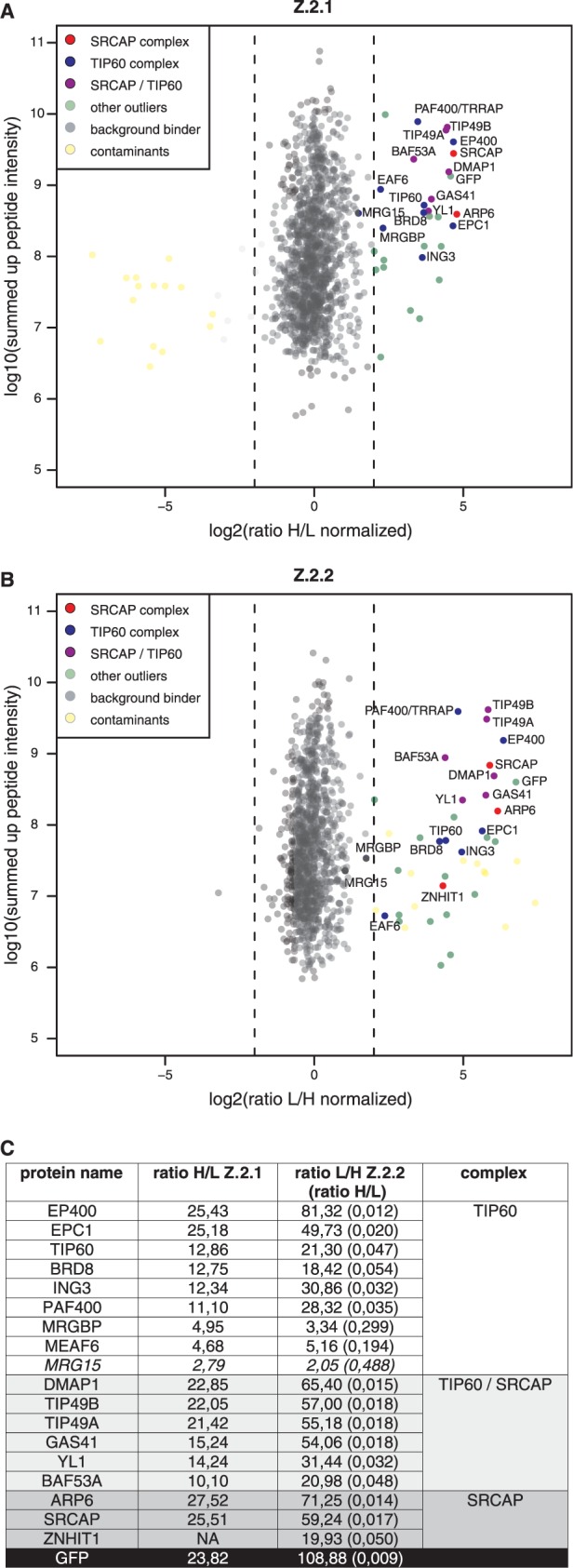
Z.2.2 associates with H2A.Z-specific SRCAP and TIP60 chaperone complexes. GFP-pull-downs for H2A.Z-specific chaperone complexes are shown. HK cells stably expressing GFP-Z.2.1 (A) and GFP-Z.2.2 (B) were SILAC-labeled and subjected to single-step affinity purifications of soluble nuclear proteins in a ‘forward’ (GFP-Z.2.1) or ‘reverse’ (GFP-Z.2.2) pull-down using GFP nanotrap beads. In each panel the ratio of the identified proteins after MS is plotted. Proteins known to interact with H2A.Z are indicated in the following way: members of the SRCAP complex in red, members of the TIP60 complex in blue and shared subunits in purple. Potential novel H2A.Z-interacting proteins are shown as green dots (‘other outliers’) and are distinguished from background binders (gray dots) and contaminants (yellow dots). See also Supplementary Table S1 for a list of all identified proteins. (C) List of the SRCAP and TIP60 complex members and their normalized binding intensity to Z.2.1 or Z.2.2. Note that for comparison reasons the obtained H/L ratios of GFP-Z.2.2 binders (numbers in brackets) were calculated in the corresponding L/H ratios. See also Supplementary Table S1 for a list of all identified proteins and their normalized H/L ratios.
