


Abstract
Eukaryotic translation initiation is a highly regulated process in protein synthesis. The principal translation initiation factor eIF4AI displays helicase activity, unwinding secondary structures in the mRNAs 5′-UTR. Single molecule fluorescence resonance energy transfer (sm-FRET) is applied here to directly observe and quantify the helicase activity of eIF4AI in the presence of the ancillary RNA-binding factor eIF4H. Results show that eIF4H can significantly enhance the helicase activity of eIF4AI by strongly binding both to loop structures within the RNA transcript as well as to eIF4AI. In the presence of ATP, the eIF4AI/eIF4H complex exhibits persistent rapid and repetitive cycles of unwinding and re-annealing. ATP titration assays suggest that this process consumes a single ATP molecule per cycle. In contrast, helicase unwinding activity does not occur in the presence of the non-hydrolysable analog ATP-γS. Based on our sm-FRET results, we propose an unwinding mechanism where eIF4AI/eIF4H can bind directly to loop structures to destabilize duplexes. Since eIF4AI is the prototypical example of a DEA(D/H)-box RNA helicase, it is highly likely that this unwinding mechanism is applicable to a myriad of DEAD-box helicases employed in RNA metabolism.
INTRODUCTION
mRNA translation in eukaryotic cells begins with the recruitment of a ribosome to the 5′-untranslated region (UTR) of the transcript. This process is regulated by a set of translation factors, and is known as the initiation step (1). In what is thought to be the rate-limiting step of translation, 43S ribosome pre-initiation complexes (40S subunits and associated factors) are recruited to the 5′ m7GpppN cap structure—a structural feature present on all nuclear-transcribed eukaryotic mRNAs. This process is mediated by the heterotrimeric cap-binding initiation complex, eukaryotic initiation factor eIF4F, which is composed of a cap-binding factor (eIF4E), an RNA helicase (eIF4A) and a scaffolding protein (eIF4G) (2). Ribosome recruitment occurs as a consequence of interactions between eIF4G and the multi-subunit, ribosome-bound eIF3 (3,4). Translation initiation of some eukaryotic mRNAs can occur independently of the 5′-end by recruitment of 40S ribosomal subunits to internal ribosome entry sites (IRESs) (5,6).
Secondary structure within the 5′-UTR poses a barrier to cap-dependent translation, requiring helicase activity to facilitate ribosome recruitment (7). eIF4A-mediated unwinding of RNA is required for ribosome recruitment to mRNA templates with even a modest amount of secondary structure (8) and is thought to allow eIF4F to gain access to the cap structure and/or to restructure cap-proximal RNA sequences (9). The importance of this step in translational regulation is highlighted by the fact that deregulated eIF4F activity, either as a consequence of altered subunit levels or perturbed eIF4E regulation, is linked to alterations in the mRNA translation profile, with profound effects on the resulting cell proteome (10). Deregulated eIF4F activity has been linked to tumor initiation and maintenance (11–14); therefore, understanding eIF4A’s helicase properties and mode of action is important for developing strategies to modulate translation initiation and provide novel therapeutic strategies to target unregulated translation in cancer cells (15,16).
eIF4A is the prototypical member of the DEA(D/H)-box RNA helicase family, and is one of the more abundant translation initiation factors, present at ∼3 copies/ribosome (17). eIF4A is present as two isoforms, eIF4AI and eIF4AII, which share 90–95% sequence identity and are thought to be functionally interchangeable (18). The helicase activity of eIF4AI is significantly stimulated by the presence of the translation initiation factors, eIF4B or eIF4H, or when eIF4A is present as a subunit of the eIF4F complex (eIF4Ac) (8,19–21). Although eIF4AI has been extensively studied using classical biochemistry approaches (19,22,23), the mechanisms underlying its ligand-dependent conformational changes, its helicase activity as an individual unit, and its significant activity enhancement in the presence of eIF4B or eIF4H, have all to date remained incompletely understood. Moreover, a direct and unambiguous measurement of eIF4AI’s position on the mRNA template during RNA unwinding has not been reported. Bulk studies of eIF4AI helicase activity have classically used gel electrophoresis oligonucleotide displacement assays, which only detect the completely unwound products and have not provided insight into unwinding dynamics.
In contrast, sm techniques, in particular, fluorescence resonance energy transfer (sm-FRET), can provide direct and highly quantitative accounts of eIF4AI location and re-positioning on its RNA substrate during unwinding, and is well suited to eIF4AI’s mechanism of action. This technique’s utility for understanding helicase activity has been highlighted by recent studies employing sm-FRET to determine the spring-loaded DNA unwinding mechanism of Hepatitis C NS3 helicase (24) and to probe a fast repetitive motion of Escherichia Coli Rep helicase (25). Here, we employ sm-FRET to probe eIF4AI’s unwinding activity. We find that eIF4AI alone can induce rare and isolated unwinding episodes of RNA duplexes. We also use sm-FRET to probe the consequences of eIF4H, another translation initiation factor shown to interact with eIF4AI (26), on the unwinding parameters of eIF4AI and find that the resulting eIF4AI/eIF4H complex exhibits an abundance of persistent, repetitive cycles of unwinding over extended periods of time. We find that eIF4AI is directed to bind to single-stranded loop structures by eIF4H, facilitating its ability to repetitively unwind duplexes in an ATP-dependent manner.
