


Abstract
The MYB proto-oncogene is expressed in most estrogen receptor-positive (ERα+) breast tumors and cell lines. Expression of MYB is controlled, in breast cancer and other cell types, by a transcriptional pausing mechanism involving an attenuation site located ∼1.7 kb downstream from the transcription start site. In breast cancer cells, ligand-bound ERα binds close to, and drives transcription beyond this attenuation site, allowing synthesis of complete transcripts. However, little is known, in general, about the factors involved in relieving transcriptional attenuation, or specifically how ERα coordinates such factors to promote transcriptional elongation. Using cyclin dependent kinase 9 (CDK9) inhibitors, reporter gene assays and measurements of total and intronic MYB transcription, we show that functionally active CDK9 is required for estrogen-dependent transcriptional elongation. We further show by ChIP and co-immunoprecipitation studies that the P-TEFb complex (CDK9/CyclinT1) is recruited to the attenuation region by ligand-bound ERα, resulting in increased RNA polymerase II Ser-2 phosphorylation. These data provide new insights into MYB regulation, and given the critical roles of MYB in tumorigenesis, suggest targeting MYB elongation as potential therapeutic strategy.
INTRODUCTION
The proto-oncogene MYB (c-MYB), which belongs to the myb family of transcription factors, has emerged as an important regulator of cell growth, survival and differentiation during hematopoiesis (1). In certain forms of aggressive leukemia, such as in MLL-associated leukemia, MYB has been shown to play critical roles in leukemogenesis (2,3). Besides its critical role in hematopoiesis and leukemia, MYB is also associated with other forms of cancer, such as colon and breast cancer. Frequent overexpression of MYB is observed in >80% of colon carcinomas and such expression correlates with poor prognosis (1,4–6). More than two-thirds of human breast cancers, most of which require estrogen for growth and survival, expresses high levels of estrogen receptor (ERα) (7). A strong correlation has been observed between high levels of MYB expression and ERα positivity in breast cancer cells (8). Reports from our laboratories have shown that MYB is essential for the proliferation of ERα+, but not for most ERα−, breast cancer cells (9), which it can suppress proliferation and apoptosis of such cells (10) and that MYB is required for mammary tumorigenesis in several mouse models (11).
Expression of MYB has been shown to be regulated by a transcriptional pausing mechanism (12–14). Genome wide studies in human ES cells and in Drosophila revealed that promoter proximal pausing is a widespread phenomenon involved in the control of expression of immediate early or developmentally regulated genes, and that >30% of gene transcription is regulated by this mechanism (15). According to the current model, soon after transcription initiation, RNA Polymerase II (Pol II) becomes associated with two sets of protein complexes: negative elongation factor (NELF) and DSIF [5,6-dichloro-1β-d-ribofuranosylbenzimidazole (DRB) sensitivity inducing factor]. The resultant transcriptional pausing, in most genes, is induced within 25–50 nt downstream from the transcription start site. Phosphorylation of the C-terminal domain of RNA Pol II on Serine 2, and of NELF and DSIF by P-TEFb [a complex of cyclinT1 and cyclin dependent kinase 9 (CDK9)] releases NELF from the Pol II complex and allows Pol II to complete the transcription (16).
In the case of MYB, unlike most genes, pausing has been detected at ∼1.7 kb downstream of the transcription initiation site (13). Nuclear run-on assays have detected transcription from the promoter and of sequences up to 1.7 kb downstream in the first intron in several cell types even when no or little MYB mRNA is expressed, indicating a block in transcriptional elongation beyond this point. This elongation block is overcome in cells that express MYB and conversely, is re-established when such cells differentiate and lose MYB expression (12,17). The region where this elongation block occurs contains a sequence that can potentially form a stem–loop (SL) structure when transcribed, associated with an adjacent poly (dT) stretch (9,13,17). In the case of colorectal cancer, a significant frequency of mutations is observed in this SL-dT region; introducing these mutations into an elongation reporter assay showed elevated levels of reporter gene activity, that correlated with the sustained high level of MYB mRNA in this type of cancer (17). However, such mutations are rare in the case of breast cancer and therefore unable to explain the high level of MYB expression in ERα+ breast cancers. In this subtype of breast cancer, MYB has emerged as a direct target of ERα (18), and Drabsch et al. (9) have shown that ligand-bound ERα acts to overcome the transcriptional elongation block imposed by the regulatory SL-dT structure. Identifying the factors or cofactors that act with ERα at this region would provide insight into the mechanism of MYB gene regulation in ERα+ breast cancer cells. Moreover, such understanding is likely to provide insight into MYB regulation in other cell types since as discussed above, expression is also regulated at the level of transcriptional elongation in hematopoietic and colon cancer cells.