Based on these results, we predicted that Z.2.1 and Z.2.2 should be incorporated into chromatin in a highly similar spatial manner. Since both SRCAP and TIP60 chaperone complexes are evolutionary conserved between different species, we tested mouse C127 cells that do not express endogenous Z.2.2 for their ability to deposit GFP-Z.2.2. Hereby we should be able to distinguish whether SRCAP and TIP60 complexes are sufficient for deposition, or if other potential primate-specific factors are needed. GFP-Z.2.1 and -Z.2.2 were transiently expressed in C127 cells, S-phase stages highlighted by EdU-incorporation and co-localization patterns visualized by fluorescence microscopy (Figure 5). GFP-Z.2.1 and -Z.2.2 showed an almost identical chromatin localization and deposition pattern, suggesting that Z.2.2 is, like Z.2.1, deposited through SRCAP and TIP60 complexes. In accordance with a recent study, we observed an enrichment of both H2A.Z variants in facultative heterochromatin regions in interphase nuclei (66). Surprisingly, although H2A.Z is expressed in all cell cycle phases (67), and GFP-Z.2.1 and -Z.2.2 expression is driven by a constitutive active promoter, chromatin deposition of both proteins is underrepresented at replication foci. This result underlines our findings that Z.2.2 interacts with all members of both TIP60 and SRCAP complexes and is actively and not passively deposited, as would have been the case during S-phase when nucleosomes are highly exchanged.
Figure 5.

Z.2.1 and Z.2.2 are actively deposited into chromatin and are under-represented at replication foci. C127 cells transiently expressing GFP-Z.2.1 (left) and -Z.2.2 (right) were pulse labeled with EdU to visualize replication foci and to identify S-phase stages. DNA was counterstained with DAPI and analyzed by wide-field deconvolution microscopy. To remove the unbound fraction in GFP-Z.2.2 expressing cells, an in situ extraction was performed prior to fixation. Cells in early, middle and late S-phases were distinguished due to their characteristic differential EdU replication labeling patterns of eu- and heterochromatic regions. Merged images in color are shown alongside (GFP: green; EdU: red; DAPI: blue). Scale bar = 5 µm.
Structural changes in Z.2.2’s C-terminus prevent histone octamer folding and enhance DNA breathing on structurally destabilized nucleosomes
Our findings thus far imply that Z.2.2 is incorporated into nucleosomes and most likely targeted by TIP60 and SRCAP complexes. Then why does a large fraction of the cellular Z.2.2 protein pool shows a high mobility and is freely dispersed in the nucleus? One plausible possibility is that Z.2.2 severely destabilizes nucleosomes due to its divergent C-terminal docking domain and is hence rapidly exchanged. To test this hypothesis, we used an in vitro reconstitution system. Recombinant human H2A variants together with H3, H2B and H4 (Supplementary Figure S4A) were refolded by dialysis, and formed complexes purified by size exclusion chromatography. As expected, both H2A and Z.2.1 containing samples readily formed histone octamers (Figure 6A, solid lines). Bbd served as a negative control, because it has been demonstrated to not form octamers under these conditions (41), a result we also observed (Figure 6A left, dotted line). Interestingly, in accordance with our FRAP data, Z.2.2 behaved like Bbd in that it only formed Z.2.2–H2B dimers, but did not complex together with (H3–H4)2 tetramers to generate octamers (Figure 6A right, dotted line), which was further confirmed by SDS–PAGE analyses of the separate fractions (Figure 6B). Thus, like for Bbd the incorporation of Z.2.2 destabilizes the interface between Z.2.2–H2B dimers and (H3–H4)2 tetramers in a C-terminal sequence dependent manner (Supplementary Figure S4B and C). In conclusion, the Z.2.2 docking domain is sufficient to prevent octamer formation.
Figure 6.

Z.2.2 does not constitute stable histone octamers with H2B, H3 and H4 in vitro. (A) Size exclusion chromatography of refolding reactions using recombinant human H3, H4 and H2B proteins together with either H2A (solid line) or Bbd (dashed line) (left overlay) or with either Z.2.1 (solid line) or Z.2.2 (dashed line) (right overlay). Peaks corresponding to aggregates, histone octamers, tetramers or dimers are labeled respectively. (B) Fractions corresponding to H2A-containing octamers, Bbd-containing tetramers and dimers (left) or Z.2.1-containing octamers and Z.2.2-containing tetramers and dimers (right) were analyzed by 18% SDS–PAGE and stained with Coomassie brilliant blue.