MATERIALS AND METHODS
RNA/DNA substrate preparation
DNA and RNA oligonucleotides were purchased from IDT (Coralville, IA, USA) and Dharmacon (Lafayette, CO, USA), respectively. The oligonucleotides were labeled using Cy3 or Cy5 (GE Healthcare, Little Chalfont, UK) according to the vendor’s recommended protocol. Oligonucleotides and dyes were mixed at a molar ratio of 1:15, respectively. The conjugation included incubating samples overnight in 0.1 M sodium tetraborate buffer (pH 8.5) at room temperature. Conjugation was confirmed using native polyacrylamide gel electrophoresis (PAGE) and quantified by UV–Vis absorption. RNA/DNA hybrid duplexes were formed by annealing the oligonucleotides in 50 mM Tris–HCl (pH 7.5), 0.2 mM EDTA and 500 mM NaCl, heating the samples to 95°C, and slowly cooling them to 4°C over a period of 2.5 h. The duplexes were then purified using native PAGE.
Hybrid hairpin structures were prepared by mixing DNA oligonucleotides, which contained a loop region, with single-stranded RNAs. The DNA oligonucleotides contained a terminal biotin moiety used for surface immobilization in the sm assays. Ligation of the DNA/RNA strands was performed with T4 RNA ligase 2 (New England Biolabs, Ipswich, MA, USA), following which substrates were purified by denaturing PAGE to remove the unligated oligonucleotides. The preparation of a blunt-ended hairpin substrate was performed by mixing the purified hairpin sample with an additional oligonucleotide (23 nt ssDNA) at a hairpin:ssDNA molar ratio of 1:10, heating to 95°C, and allowing to slowly cool to 4°C over a period of 2.5 h. The blunt-end hairpin was purified using native PAGE. The sequences of all oligonucleotides used in this study are shown in the Supplementary Figure S1.
Protein expression and purification
Recombinant His6-eIF4AI and His6-eIF4H were expressed in E. coli BL21-Gold (DE3) cells (Stratagene, La Jolla, CA, USA). Following IPTG-induction of expression, lysis of cells by sonication and clearing of the supernatant by centrifugation, His6-eIF4AI-containing lysate was loaded on a Ni++-Sepharose 6 Fast Flow resin (GE Healthcare, Piscataway, NJ, USA). The matrix was washed, eluted with A300 buffer [20 mM Tris (pH 7.5), 10% glycerol, 300 mM KCl, 0.1 mM EDTA] containing 0.2 M imidazole. Following dialysis against A100 buffer [20 mM Tris (pH 7.5), 10% glycerol, 100 mM KCl, 0.1 mM EDTA and 2 mM DTT], eIF4AI was loaded onto a Q-Sepharose fast flow matrix (GE Healthcare). The resin was washed with 10 column volumes of A100 and protein eluted using a linear salt gradient from 100 mM to 500 mM KCl in buffer A (20 mM Tris (pH 7.5), 10% glycerol, 0.1 mM EDTA, 2 mM DTT). The eluted fractions were analyzed by SDS–PAGE and those containing eIF4AI were pooled and dialyzed against buffer A.
His6-eIF4H was first enriched on a Ni++-Sepharose column then further purified on a Heparin-Sepharose matrix (GE Healthcare). After applying the sample, the resin was washed with 1 column volume of A100 containing 2 mM DTT, and eIF4H eluted with a linear salt gradient from 100 to 500 mM KCl in buffer B [20 mM Tris (pH 7.5), 10% glycerol, 0.1 mM EDTA and 2 mM DTT]. The eluted fractions were analyzed by SDS–PAGE and those containing t His6-eIF4H were pooled and dialyzed against storage buffer [20 mM HEPES (pH 7.5), 25% glycerol, 50 mM KCl, 0.1 mM EDTA and 1 mM DTT].
Bulk FRET assay
In the bulk measurements, the donor (Cy3) and the acceptor (Cy5) signals were collected using a home-build confocal setup as previously described (27). Cy3 was excited by a 514.5-nm line of Ar+ laser (Spectra-Physics, Santa Clara, CA, USA). Emissions from donor and acceptor channel were discriminated using dichroic mirrors (Chroma Technology, Bellows Falls, VT, USA) and measured by two avalanche photodiodes (Perkin-Elmer, Wellesley, MA, USA). The exposure time is 1 s and the sampling rate is 2 Hz. In all bulk experiments, 1 nM of duplex substrate molecules were used to minimize reannealing of the strands upon unwinding.
sm-FRET assays
Sm measurements were performed using a homemade microfluidic chamber equipped with inlet and outlet flow tubing as described in Di Fiori and Meller (28). The quartz coverslip (Esco Products, Oak Ridge, NJ, USA) of the cell was first coated with biotinylated BSA (Sigma, St Louis, MO, USA) and then with streptavidin (Thermo, Rockford, IL, USA). Biotinylated samples were immobilized on the biotinylated BSA-anchored streptavidin as previously described (28).