In this report, we show that functionally active transcriptional elongation factor P-TEFb is required to relieve the transcriptional attenuation mediated by the SL-dT region between exons 1 and 2 of the MYB gene. We find that ERα forms a complex with both CyclinT1 and CDK9 in MCF-7 cells, which is independent of the presence of estrogen. Using chromatin-immunoprecipitation (ChIP), we also show that upon β-estradiol activation, ligand-bound ER recruits the P-TEFb complex to a region near the regulatory SL-dT motif, and that P-TEFb kinase activity is necessary for MYB transcription to continue beyond this motif.
MATERIALS AND METHODS
Cell Culture
HEK293T cells were maintained routinely in DMEM high glucose medium with 10% FBS, l-Glutamine and Penicillin–Streptomycin (GIBCO/BRL, Grand Island, NY, USA) at 37°C incubator in the presence of 5% CO2. MCF-7 cells were maintained regularly as a monolayer in DMEM supplemented with 10% FBS, l-Glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids (GIBCO/BRL, Grand Island, NY), 10 µg/ml insulin (Sigma, St Louis, MO, USA) and Penicillin–Streptomycin (GIBCO/BRL, Grand Island, NY, USA). In order to observe the effect of estrogen, MCF-7 cells were sub-cultured and maintained in a phenol red free DMEM (GIBCO/BRL, Grand Island, NY, USA) with 10% charcoal stripped serum (CSS; GIBCO/BRL, Grand Island, NY, USA) and all other supplements mentioned above for 48 h before 17-β-estradiol (Sigma, St Louis, MO, USA) was added to the final concentration of 10 nM.
Antibodies
For ChIP assays, immunoprecipitation and western blotting, anti-ERα HC20 rabbit polyclonal, anti-CDK9 C20 rabbit polyclonal, anti-CyclinT1 T-18 goat polyclonal antibodies (Santa Cruz Biotechnology, USA) and mouse monoclonal α-Tubulin antibody (Sigma, St Louis, MO, USA) were used. Whole RNA pol II or Ser-2 phosphorylated RNA pol II was detected in ChIP assays by anti-pol II 8WG1b mouse monoclonal or anti-RNA pol II phospho CTDS2 rabbit polyclonal antibodies, respectively (Abcam, UK).
Plasmid DNA, transfection and reporter gene assay
WT and ΔSL-CAT constructs, used in this assay, were described previously (11). In WT-CAT construct, promoter, exon 1 and intron 1 sequences of c-MYB gene were cloned upstream of the CAT. In order to generate the SL deletion construct (ΔSL-CAT), a stretch of 76 bp was deleted from WT-CAT, which ends at the poly T stretch. CyclinT1 and CDK9 and dominant negative CDK9 (DN--CDK9; containing a point mutation at nucleotide 563 which converts Asp into Asn (29) were cloned in pcDNA3.1 vector and expression of all proteins were confirmed by western blot. β-Actin-promoter-luciferase expression vector was used as an internal control in the reporter assay obtained as a generous gift from C. Popa, University of Queensland, Queensland, Australia. The ERα expression construct pAER in which ERα c DNA expression is driven by the human β-actin-promoter, was obtained from the laboratory of Professor Christine Clarke, Department of Medicine, University of Sydney. Approximately 2.0 µg of total plasmid DNA was transfected into HEK293T cells by using Lipofectamine 2000. Briefly, 0.3 × 106 cells/well of a six-well plate was seeded and transfections were carried out according to manufacturer protocol. After 24 h transfection, cells were incubated with fresh DMEM supplemented with 10 nM estrogen for 16 h. Cells were lysed using 300 µl of lysis buffer/well of a six-well plate and 20 µl of extract was used to estimate the reporter gene activity using a CAT-ELISA assay kit (Roche, Indianapolis, IN, USA). The same extracts were utilized to estimate the control β-actin-promoter-luciferase activity using LUC-assay kit (Promega, WI, USA).