Although no Z.2.2 containing histone octamers could be generated in vitro, our results using GFP-Z.2.2 strongly suggest that Z.2.2 can be part of nucleosomes. To test this in vitro and to evaluate the effect of Z.2.2 on nucleosome stability, we reconstituted mononucleosomes by mixing Z.2.2–H2B dimers, (H3–H4)2 tetramers and DNA containing a ‘Widom 601’ DNA positioning sequence in a 2:1:1 ratio. As controls, we reconstituted H2A or Z.2.1 containing nucleosomes by mixing octamers and DNA in a 1:1 ratio. As expected, analysis of all nucleosomes by native PAGE showed a single band before and after heat shift (Figure 7A), indicating a unique position on the ‘Widom 601’ DNA template. Purification of nucleosomes from a native gel and analysis of the protein content by SDS–PAGE (Coomassie staining and immunoblot) showed that Z.2.2 was indeed incorporated into nucleosomes (Figure 7B). All nucleosomes were further evaluated for their resistance to MNase cleavage as an indicator of stably organized nucleosomes and to determine nucleosomal DNA length (Figure 7C and Supplementary Figure S5). We observed fragments corresponding to protected nucleosomal DNA with the length of 146 bp for all variant nucleosomes tested. The appearance of smaller, subnucleosomal fragments indicates that DNA breathing occurred (68). Interestingly, DNA of Z.2.2 nucleosomes is less protected, since subnucleosomal fragments were obtained at lower MNase concentrations than with H2A or Z.2.1 nucleosomes. Additionally, at higher MNase concentrations a stable DNA fragment of about 120 bp was most abundant for Z.2.2 nucleosomes (Supplementary Figure S5), indicating that this might be the preferred DNA length wrapped around this octamer. These data suggest that increased DNA breathing occurs in Z.2.2 nucleosomes, which as a result might be less stable. To quantify nucleosome stability in vitro we measured salt-dependent changes in nucleosome structure using smFRET (69). In line with the results presented above, Z.2.2 containing recombinant nucleosomes lost their compact structure at lower salt concentrations than Z.2.1 or H2A-containing ones (Figure 7D). To investigate whether the observed Z.2.2-dependent nucleosome destabilization is true in the context of chromatin, we isolated chromatin from HK cells expressing GFP-H2A variants and incubated it with buffer containing increasing amounts of salt. Histones that remained stable chromatin components were precipitated and detected by IB (Figure 7E). As observed with FRET techniques, Z.2.2-containing nucleosomes disintegrated between 200 and 400 mM NaCl, and were therefore even less stable than Bbd-containing ones. In summary, incorporation of Z.2.2 leads to a severely reduced nucleosome stability due to C-terminal sequence dependent changes in its docking domain and subsequent loss of its interaction with histone H3.
Figure 7.

Z.2.2-containing nucleosomes are less resistant to MNase digestion and increased ionic strength. (A) H2A, Z.2.1 or Z.2.2 containing nucleosomes were assembled on DNA by salt gradient deposition, incubated at 4°C or 37°C to evaluate DNA positioning and separated by a native 5% PAGE gel. (B) Agarose-gel-electro-eluted material from (A) was analyzed by 18% SDS–PAGE and Coomassie stained to evaluate stoichiometry of histones after nucleosome assembly (top). Stars indicate H2A variants that were used for assembly. Further evaluation of histone stoichiometry after nucleosome assembly was done by IB using a LI-COR instrument (bottom). Assembled nucleosomes containing Z.2.1 or Z.2.2 were immunoblotted and the amount of histones was visualized using an αH3 antibody (top) and an N-terminal αZ antibody (recognizes all H2A.Z variants, bottom). (C) Mononucleosomes containing either H2A, Z.2.1 or Z.2.2 were digested with increasing concentrations of MNase and extracted DNA was separated using Bioanalyzer. Stars indicate DNA length of 146 bp. For detailed electropherogram analyses of fragment lengths in each sample see Supplementary Figure S5. (D) Mononucleosomes containing either H2A, Z.2.1 or Z.2.2 histones together with double dye labeled DNA were incubated with increasing amounts of salt. smFRET measurement values of each salt concentration were normalized to 0 mM NaCl. Error bars represent SEM of six measurements. (E) Chromatin from HK-GFP cells was isolated and incubated with increasing amounts of salt. Chromatin-bound histones were precipitated and detected by IB using αGFP antibody. Staining with αH2A was used as loading control.
Our FRAP data suggest that the Z.2.2 C-terminal amino acids might have a direct influence on the nucleosomal structure by affecting interactions with DNA and/or adjacent histones. Based on the existing structural data (50), we performed MD simulations of nucleosomes containing Z.1 (Supplementary Figure S7) or Z.2.2. In addition, we also included the deletion mutant Z.2.1113, which did not show any change in short-term FRAP (Figure 3C), but some increase in mobility in long-term FRAP (Supplementary Figure S3B) in our assay. These in silico models revealed that changes in the C-terminus of H2A.Z strongly affect its protein structure (Figure 8A). Strikingly, different statistical descriptors over the MD-trajectory like distance and mobility (B-factor) show in contrast to Z.1 and Z.2.1113 unique properties for the Z.2.2 tail. Only Z.2.2 leads to a substantial structural change in the C-terminus resulting in an increased distance to histone H3, which in turn makes a hydrogen bond interaction between peptide backbone NH of Cys112 in Z.2.2 and the oxygen in the Gln55 side chain in H3 impossible (Figure 8B). Additionally, an increase in the B-factor for Z.2.2 indicates a substantially enhanced mobility of Z.2.2’s C-terminus (Figure 8C). We also calculated the Z.2.2–H3 interaction energy and observed a switch from negative to positive values in the case of Z.2.2 suggesting that this histone variant destabilizes the nucleosome (Figure 8D). In summary, these data suggest that the C-terminal sequence of Z.2.2 leads to a more dynamic structure that in turn loses binding to histone H3 and destabilizes the nucleosomal structure, providing a reasonable explanation for the observed in vivo and in vitro data.