In a typical experiment, a 10 pM RNA/DNA hybrid hairpin solution was flowed through the cell, immobilizing roughly 100 molecules in a single field of view of the microscope. In order to remove unattached hybrids, the cell was washed 10 times with imaging buffer, complemented with an oxygen scavenging system [50 mM Tris (pH 7.5), 50 mM NaCl, saturated Trolox (>2 mM) (Sigma), 5 mM Protocatechusate (Sigma) and 0.15 μM Protocatechuate 3,4-Dioxygenase(Sigma)] (29). eIF4AI or eIF4AI/eIF4H (1:1) complex (1 µM) was incubated with the immobilized hybrids for 10 min at 22°C in imaging buffer containing 1 mM ATP (Roche Applied Science, Indianapolis, IN, USA) and 1 mM MgCl2.
sm data acquisition and analysis
The FRET donor (Cy3) and acceptor (Cy5) signals were collected using a modified objective-type total internal reflection (TIRF) microscope (IX71, Olympus, Japan). Cy3 was excited by a 532-nm laser (Millennia V, Spectra-Physics, Santa Clara, CA, USA). Emissions from donor and acceptor were discriminated using dichroic mirrors (Chroma Technology, Bellows Falls, VT, USA) and imaged by an EMCCD camera (iXon DV 897, Andor Technology, Belfast, Ireland) at 10 frames/s, except where otherwise noted. A custom image acquisition program written using LabVIEW (National Instruments, Austin, TX, USA) was used to acquire movies and stream data to the computer’s hard-drive. sm traces were extracted and analyzed using programs written in Matlab (Mathworks, Natick, MA, USA). FRET values were calculated as IA/(IA + ID), where IA and ID are the fluorescence intensity of the acceptor and donor channels, respectively, following correction of background noise. The two-state FRET oscillations were analyzed using Hidden Markov Modeling (HAMMY).
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSA) reactions were performed in a binding buffer (20 mM Tris–Cl, pH 7.4, 80 mM KCl, 2.5 mM MgCl2, 1 mM DTT and 1% glycerol). Reactions containing the indicated proteins, fluorophore-labeled RNA, BSA, PEG and ATP were performed at RT over the course of 30 min. RNA/DNA hairpin: substrates included RNA/DNA hybrid hairpins carrying 3 - to 12-nt long loops, with a 12-bp stem region. The single-stranded overhang region consisted of 23 nt. A Cy3 fluorophore was attached internally to the DNA sequence. Samples were loaded onto 10% non-denaturing polyacrylamide gel and following electrophoresis, gels were scanned for Cy3 emission using a Pharos FX Scanner (BioRad, Hercules, CA, USA).
ATPase activity
Phosphate (Pi) release from eIF4AI (and eIF4AI/eIF4H complex) upon addition of ATP (10–100 µM) was measured using a fluorescently labeled mutant of phosphate-binding protein (MDCC-PiBP, Invitrogen PV4407). Background Pi was removed from the reaction buffer [20 mM Tris–Cl (pH 7.5), 80 mM KCl, 2.5 mM MgCl2] using a MOP solution containing 7-methylguanosine (200 µM) and purine nucleoside phosphorylase (Sigma-Aldrich).
RESULTS
Bulk and sm characterization of eIF4AI unwinding activity using FRET
Before undertaking sm-FRET measurements, we verified that FRET could be used to assess eIF4AI helicase activity in bulk. As previously reported (8), the helicase activity of eIF4A was observed using a radiometric-based gel assay (Supplementary Figure S2). We then developed a bulk FRET assay for eIF4AI, using an RNA/DNA hybrid duplexes since our plan was to immobilize RNA substrates to solid support matrices using DNA for sm-FRET experiments (see below) and eIF4AI is able to unwind such substrates (30). Substrates consisting of 18-mer RNA/DNA hybrid duplexes (Tm = 51.1°C, ΔG = −21.4 kcal/mol) labeled using a FRET pair (Cy3–Cy5) located on opposite strands displayed a stable, high FRET level (Figure 1). Upon addition of 1 mM ATP and increasing concentrations of eIF4AI, we observed a gradual decrease in FRET signal, followed by a steady lower FRET state determined by eIF4AI’s concentration (Figure 1). The characteristic half-life for FRET kinetics was ∼3 min. In this assay, we used an extremely low RNA/DNA duplex concentration (1 nM) to avoid re-annealing of the two nucleic acids strands within the experimental timeframe. No FRET change was observed in the absence of ATP and the FRET signal was eIF4AI concentration dependent (Figure 1). Gel electrophoresis analysis of radio-labeled RNA duplexes showed that in the presence of ATP, eIF4AI unwinds RNA duplexes, in agreement with the FRET measurements, and consistent with previous studies (19). These experiments allow us to conclude that increasing eIF4AI concentrations lead to an increase in unwound RNA:DNA duplexes, although we can make no inferences on the linearity of the response from these experiments.
Figure 1.