Drug treatment
DRB, flavopiridol and anti-estrogen ICI 182780 were dissolved in appropriate solvent as recommended by the manufacturer to make 1000-fold concentrated stocks. MCF-7 cells were seeded at the density of 0.3 × 106 cells/well of a six-well plate and incubated overnight at 37°C. Drugs were diluted in complete DMEM medium, added to the plate and incubation continued for 4 h before harvest. To estimate the effect of P-TEFb inhibitory drugs in reporter gene assays, HEK293T cells were transfected 24 h before drug treatment.
Immunoprecipitation and western blot
For transient transfection followed by immunoprecipitation experiments, ∼3 × 106 cells were seeded in a 100 mm dish and transfected using Lipofectamine 2000 as described above. Cell were harvested 48 h after transfection and extracts were prepared with RIPA lysis buffer (50 mM Tris–Cl, pH 8.3, 150 mM NaCl, 1% NP40, 1% Deoxycholic acid, 0.02% SDS, Bio-Rad, USA, 1 mM PMSF, 1 mM DTT, 5% glycerol, Protease inhibitor cocktail, Roche Applied Science). Extracts were centrifuged for 10 min at 10 000 g and supernatants were used for immunoprecipitation. Extracts were incubated overnight at 4°C, with primary antibody or corresponding control IgG. Immunocomplexes were precipitated by adding 20 µl Protein A/G-Sephadex bead suspension (Santacruz Biotech, USA). Beads were washed thrice with 1.0 ml of RIPA lysis buffer without protease inhibitor cocktail. Immunocomplexes were boiled 5 min with 20 µl of 2× SDS–PAGE sample buffer and analyzed by SDS–PAGE gel followed by western blotting. Blots were incubated with goat anti-mouse IgG coupled with horseradish peroxide and the detections were carried out by enhanced chemiluminescence reagent (Thermo Fischer Scientific, USA). Images were recorded and analyzed by CHEMIDOCK western imaging machine (Fischer Biotech, USA).
RT-PCR
MCF-7 cells were treated with drug for 4 h and total RNA extracted using an RNeasy kit (Qiagen, GmBH, Germany). Approximately, 2.0 µg of RNA was used to synthesize cDNA for RT-PCR using Super ScriptR III First-Strand Synthesis System (Invitrogen, USA). cDNA was diluted 2-fold and 2 µl was used in the RT-PCR. In a 25 µl reaction mixture, 12.5 µl of 2× iQ-SYBER green PCR mix (Bio-Rad, Hercules, CA, USA) was added to 12.5 µl solution containing cDNA and 100 nM of PCR primer. Real-time qPCR to measure MYB mRNA, cyclophilin mRNA and 18S rRNA was carried out at 90°C for 10 s, 56°C for 15 s and 72°C for 20 s for 40 cycles in a Rotor Gene 3000 machine (Corbett Research, Sydney, Australia). Quantitative gene expression was analyzed by the RotorGene 3000 software using cyclophilin A as an internal control and 18S rRNA as a loading control. The following sets of primers were used: c-MYB forward, 5′-GCC AAT TAT CTC CCG AAT CGA-3′; c-MYB reverse, 5′-ACC AAC GTT TCG GAC CGT A-3′; cyclophilin A forward, 5′-GGC AAA TGC TGG ACC CAA CAC AAA-3′; cyclophilin A reverse, 5′-CTA GGC ATG GGA GGG AAC AAG GAA-3′. Human 18S rRNA forward: 5′-GAG GTA GTG ACG AAA AAT AAC AAT-3′, 18S rRNA reverse: 5′-TTG CCC TCC AAT GGA TCCT-3′ (19).