Figure 8.

Unique Z.2.2 C-terminal amino acids cause significant changes in protein and nucleosome structure. (A) In silico models of Z.1, Z.2.1113 and Z.2.2 C-terminal C-chains (yellow; from amino acids 84 to C-terminus, including the complete docking domain) together with the E-chain of histone H3 (blue; amino acids 33–60, including αN-helix). Side (left) and frontal views (right) of four MD simulations are shown respectively. See Supplementary Figure S7 for complete in silico model of H2A.Z-containing nucleosome. (B) Simulated distances between peptide backbone NH of amino acids 112 in H2A.Z (His or Cys, respectively) variants and the oxygen in the Gln55 sidechain in H3 based on in silico nucleosome models containing either Z.1 (white), Z.2.1113 (light gray) or Z.2.2 (dark gray) proteins. Error bars represent SD of four independent simulations. (C) Simulated mobility measuring B-factor values between amino acids 108 and 113 in respective H2A.Z variant C-termini. Error bars represent SD of four independent simulations. (D) Simulated interaction energy between tetramer versus respective H2A.Z variant-containing dimers.
DISCUSSION
In this work, we have identified a previously unknown histone H2A.Z variant and provide a comprehensive characterization of its nucleosomal properties. This alternatively spliced variant, Z.2.2, is present to different degrees in all human cell lines and tissues investigated, with a significant enrichment in brain. Z.2.2 contains a shortened and in six amino acids divergent C-terminus compared to Z.1 and Z.2.1 that is necessary, but not sufficient, to weaken chromatin association. Only in the context of the unique Z.2.2 docking domain does the C-terminal sequence negatively affect nucleosome stability in vitro and in vivo. To our knowledge, Z.2.2 has the strongest destabilizing effect on nucleosomal structure compared to other histone H2A variants reported to date.
Only one other histone variant, macroH2A, has been shown thus far to be alternatively spliced (70). Here, like our observation with H2A.Z, two independent genes mH2A1 and mH2A2 exist in mammals, with only mH2A1 being alternatively spliced resulting in functional different proteins (71). In our study, we demonstrate that the human H2A.Z.2 (H2AFV) primary transcript is alternatively spliced generating Z.2.1 and Z.2.2 mRNAs and proteins. These observations suggest that Z.2.2 is tightly regulated in a tissue-specific manner through alternative splicing and/or RNA stability. Our findings now raise the intriguing possibility that alternative splicing of histone variants might not be rare but more common than previously thought. If true, it will be of interest to reevaluate other intron-containing histone variant genes with regard to their possible alternative transcripts and protein products.
Bioinformatic genome analyses revealed the existence of Z.2.2-specific sequences only in humans, old and new world primates and to some extend in other mammals, with the exclusion of mouse, rat and even lower eukaryotes. It remains to be seen, whether Z.2.2’s evolution is indeed limited to primates only. Primate-specific gene products have been often identified in human brain and reproductive tissues (72), supporting the notion that their RNAs and proteins might be essential to adaptive changes leading to human development and further speculates that primate-specific genes might be important in reproductive function and disease. Since we have found Z.2.2 transcripts to be strongly enriched in brain samples of higher brain function in comparison to other tissues and cell types, it will be of great interest to determine in future studies, if this novel variant might play an important functional role in this particular organ. These observations also raise the interesting question of how alternative splicing and/or differential stability of H2AFV transcripts are tissue specifically regulated.
Another intriguing feature of Z.2.2 is its influence on nucleosome stability. Although Z.2.2 localizes exclusively to the nucleus, only a minor proportion is stably incorporated into chromatin. The only other exception in humans known thus far is Bbd, which has previously been demonstrated to destabilize the nucleosome structure (41,53,73). Bbd, similar to Z.2.2, is a shorter H2A variant with an unusual C-terminus and a considerable different primary histone fold sequence that might explain its ability to destabilize nucleosomes. In agreement, a recent study demonstrated that the incomplete C-terminal docking domain of Bbd results in structural alterations in nucleosomes and that those are in turn associated with an inability of the chromatin remodeler RSC to both remodel and mobilize nucleosomes (8). Z.2.2, on the other hand, is identical to Z.2.1, except that its C-terminus is 14 amino acids shorter and in six amino acids altered. How can this small change in Z.2.2’s primary sequence lead to such drastic effects on chromatin association?