Bulk FRET assay displays an eIF4AI-dependent decrease in the steady-state FRET level. (A) A schematic diagram showing the substrate molecules for bulk FRET assay. An 18 base pair RNA/DNA duplex labeled with a FRET pair introduced at a very low concentration (1 nM) is unwound only in the presence of ATP and eIF4AI, reaching a lower steady-state FRET level. (B) Results from bulk FRET assays monitoring eIF4AI helicase activity at the indicated eIF4AI concentrations and 1 mM ATP.
We next sought to characterize eIF4AI helicase activity using sm-FRET. Initial attempts to use hybrid duplexes of the type shown in Figure 1A as substrates were found to be ineffective for sm-FRET since following unwinding of the duplexes by eIF4AI, non-biotinylated strands (carrying either the donor or the acceptor dye) diffused away from the coverslip surface resulting in loss of FRET signal. Hence, we developed a series of RNA/DNA hybrid hairpin structures in which we designed loops of various sizes, ranging from 3 to 12 nt long (a hairpin molecule with a 12-nt long loop is shown schematically in Figure 2A). All the hairpin substrate molecules used in this study have ΔG values within the range of −16 to −15 kcal/mol (Supplementary Figure S1), which according to Figure 1 and previous reports (30), should be completely unwound by eIF4AI. The loop structure facilitated multiple observations of repeated unzipping/re-annealing kinetics for the same molecule. Figure 2B displays a sm time-trace of the donor (Cy3 green line, ID) and acceptor (Cy5 red line, IA) intensities and their corresponding FRET value (IA/(ID+IA), blue line) as a function of time. When both ATP (1 mM) and eIF4AI (2 µM) were added, rare but distinct episodes of FRET transitions were observed, characteristically moving from an initial FRET value of 0.90 (corresponding to a closed hairpin structure) down to a value of 0.60, then returning to 0.90. Statistical analysis was performed on a set of 1000 individual molecules, all of which exhibited stable donor and acceptor emission for a minimum of 2 min. We found that traces containing at least one FRET transition of any magnitude composed only 3.8% of all traces. This subset of events was used to construct a histogram of FRET values that displays two clear peaks at FRET levels of 0.9 and 0.6, corresponding to two well-defined FRET states that can be approximated by a double Gaussian fit (black line; Figure 2C). Photophysical effects could be ruled out as the source of the FRET transitions for two reasons: (i) Control experiments showed that in the absence of either ATP or eIF4AI no FRET transitions were observed (Supplementary Figure S3). (ii) It is well established that fluorophore ‘blinking’ involves complete ‘switching off’ of one of the dyes, rather than a transition to an intermediate FRET level (31,32). We observed that even at low FRET values we still detect substantial photon emissions from both dyes (Figure 2), making it unlikely that these transitions are caused by photophysical effects.
Figure 2.

sm-FRET recordings of hairpin substrates in the presence of eIF4AI. (A) Schematic diagram illustrating DNA/RNA hairpin substrate harboring a 12-nt loop and 12 nt duplex region, labeled with a FRET pair (Cy3–Cy5). The substrate is immobilized to a glass surface via the 3′-end biotin. (B) sm-FRET of the immobilized RNA/DNA hairpin in the presence of eIF4AI, displaying episodes consisting of decreased FRET signals followed by return to original values. (C) FRET distribution from individual sm traces (N = 38 out of 1000) showing two distinct peaks at 0.9 and 0.6.
Repetitive unwinding is induced in the presence of eIF4H
Next, we wished to assess the consequences of eIF4H on the unwinding parameters of eIF4AI, given that previous bulk studies have reported elevated eIF4AI ATPase activity in the presence of eIF4H (20,33), as well as a stimulation of helicase activity (20,34) (Supplementary Figure S4). The addition of 1 µM eIF4H to eIF4AI (at a 1:1 ratio) into the sm assay produced a striking change in the FRET dynamics. The FRET levels repeatedly and rapidly varied between two discrete values (0.9 and 0.4) for periods lasting several minutes (Figure 3A). Moreover, in sharp contrast to the isolated FRET transitions caused by eIF4AI alone (Figure 2B), the oscillating two-level transition pattern for the 12-base hairpin loop accounted for >50% of all sm traces in a population of more than 2000 molecules (Figure 3B). Since the transitions in FRET value from 0.9 to 0.4 reflect roughly a 3 nm change in donor–acceptor distance (approximately the length of the eIF4AI dumbbell configuration at its widest point) (35), and given that the FRET transitions are ATP-dependent, the results are consistent with previous results that eIF4H, which does not possess inherent helicase activity, stimulates the unwinding activity of eIF4AI. The eIF4AI/eIF4H complex leads to reversible unwinding of structured RNA, while simultaneously preventing fast reannealing of the RNA template. Each oscillation of the FRET trace between the high and low level represents a single unwinding cycle. A Hidden–Markov Model (HMM) analysis of the observed two-state fluctuations (36) was used to extract the dynamics of this process, characterized by ‘waiting time’ (high FRET level) and ‘unwinding time’ (low FRET level). Statistical analysis of thousands of these transitions revealed exponential distributions of the dwell times for each state, where the characteristic unwinding time was found to be τU = 0.71 ± 0.02 s and the characteristic waiting time was found to be τW = 1.35 ± 0.05 s (Figure 3C).