The real-time qPCR conditions to measure intronic transcripts were 95°C for 20 s, 95°C for 10 s, 60°C for 20 s for 40 cycles. The primer sequences used to quantitate these PCR products were Pre-I forward: 5′-GAA ATC CTC GTC CGA ACT GTC AG-3′, Pre-I reverse: 5′-GCG TGT GCT GCT GGG AAA G-3′; Post-III forward: 5′-CCT CCG AAT CAC AGT AGC-3′, Post-III reverse: 5′-TTC TGT CAA GGA AAC AAA CC-3′; Post-IV forward: 5′-GTG GAG GCT AGA CTA G:AA CC-3′, Post-IV reverse: 5′-ACC CAG GAA CAA GCA ACC-3′; cyclophilin A intron forward: 5′ ATT GTC CCT CTG CC-3′ and cyclophilin A intron reverse: 5′- AGG AAA TCG CTC TGT CCT CA-3′.
ChIP assays
MCF-7 cells were grown in T175 flasks to 70–80% confluence (∼3 × 107 cells). Cells were washed thrice with PBS and incubated 20 min at room temperature with 1% formaldehyde. The cross-linking was stopped by the addition of 0.1 M glycine for 5 min. Cells were washed and resuspended in 1.0 ml of lysis buffer (50 mM Tris–HCl pH 8.1, 1.0% SDS, 10.0 mM EDTA and protease inhibitor cocktail). Chromatin was sonicated (Sonics & Materials, Inc., Newtown, CT, USA) for eight cycles of 20 s sonication utilizing 40% efficiency and 2 min incubation at 4°C. Extracts were centrifuged for 16,000 g for 10 min and the supernatant was diluted 1:10 with IP dilution buffer (20 mM Tris–HCl, pH 8.1, 1.0% TritonX-100, 2.0 mM EDTA, 150 mM NaCl). After pre-clearing it with salmon sperm DNA (2 µg, SIGMA, USA) and protein-Sepharose (45 µl of 50% slurry, Santa Cruz Biotech, USA), the extract (100 µl) was used for immunoprecipitation for overnight at 4°C with 1 µg of antibody and corresponding control IgG. Immunoprecipitants were collected by adding 45 µl protein A/G-Sepharose slurry and 2 µg salmon sperm DNA after incubating 1 h at room temperature and washed extensively once with TSEI (20 mM Tris–HCl pH 8.1, 0.1% SDS, 1.0% TritonX-100, 2.0 mM EDTA and 150 mM NaCl), four times with TSEII (20 mM Tris–HCl pH 8.1, 0.1% SDS, 1.0% TritonX-100, 2.0 mM EDTA, 500 mM NaCl, once with Buffer III (10 mM Tris–HCl, pH 8.1, 1 mM EDTA, 1.0% deoxycholate, 1.0% NP-40, 0.25 M LiCl) and thrice with TE. The samples were eluted with 500 µl elution buffer (1.0% SDS, 0.1 M NaHCO3). Eluates were mixed with 0.2 M NaCl and kept overnight at 65°C to reverse the cross links. DNA samples were isolated by phenol/chloroform/isoamyl alcohol extraction and resuspended in 40 µl TE and analyzed by qPCR. Primers used for pS2 gene are forward: 5′-GGC CAT CTC TCA CTA TGA ATC ACT TCT GC-3′, reverse: 5′-GGC AGG CTC TGT TTG CTT AAA GAG CG-3′ and for Myb intron-1 are forward: 5′ AAA GAG CGT GGG TGG AGA C 3′ and reverse: 5′GCA GTC GGG TTT CTC TTC C 3′.