We show that Z.2.2 can be part of a bona fide nucleosome and that it interacts with the H2A.Z-specific TIP60 and SRCAP chaperone complexes. These complexes have been shown to catalyze the exchange of H2A–H2B dimers with H2A.Z–H2B dimers in nucleosomes and our finding therefore suggests that both complexes are also involved in an active chromatin incorporation of Z.2.2. Supporting this idea is the observation that both Z.2.1 and Z.2.2 are incorporated into chromatin in a replication-independent manner, even in mouse cells that do not express endogenous Z.2.2. Both H2A.Z variants are not primarily deposited at replication foci, not even in middle S-phase when facultative heterochromatin is replicated, where the majority of the H2A.Z protein pool is found in interphase cells (66). Our findings are in agreement with a model proposed by Hardy and Robert, in which H2A.Z variants are randomly deposited into chromatin by specific chaperone complexes in a replication-independent manner coupled to a subsequent targeted H2A.Z depletion (74). As a consequence, an enrichment of H2A.Z at non-transcribed genes and heterochromatin regions over several cell generations can be observed (74). It might be possible that INO80 facilitates this eviction function, as it has been shown to exchange nucleosomal H2A.Z–H2B dimers with free H2A–H2B dimers (75). It will be of interest in future studies to determine whether Z.2.2 exchange is subjected to a similar mechanism. Taken together, our findings strongly imply that Z.2.2 is actively deposited into chromatin through the interaction with evolutionary conserved chaperone complexes. Nevertheless, a large fraction of Z.2.2 protein is not chromatin bound and we have mapped the region crucial for high FRAP mobility to its docking domain. In addition to Z.2.2’s unique C-terminal amino acids this region spans the highly conserved acidic patch responsible for deposition (76), the M6 region that is functionally essential in fly H2A.Z (60) and required for the interaction with the SWR1 complex in yeast (77). Strikingly, in silico simulation of Z.2.2 predicted dynamic structural changes that in turn weaken interaction with histone H3 and destabilize the nucleosome structure. Such a gross structural alteration explains why Z.2.2 is not able to form stable octamers in vitro and leads to enhanced DNA breathing in a nucleosomal context. Hence, Z.2.2 incorporation into chromatin disrupts nucleosomes more easily and supports a model in which Z.2.2 is more rapidly exchanged than Z.2.1.
What functional outcome might Z.2.2 cause when incorporated into chromatin? And how is the variant composition of Z.2.2 containing nucleosomes? It has been shown that a special class of nucleosomes containing both H2A.Z and H3.3 variants exists in humans (78). These nucleosomes are enriched at promoters, enhancers and insulator region and promote the accessibility of transcription factors to these DNA regions (78), most likely due to their extreme sensitivity to disruption (79). Since these studies nicely demonstrate that differential nucleosome stabilities due to the incorporation of different histone variants influence transcriptional regulation, it is tempting to speculate that Z.2.2 might also affect chromatin-related processes. Future experiments will shed light on Z.2.2 function(s), especially with regard to its increased expression in human brain areas, and explain why and where nucleosomal destabilization is needed. This is of particular interest, since Bbd that also leads to nucleosomal destabilization is almost exclusively present in testis (80–82) in contrast to the apparently ubiquitously expressed Z.2.2, possibly pointing toward distinct roles of both destabilizing H2A variants in different tissues. Our data suggest that additional interesting, yet unidentified, histone variants may exist and await their discovery.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table S1, Supplementary Figures S1–S7 and Supplementary Materials and Methods.
FUNDING
German Research Foundation (DFG), SFB Transregio 5 (to S.B.H., E.K., H.L. and L.S.); the BioImaging Network Munich and the German Ministry for Education and Research (BMBF) (to H.L.); the Center for Integrated Protein Science Munich (CIPSM) (to H.L., J.M., M.M. and S.B.H.); the European Research Council (to J.M.); the International Max-Planck Research School for Molecular and Cellular Life Sciences (IMPRS-LS) (to S.P., S.M.W. and K.S.); the International Doctorate Program NanoBio Technology (IDK-NBT) (to K.S.). Funding for open access charge: DFG.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank S. Dambacher, M. Kador, H. Klinker, C. Mehlhorn, M. Holzner and F. Müller-Planitz for advice and help. The authors also thank U. Rothbauer for providing various GFP-Trap beads to initially test GFP pull-down efficiency. The authors are grateful to D. Rhodes, E. Bernstein, R. Schneider, M. Hendzel, T. Misteli and S. Matsunaga for reagents. The authors are indebted to E. Bernstein for critical examination of the manuscript. The authors thank especially the Hake lab and members of the Adolf-Butenandt Institute for constructive discussions.