Figure 3.

sm-FRET recordings of hairpin substrates in the presence of eIF4AI/eIF4H. (A) A representative sm time trace of an RNA/DNA hairpin structure (Schematic Illustration in Figure 2A). The hairpin molecule is immobilized to the cover-slip surface using a biotin moiety. eIF4AI/eIF4H are introduced at a concentration of 200 nM and ATP concentration is 0.1 mM. Donor ( ) and acceptor (
) and acceptor (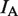 ) intensities (green and red, respectively) are measured and the corresponding FRET trace is calculated by:
) intensities (green and red, respectively) are measured and the corresponding FRET trace is calculated by:  (blue). (B) Histogram of FRET levels obtained from N > 2000 individual molecules under the same conditions as in A, showing two prominent peaks at 0.9 and 0.4. The oscillating FRET behavior is observed in over 50% of the sm events and is ATP dependent. (C) Dwell times of the low- and high-FRET states are used to construct the time histograms of the unwinding time and waiting time intervals (blue and green, respectively). Single exponential models are used to fit the data (black lines), yielding the characteristic timescales
(blue). (B) Histogram of FRET levels obtained from N > 2000 individual molecules under the same conditions as in A, showing two prominent peaks at 0.9 and 0.4. The oscillating FRET behavior is observed in over 50% of the sm events and is ATP dependent. (C) Dwell times of the low- and high-FRET states are used to construct the time histograms of the unwinding time and waiting time intervals (blue and green, respectively). Single exponential models are used to fit the data (black lines), yielding the characteristic timescales  and
and 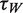 .
.
To exclude the possibility that the observed FRET transitions stem from binding/unbinding events of multiple eIF4AI/eIF4H complexes to the nucleic acid substrates, we documented FRET activity (in the presence of eIF4AI, eIF4H and ATP) for a set of substrate molecules, followed by flushing the flow cell with more than 5 volumes of buffer lacking eIF4AI, eIF4H or ATP and monitoring FRET activity for 60 s (Figure 4). This treatment, which resulted in a loss of FRET fluctuations (Figure 4B), was then followed by re-introduction of only ATP into the system. We noted a resumption of repetitive FRET fluctuations (Figure 4C) for the majority of the observed sm (some molecules were either photo-bleached during the process or washed away). We interpret these results to indicate that the FRET activity observed post-ATP reintroduction is due to the presence of eIF4AI/eIF4H proteins that have remained bound to the duplex strand during all washes. Taken together with the results above illustrating that the observed repetitive FRET transitions are ATP-dependent and reflect a change in donor–acceptor distance of ∼3 nm, they most likely result from an oscillatory unwinding/reannealing pattern of the same eIF4AI/eIF4H complex that remained bound to its substrate.
Figure 4.

eIF4AI/eIF4H complex remains bound to the hairpin substrate during cycles of repetitive FRET signal oscillations. A typical FRET trace for a single substrate molecule acquired in reactions containing: (A) both eIF4AI, eIF4H (1 µM) and ATP (1 mM); (B) after>5 × volume flushing of the chamber with buffer lacking eIF4AI, eIF4H and ATP (‘Wash 1’); (C) following reintroduction of only ATP at 1 mM (‘Wash 2’).
The eIF4AI/eIF4H complex preferably binds to the loop region of hairpins
To further evaluate the stimulatory role of eIF4H on eIF4AI unwinding dynamics, we explored how substrate design could affect eIF4AI activity. It has been reported that eIF4AI unwinds RNA substrates with single-strand overhangs slightly more efficiently than blunt ended RNA duplexes (30). By analogy, we speculated that eIF4H facilitates duplex unwinding activity of eIF4AI by binding first to single-stranded portions of the RNA. This would serve two goals, both increasing the affinity of the eIF4AI/eIF4H complex for single-stranded domains of the RNA transcript as compared to the affinity of eIF4AI alone for such substrates, and also allowing for a more processive unwinding mechanism. In order to explore the binding affinity of eIF4AI to the RNA substrate in the absence or presence of eIF4H, we performed a series of electrophoretic mobility shift assays (Supplementary Figure S5). A distinct complex is not apparent upon incubation of eIF4AI or eIF4H with RNA substrate. However, both proteins together are capable of forming a distinct complex in the presence of ATP or ATP-γS (Supplementary Figure S5, lanes 7 and 8). Furthermore, we found that eIF4AI and eIF4H formed more stable complexes when the loop size of the hairpin substrates exceeded 6 nt (Supplementary Figure S6).