REFERENCES
- 1.van Holde KE. Chromatin. New York: Springer; 1988. [Google Scholar]
- 2.Bonisch C, Nieratschker SM, Orfanos NK, Hake SB. Chromatin proteomics and epigenetic regulatory circuits. Exp. Rev. Proteomics. 2008;5:105–119. doi: 10.1586/14789450.5.1.105. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem. Cell Biol. 2006;84:505–517. doi: 10.1139/o06-085. [DOI] [PubMed] [Google Scholar]
- 4.Boulard M, Bouvet P, Kundu TK, Dimitrov S. Histone variant nucleosomes: structure, function and implication in disease. Subcell. Biochem. 2007;41:71–89. [PubMed] [Google Scholar]
- 5.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr. Opin. Genet. Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 6.Ausio J, Abbott DW. The many tales of a tail: carboxyl-terminal tail heterogeneity specializes histone H2A variants for defined chromatin function. Biochemistry. 2002;41:5945–5949. doi: 10.1021/bi020059d. [DOI] [PubMed] [Google Scholar]
- 7.Wang AY, Aristizabal MJ, Ryan C, Krogan NJ, Kobor MS. Key functional regions in the histone variant H2A.Z C-terminal docking domain. Mol. Cell. Biol. 2011;31:3871–3884. doi: 10.1128/MCB.05182-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shukla MS, Syed SH, Goutte-Gattat D, Richard JL, Montel F, Hamiche A, Travers A, Faivre-Moskalenko C, Bednar J, Hayes JJ, et al. The docking domain of histone H2A is required for H1 binding and RSC-mediated nucleosome remodeling. Nucleic Acids Res. 2011;39:2559–2570. doi: 10.1093/nar/gkq1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogler C, Huber C, Waldmann T, Ettig R, Braun L, Izzo A, Daujat S, Chassignet I, Lopez-Contreras AJ, Fernandez-Capetillo O, et al. Histone H2A C-terminus regulates chromatin dynamics, remodeling, and histone H1 binding. PLoS Genet. 2010;6:e1001234. doi: 10.1371/journal.pgen.1001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Billon P, Cote J. Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim Biophys Acta. 2011;1819:290–302. doi: 10.1016/j.bbagrm.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Marques M, Laflamme L, Gervais AL, Gaudreau L. Reconciling the positive and negative roles of histone H2A.Z in gene transcription. Epigenetics. 2010;5:267–272. doi: 10.4161/epi.5.4.11520. [DOI] [PubMed] [Google Scholar]
- 12.Draker R, Cheung P. Transcriptional and epigenetic functions of histone variant H2A.Z. Biochem. Cell Biol. 2009;87:19–25. doi: 10.1139/O08-117. [DOI] [PubMed] [Google Scholar]
- 13.Svotelis A, Gevry N, Gaudreau L. Regulation of gene expression and cellular proliferation by histone H2A.Z. Biochem. Cell Biol. 2009;87:179–188. doi: 10.1139/O08-138. [DOI] [PubMed] [Google Scholar]
- 14.Brickner DG, Cajigas I, Fondufe-Mittendorf Y, Ahmed S, Lee PC, Widom J, Brickner JH. H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007;5:e81. doi: 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, Grewal SI. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature. 2009;461:419–422. doi: 10.1038/nature08321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rangasamy D, Berven L, Ridgway P, Tremethick DJ. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003;22:1599–1607. doi: 10.1093/emboj/cdg160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rangasamy D, Greaves I, Tremethick DJ. RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat. Struct. Mol. Biol. 2004;11:650–655. doi: 10.1038/nsmb786. [DOI] [PubMed] [Google Scholar]
- 18.Zlatanova J, Thakar A. H2A.Z: view from the top. Structure. 2008;16:166–179. doi: 10.1016/j.str.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Dryhurst D, McMullen B, Fazli L, Rennie PS, Ausio J. Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 2012;315:38–47. doi: 10.1016/j.canlet.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Valdes-Mora F, Song JZ, Statham AL, Strbenac D, Robinson MD, Nair SS, Patterson KI, Tremethick DJ, Stirzaker C, Clark SJ. Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 2012;22:307–321. doi: 10.1101/gr.118919.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010;20:1383–1390. doi: 10.1101/gr.106542.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svotelis A, Gevry N, Grondin G, Gaudreau L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle. 2010;9:364–370. doi: 10.4161/cc.9.2.10465. [DOI] [PubMed] [Google Scholar]
- 23.Hua S, Kallen CB, Dhar R, Baquero MT, Mason CE, Russell BA, Shah PK, Liu J, Khramtsov A, Tretiakova MS, et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008;4:188. doi: 10.1038/msb.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eirin-Lopez JM, Gonzalez-Romero R, Dryhurst D, Ishibashi T, Ausio J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 2009;9:31. doi: 10.1186/1471-2148-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beck HC, Nielsen EC, Matthiesen R, Jensen LH, Sehested M, Finn P, Grauslund M, Hansen AM, Jensen ON. Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell Proteomics. 2006;5:1314–1325. doi: 10.1074/mcp.M600007-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Bonenfant D, Coulot M, Towbin H, Schindler P, van Oostrum J. Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol. Cell Proteomics. 2006;5:541–552. doi: 10.1074/mcp.M500288-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Dryhurst D, Ishibashi T, Rose KL, Eirin-Lopez JM, McDonald D, Silva-Moreno B, Veldhoen N, Helbing CC, Hendzel MJ, Shabanowitz J, et al. Characterization of the histone H2A.Z-1 and H2A.Z-2 isoforms in vertebrates. BMC Biol. 2009;7:86. doi: 10.1186/1741-7007-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarcinella E, Zuzarte PC, Lau PN, Draker R, Cheung P. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol. Cell. Biol. 2007;27:6457–6468. doi: 10.1128/MCB.00241-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiedemann SM, Mildner SN, Bonisch C, Israel L, Maiser A, Matheisl S, Straub T, Merkl R, Leonhardt H, Kremmer E, et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell. Biol. 2010;190:777–791. doi: 10.1083/jcb.201002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat. Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 31.Wysocka J, Reilly PT, Herr W. Loss of HCF-1-chromatin association precedes temperature-induced growth arrest of tsBN67 cells. Mol. Cell. Biol. 2001;21:3820–3829. doi: 10.1128/MCB.21.11.3820-3829.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Roberts DN, Cairns BR. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005;123:219–231. doi: 10.1016/j.cell.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hake SB, Garcia BA, Kauer M, Baker SP, Shabanowitz J, Hunt DF, Allis CD. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc. Natl Acad. Sci. USA. 2005;102:6344–6349. doi: 10.1073/pnas.0502413102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 36.Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 37.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 38.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl Acad. Sci. USA. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- 40.Thastrom A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 41.Bao Y, Konesky K, Park YJ, Rosu S, Dyer PN, Rangasamy D, Tremethick DJ, Laybourn PJ, Luger K. Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J. 2004;23:3314–3324. doi: 10.1038/sj.emboj.7600316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gansen A, Valeri A, Hauger F, Felekyan S, Kalinin S, Toth K, Langowski J, Seidel CA. Nucleosome disassembly intermediates characterized by single-molecule FRET. Proc. Natl Acad. Sci. USA. 2009;106:15308–15313. doi: 10.1073/pnas.0903005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller BK, Zaychikov E, Brauchle C, Lamb DC. Pulsed interleaved excitation. Biophys. J. 2005;89:3508–3522. doi: 10.1529/biophysj.105.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nir E, Michalet X, Hamadani KM, Laurence TA, Neuhauser D, Kovchegov Y, Weiss S. Shot-noise limited single-molecule FRET histograms: comparison between theory and experiments. J. Phys. Chem. B. 2006;110:22103–22124. doi: 10.1021/jp063483n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee NK, Kapanidis AN, Wang Y, Michalet X, Mukhopadhyay J, Ebright RH, Weiss S. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 2005;88:2939–2953. doi: 10.1529/biophysj.104.054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 48.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 49.Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 50.Suto RK, Clarkson MJ, Tremethick DJ, Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000;7:1121–1124. doi: 10.1038/81971. [DOI] [PubMed] [Google Scholar]
- 51.Krieger E, Nielsen JE, Spronk CA, Vriend G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph Model. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Higashi T, Matsunaga S, Isobe K, Morimoto A, Shimada T, Kataoka S, Watanabe W, Uchiyama S, Itoh K, Fukui K. Histone H2A mobility is regulated by its tails and acetylation of core histone tails. Biochem. Biophys. Res. Commun. 2007;357:627–632. doi: 10.1016/j.bbrc.2007.03.203. [DOI] [PubMed] [Google Scholar]
- 53.Gautier T, Abbott DW, Molla A, Verdel A, Ausio J, Dimitrov S. Histone variant H2ABbd confers lower stability to the nucleosome. EMBO Rep. 2004;5:715–720. doi: 10.1038/sj.embor.7400182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lever MA, Th'ng JP, Sun X, Hendzel MJ. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000;408:873–876. doi: 10.1038/35048603. [DOI] [PubMed] [Google Scholar]
- 55.Th'ng JP, Sung R, Ye M, Hendzel MJ. H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J. Biol. Chem. 2005;280:27809–27814. doi: 10.1074/jbc.M501627200. [DOI] [PubMed] [Google Scholar]
- 56.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408:877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- 57.Stasevich TJ, Mueller F, Brown DT, McNally JG. Dissecting the binding mechanism of the linker histone in live cells: an integrated FRAP analysis. EMBO J. 2010;29:1225–1234. doi: 10.1038/emboj.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmiedeberg L, Weisshart K, Diekmann S, Meyer Zu Hoerste G, Hemmerich P. High- and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol. Biol. Cell. 2004;15:2819–2833. doi: 10.1091/mbc.E03-11-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–725. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- 60.Clarkson MJ, Wells JR, Gibson F, Saint R, Tremethick DJ. Regions of variant histone His2AvD required for Drosophila development. Nature. 1999;399:694–697. doi: 10.1038/21436. [DOI] [PubMed] [Google Scholar]