The preferred binding location of the eIF4AI/eIF4H complex to the RNA substrates used in these experiments was determined by two additional sm-FRET experiments. First, a complementary DNA oligonucleotide located adjacent to the biotin residue was used to block the single-stranded portion of the RNA substrate (Supplementary Figure S7). For these ‘blocked’ samples, repetitive FRET fluctuations were observed and persisted to the same extent as reported above, indicating that a single-stranded overhang does not contribute significantly to eIF4AI/eIF4H’s repetitive unwinding capability. Next, under the assumption that that eIF4H must preferentially bind to the loop region, we tested substrates with different loop sizes, ranging from 3 to 12 nt long. In each of these experiments, we collected at least 1500 sm traces and calculated the fraction of traces displaying the oscillating FRET pattern in each experiment. The results indicate that FRET oscillations are practically non-existent when the loop size is shorter than 6 nt (Figure 5). Furthermore, the FRET assays showed a distinct increase in the percent of substrate molecules undergoing oscillations for longer loop sizes, reaching ∼65% when the loop size is 12 nt long. The results suggest that the critical minimum loop size required for the eIF4AI/eIF4H complex to bind and initiate repetitive unwinding activity upon an RNA substrate is 6 nt.
Figure 5.

eIF4AI/eIF4H complex requires hairpin substrates containing minimum loop sizes of 6 nt for repetitive unwinding. The fraction of sm traces showing > 3 oscillations in a 60 s monitoring time window is plotted as a function of loop length (in nucleotides). Each data point consists of at least 1500 sm traces.
Sm measurements suggest that the repetitive RNA unwinding involves a single ATP hydrolysis per FRET cycle
To explore the role of ATP hydrolysis in the repetitive unwinding process, we reduced the ATP concentration from 1 mM down to 30 µM and measured the resulting sm-FRET dynamics (Figure 6A). If the ‘waiting time’ τw, represents the time required for the recruitment of a fresh ATP molecule from bulk, then reducing ATP concentration to the point where ATP diffusion time becomes the rate-limiting step is expected to affect τw but not ‘unwinding time’, τU. This prediction is confirmed by our data. A 60% increase in τw (circles) was observed upon reduction of the ATP concentration below 0.1 mM, whereas the unwinding time (triangles) remained unaffected (τU = 0.64 ± 0.15 s) for all ATP concentrations measured (the dwell-time distributions for each data point are shown in Supplementary Figure S8). Moreover, the fact that τU does not increase upon a decreasing the ATP concentration, while τw does increase, suggests that a single ATP molecule is consumed per unwinding cycle since consumption of two or more ATP molecules per cycle would have resulted in a concurrent increase in τU when the ATP concentration falls below ∼0.1 mM.
Figure 6.

Dependence of the hairpin substrate unwinding dynamics by the eIF4AI/eIF4H complex on ATP or ATP-γS concentrations. In these experiments, 12-nt hairpin loop structures were used, and 1 µM of both eIF4AI and eIF4H. (A) Relationship of the waiting times (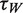 , circles) and the unwinding times (
, circles) and the unwinding times (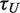 , triangles) on the ATP concentration. Each data point represents statistics of at least 1000 sm traces (detailed distributions are provided in the Supporting Information file). (B) The dependence of waiting times (
, triangles) on the ATP concentration. Each data point represents statistics of at least 1000 sm traces (detailed distributions are provided in the Supporting Information file). (B) The dependence of waiting times (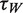 , circles) and unwinding times (
, circles) and unwinding times ( , triangles) on the relative ATP-γS concentration, with a fixed ATP concentration of 0.1 mM. The abscissa
, triangles) on the relative ATP-γS concentration, with a fixed ATP concentration of 0.1 mM. The abscissa  . Each data point represents statistics of at least 500 sm traces.
. Each data point represents statistics of at least 500 sm traces.
We also tested whether or not the non-hydrolysable ATP analog, ATP-γS, could substitute for ATP in the unwinding reaction (Figure 6B). By keeping the total concentration of [ATP]+[ATP-γS] fixed, we found that increasing the fractional amount of ATP-γS up to 15% had little effect on the waiting time or the unwinding time (the dwell-time distributions for each data point are shown in Supplementary Figure S9). However, we find that both the fraction of sm traces showing consecutive sm-FRET oscillations within a fixed 60 s monitoring interval, as well as the average number of repetitive FRET cycles observed for any given molecule, displayed a 3- to 4-fold decrease with increasing x values (Supplementary Figure S10). This experiment suggests that ATP-γS can substituted for ATP in the initial eIF4AI/eIF4H complex formation, but irreversibly terminates the FRET oscillations since these require a hydrolytic event.
DISCUSSION
Sm approaches can be used to directly quantify a myriad of biomolecular processes, providing observational access to, and information about, their detailed dynamics under physiological conditions, especially when used in combination with classical biochemical assays. Here, we utilized sm-FRET to probe the unwinding dynamics of eukaryotic initiation factor eIF4AI in the presence of the RNA-binding factor eIF4H. Our results indicate that the assembly of the eIF4AI/eIF4H complex on the RNA transcript, and subsequent unwinding of its duplex regions can be significantly enhanced by long-lived binding of the complex to single-stranded loop domains longer than a critical minimum length of 6 nt. In accordance with previous studies indicating that eIF4H binds to RNA via the RNA recognition motif (RRM) domain, our results support a simple RNA unwinding model, where repetitive zipper-like unwinding/re-annealing cycles can be efficiently executed by the eIF4AI/eIF4H complex when bound to a loop domain within the secondary structure. Upon ATP hydrolysis, unwinding of the duplex region induces rapid high-to-low FRET level transitions (Figures 3 and 4). This mode of short distance helicase activity is quite efficient, consuming a single ATP molecule per cycle of unwinding. The unwinding process can be terminated upon binding of the non-hydrolysable ATP analog, ATP-γS. The ATP dependence of the unwinding activity of eIF4AI/eIF4H is reminiscent of other DEAD-box helicases such as CYT-19 and DbpA, which have been shown to achieve complete strand separation of RNA duplexes via hydrolysis of a single ATP molecule (37–39).