- 61.Lu PY, Levesque N, Kobor MS. NuA4 and SWR1-C: two chromatin-modifying complexes with overlapping functions and components. Biochem. Cell Biol. 2009;87:799–815. doi: 10.1139/O09-062. [DOI] [PubMed] [Google Scholar]
- 62.Cai Y, Jin J, Florens L, Swanson SK, Kusch T, Li B, Workman JL, Washburn MP, Conaway RC, Conaway JW. The mammalian YL1 protein is a shared subunit of the TRRAP/TIP60 histone acetyltransferase and SRCAP complexes. J. Biol. Chem. 2005;280:13665–13670. doi: 10.1074/jbc.M500001200. [DOI] [PubMed] [Google Scholar]
- 63.Ruhl DD, Jin J, Cai Y, Swanson S, Florens L, Washburn MP, Conaway RC, Conaway JW, Chrivia JC. Purification of a human SRCAP complex that remodels chromatin by incorporating the histone variant H2A.Z into nucleosomes. Biochemistry. 2006;45:5671–5677. doi: 10.1021/bi060043d. [DOI] [PubMed] [Google Scholar]
- 64.Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Auger A, Galarneau L, Altaf M, Nourani A, Doyon Y, Utley RT, Cronier D, Allard S, Cote J. Eaf1 is the platform for NuA4 molecular assembly that evolutionarily links chromatin acetylation to ATP-dependent exchange of histone H2A variants. Mol. Cell. Biol. 2008;28:2257–2270. doi: 10.1128/MCB.01755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hardy S, Jacques PE, Gevry N, Forest A, Fortin ME, Laflamme L, Gaudreau L, Robert F. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 2009;5:e1000687. doi: 10.1371/journal.pgen.1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu RS, Tsai S, Bonner WM. Patterns of histone variant synthesis can distinguish G0 from G1 cells. Cell. 1982;31:367–374. doi: 10.1016/0092-8674(82)90130-1. [DOI] [PubMed] [Google Scholar]
- 68.Ramaswamy A, Bahar I, Ioshikhes I. Structural dynamics of nucleosome core particle: comparison with nucleosomes containing histone variants. Proteins. 2005;58:683–696. doi: 10.1002/prot.20357. [DOI] [PubMed] [Google Scholar]
- 69.Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl Acad. Sci. USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rasmussen TP, Huang T, Mastrangelo MA, Loring J, Panning B, Jaenisch R. Messenger RNAs encoding mouse histone macroH2A1 isoforms are expressed at similar levels in male and female cells and result from alternative splicing. Nucleic Acids Res. 1999;27:3685–3689. doi: 10.1093/nar/27.18.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005;12:624–625. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- 72.Tay SK, Blythe J, Lipovich L. Global discovery of primate-specific genes in the human genome. Proc. Natl Acad. Sci. USA. 2009;106:12019–12024. doi: 10.1073/pnas.0904569106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Angelov D, Verdel A, An W, Bondarenko V, Hans F, Doyen CM, Studitsky VM, Hamiche A, Roeder RG, Bouvet P, et al. SWI/SNF remodeling and p300-dependent transcription of histone variant H2ABbd nucleosomal arrays. EMBO J. 2004;23:3815–3824. doi: 10.1038/sj.emboj.7600400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hardy S, Robert F. Random deposition of histone variants: a cellular mistake or a novel regulatory mechanism? Epigenetics. 2010;5:368–372. doi: 10.4161/epi.5.5.11787. [DOI] [PubMed] [Google Scholar]
- 75.Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 2011;144:200–213. doi: 10.1016/j.cell.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jensen K, Santisteban MS, Urekar C, Smith MM. Histone H2A.Z acid patch residues required for deposition and function. Mol. Genet. Genomics. 2011;285:287–296. doi: 10.1007/s00438-011-0604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu WH, Alami S, Luk E, Wu CH, Sen S, Mizuguchi G, Wei D, Wu C. Swc2 is a widely conserved H2AZ-binding module essential for ATP-dependent histone exchange. Nat. Struct. Mol. Biol. 2005;12:1064–1071. doi: 10.1038/nsmb1023. [DOI] [PubMed] [Google Scholar]
- 78.Jin C, Zang C, Wei G, Cui K, Peng W, Zhao K, Felsenfeld G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21:1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chadwick BP, Willard HF. A novel chromatin protein, distantly related to histone H2A, is largely excluded from the inactive X chromosome. J. Cell. Biol. 2001;152:375–384. doi: 10.1083/jcb.152.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishibashi T, Li A, Eirin-Lopez JM, Zhao M, Missiaen K, Abbott DW, Meistrich M, Hendzel MJ, Ausio J. H2A.Bbd: an X-chromosome-encoded histone involved in mammalian spermiogenesis. Nucleic Acids Res. 2010;38:1780–1789. doi: 10.1093/nar/gkp1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soboleva TA, Nekrasov M, Pahwa A, Williams R, Huttley GA, Tremethick DJ. A unique H2A histone variant occupies the transcriptional start site of active genes. Nat. Struct. Mol. Biol. 2011;19:25–30. doi: 10.1038/nsmb.2161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.