Linking eIF4AI activity to eIF4H function establishes a crucial means by which eIF4AI activity could be regulated. Generally, eIF4H activity involves directing other cellular proteins to specific mRNA structures to enhance their activity. For example, eIF4H binding is required for the herpes simplex virus virion host shutoff (Vhs) protein-induced degradation of many mRNAs, where it targets Vhs to preferred sites within mRNAs (40). siRNA-mediated depletion of eIF4H, but not eIF4B, in HeLa cells impedes Vhs-mediated mRNA degradation (40). We find that eIF4H plays a similar role in stimulating eIF4AI activity.
The marked increase in unwinding activity observed for hairpin substrates, as compared to a simple duplex with an overhang, suggests that the eIF4AI/eIF4H complex binds to hairpin loops with higher affinity than to single-stranded overhangs. This configuration allows it to maintain close interaction with both strands in order to initiate multiple unwinding episodes, which in turn keeps the RNA in its unwound state for a longer total period of time. Another conceivable advantage of this configuration is that the eIF4AI/eIF4H repeated unwinding activity augments downstream steps, ultimately and indirectly stimulating ribosome binding, and consequently protein synthesis. eIF4H, like eIF4B, enhances the helicase activity of eIF4A and contains an RRM that shows sequence homology to eIF4B in this region—however, the two proteins are dissimilar in their mechanism of action and their influence on eIF4A RNA binding and ATPase activity (20). It remains to be determined whether our results with eIF4H can be extrapolated to eIF4B and will exhibit a complementary function in our assay. The mRNA 5′-UTRs present a complex landscape of short stem and loop regions. Our data suggests that if eIF4H could be delivered internally to such loops, in conjunction with eIF4AI, RNA remodeling should rapidly occur. This might play an important role in IRES-dependent translation initiation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–10 and Supplementary References [41,42].
FUNDING
Human Frontiers Science Program (HFSP) award RGP0036-2005 (to A.M. and N.S.); Canadian Institutes of Health Research (MOP-106530); Ministere du Développement Economique, Innovation et Exportation (to J.P.). Funding for open access charge: HFSP award [RGP0036-2005].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonenberg N. mRNA 5′ cap-binding protein elF4E and control of cell growth. In: Hershey JWB, Matthews MB, Sonenberg N, editors. Translational Control. Cold Spring Harbour, NY: Cold Spring Harbour Laboratory Press; 1996. pp. 245–269. [Google Scholar]
- 3.Lamphear BJ, Kirchweger R, Skern T, Rhoads RE. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. J. Biol. Chem. 1995;270:21975–21983. doi: 10.1074/jbc.270.37.21975. [DOI] [PubMed] [Google Scholar]
- 4.Merrick WC, Hershey JWB. The pathway and mechanism of eukaryotic protein synthesis. In: Hershey JWB, Matthews BF, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 31–70. [Google Scholar]
- 5.Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic messenger RNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 6.Jang SK, Davies MV, Kaufman RJ, Wimmer E. Initiation of protein-synthesis by internal entry of ribosomes into the 5’ nontranslated region of encephalomyocarditis virus-RNA in vivo. J. Virol. 1989;63:1651–1660. doi: 10.1128/jvi.63.4.1651-1660.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pestova TV, Kolupaeva VG. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002;16:2906–2922. doi: 10.1101/gad.1020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozen F, Edery I, Meerovitch K, Dever TE, Merrick WC, Sonenberg N. Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol. Cell. Biol. 1990;10:1134–1144. doi: 10.1128/mcb.10.3.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray BK, Lawson TG, Kramer JC, Cladaras MH, Grifo JA, Abramson RD, Merrick WC, Thach RE. Atp-dependent unwinding of messenger-Rna structure by eukaryotic initiation-factors. J. Biol. Chem. 1985;260:7651–7658. [PubMed] [Google Scholar]
- 10.Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 11.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by eukaryotic initiation-factor subunit that binds to messenger-RNA 5’ cap. Nature. 1990;345:544–547. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 12.Wendel HG, de Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 13.Mavrakis KJ, Wendel HG. Translational control and cancer therapy. Cell Cycle. 2008;7:2791–2794. doi: 10.4161/cc.7.18.6683. [DOI] [PubMed] [Google Scholar]
- 14.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat. Rev. Cancer. 2010;10:254–266. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 15.Malina A, Cencic R, Pelletier J. Targeting translation dependence in cancer. Oncotarget. 2011;2:76–88. doi: 10.18632/oncotarget.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bordeleau ME, Matthews J, Wojnar JM, Lindqvist L, Novac O, Jankowsky E, Sonenberg N, Northcote P, Teesdale-Spittlet P, Pelletier J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc. Natl Acad. Sci. USA. 2005;102:10460–10465. doi: 10.1073/pnas.0504249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duncan R, Hershey JWB. Identification and quantitation of levels of protein-synthesis initiation-factors in crude Hela-cell lysates by two-dimensional polyacrylamide-gel electrophoresis. J. Biol. Chem. 1983;258:7228–7235. [PubMed] [Google Scholar]
- 18.Rogers GW, Komar AA, Merrick WC. ElF4A: The godfather of the DEAD box helicases. Prog. Nucleic Acid Res. Mol. Biol. 2002;72:307–331. doi: 10.1016/s0079-6603(02)72073-4. [DOI] [PubMed] [Google Scholar]
- 19.Rogers GW, Richter NJ, Merrick WC. Biochemical and kinetic characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem. 1999;274:12236–12244. doi: 10.1074/jbc.274.18.12236. [DOI] [PubMed] [Google Scholar]
- 20.Rogers GW, Richter NJ, Lima WF, Merrick WC. Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. J. Biol. Chem. 2001;276:30914–30922. doi: 10.1074/jbc.M100157200. [DOI] [PubMed] [Google Scholar]
- 21.Richter NJ, Rogers GW, Hensold JO, Merrick WC. Further biochemical and kinetic characterization of human eukaryotic initiation factor 4H. J. Biol. Chem. 1999;274:35415–35424. doi: 10.1074/jbc.274.50.35415. [DOI] [PubMed] [Google Scholar]
- 22.Lorsch JR, Herschlag D. The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry. 1998;37:2180–2193. doi: 10.1021/bi972430g. [DOI] [PubMed] [Google Scholar]
- 23.Lorsch JR, Herschlag D. The DEAD box protein eIF4A. 2. A cycle of nucleotide and RNA-dependent conformational changes. Biochemistry. 1998;37:2194–2206. doi: 10.1021/bi9724319. [DOI] [PubMed] [Google Scholar]
- 24.Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513–516. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myong S, Rasnik I, Joo C, Lohman TM, Ha T. Repetitive shuttling of a motor protein on DNA. Nature. 2005;437:1321–1325. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- 26.Feng P, Everly DN, Jr, Read GS. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J. Virol. 2005;79:9651–9664. doi: 10.1128/JVI.79.15.9651-9664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabanayagam CR, Eid JS, Meller A. High-throughput scanning confocal microscope for single molecule analysis. Appl. Phys. Lett. 2004;84:1216–1218. [Google Scholar]
- 28.Di Fiori N, Meller A. The effect of dye-dye interactions on the spatial resolution of single-molecule FRET measurements in nucleic acids. Biophys. J. 2010;98:2265–2272. doi: 10.1016/j.bpj.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aitken CE, Marshall RA, Puglisi JD. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers GW, Lima WF, Merrick WC. Further characterization of the helicase activity of eIF4A - Substrate specificity. J. Biol. Chem. 2001;276:12598–12608. doi: 10.1074/jbc.M007560200. [DOI] [PubMed] [Google Scholar]
- 31.Bates M, Blosser TR, Zhuang XW. Short-range spectroscopic ruler based on a single-molecule optical switch. Phys. Rev. Lett. 2005;94:108101. doi: 10.1103/PhysRevLett.94.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rasnik I, McKinney SA, Ha T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 33.Rozovsky N, Butterworth AC, Moore MJ. Interactions between eIF4AI and its accessory factors eIF4B and eIF4H. RNA. 2008;14:2136–2148. doi: 10.1261/rna.1049608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozes AR, Feoktistova K, Avanzino BC, Fraser CS. Duplex unwinding and ATPase activities of the DEAD-box helicase eIF4A are coupled by eIF4G and eIF4B. J. Mol. Biol. 2011;412:674–687. doi: 10.1016/j.jmb.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caruthers JM, Johnson ER, McKay DB. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl Acad. Sci. USA. 2000;97:13080–13085. doi: 10.1073/pnas.97.24.13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Potratz JP, Tijerina P, Del Campo M, Lambowitz AM, Russell R. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc. Natl Acad. Sci. USA. 2008;105:20203–20208. doi: 10.1073/pnas.0811075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Bio. 2011;12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 39.Henn A, Cao W, Licciardello N, Heitkamp SE, Hackney DD, De La Cruz EM. Pathway of ATP utilization and duplex rRNA unwinding by the DEAD-box helicase, DbpA. Proc. Natl Acad. Sci. USA. 2010;107:4046–4050. doi: 10.1073/pnas.0913081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarma N, Agarwal D, Shiflett LA, Read GS. Small interfering RNAs that deplete the cellular translation factor eIF4H impede mRNA degradation by the virion host shutoff protein of herpes simplex virus. J. Virol. 2008;82:6600–6609. doi: 10.1128/JVI.00137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.