RESULTS
A functionally active P-TEFb complex is required for ERα-mediated promotion of MYB transcriptional elongation
As discussed above, differential expression of MYB in several mammalian cell lines and lymphomas has been shown to be due to blocking of transcriptional elongation within the first intron (14,20,21). Previous data from our laboratory established that in the case of ERα+ breast cancer cells, ligand-bound ERα plays a critical role in relieving this block (9). However, the factors or cofactors which are specifically involved in this process are not known. In general, the complex involved in relieving the stalling of RNA Pol II is comprised of several proteins besides P-TEFb, such as Brd4, HEXIM1 (and/or HEXIM2), LARP7 and MePCE (16,22–28). Since P-TEFb is known to play the central role in this complex, and can be targeted by compounds that inhibit its CDK9 component, we focused initially on the role of this heterodimeric complex in relieving the transcriptional pausing of MYB. To understand the regulatory process, we have used a reporter gene construct where the human MYB promoter, exon 1 and intron 1 sequences were cloned upstream of a CAT reporter gene (WT-MYB reporter construct) as described previously (17). HEK293 cells were transiently transfected with the WT MYB reporter construct along with ERα, cyclinT1 and CDK9 in the presence or absence of estrogen. The results shown in Figure 1A indicate that maximal reporter activity requires co-expression of P-TEFb (CyclinT1 and CDK9) and ERα in the presence of estrogen. We also observed a dose-dependent effect of P-TEFb expression on the activation of the WT-MYB reporter construct. To demonstrate that these effects specifically reflected overcoming inhibition imposed by the SL-dT motif, HEK293T cells were co-transfected with a mutant reporter construct in which the SL-dT sequence was deleted (ΔSL-dT MYB reporter) (9,17). As expected, the activity of this reporter gene was higher than that of the WT-MYB reporter in the absence of ERα, P-TEFb or estrogen, and it remained unresponsive to the over-expression of P-TEFb, ERα and estrogen. To demonstrate the requirement for catalytically active P-TEFb, HEK293 cells were co-transfected with CyclinT1 and either WT or dominant negative (‘kinase dead’) (29) CDK9 (DNCDK9) to generate functionally active or inactive P-TEFb complexes, respectively. With respect to WT CDK9, a 3-fold down-regulation in WT-Myb reporter gene activity was observed when cells were co-transfected with DNCDK9, whereas under same conditions activity of the mutant SL-dT MYB-reporter construct remained unaffected (Figure 1B). To confirm these findings we treated cells with the CDK9 inhibitor DRB for 4 h. In Figure 1C, the activity of the WT-Myb reporter was down-regulated 3-fold in the presence of DRB whereas no significant change was observed in the activity of the mutant reporter construct. From these results, we infer that functionally active P-TEFb complex is required, and ERα and P-TEFb act coordinately, to overcome the pausing of RNA Pol II in the vicinity of the SL-dT region.
Figure 1.

P-TEFb and an intact SL region of intron I are required for the transcriptional regulation of MYB reporter constructs by estrogen. (A) HEK293 cells are transfected (∼0.5 × 106/well of six-well plate) with 1.0 µg of WT-MYB CAT (Left panel) or 1.0 µg ΔSL-MYB CAT construct (Right panel) along with 0.1 or 0.2 µg each of CyclinT1 and CDK9 and 10 ng pCMV β-actin-promoter-luciferase expression plasmid. After 24 h, cells were incubated with fresh medium supplemented with or without 10 nM estradiol for 16 h. (B) Transient transfections in HEK293 cells were carried out, as described above, in the presence of either WT P-TEFb (0.1 µg each of CyclinT1 and CDK9) or dominant negative CDK9 expression construct (0.1 µg) along with WT P-TEFb complex before adding estrogen to the medium. (C) After transient transfection, cells were either exposed to estrogen or to 25 µM DRB as indicated for 16 h. In each case, CAT activity was normalized to β-actin-promoter-Luc activity as an internal control. Experiments were done in triplicate and repeated at least thrice.
Next, we were interested to determine whether the P-TEFb complex is also involved in the regulation of endogenous MYB transcription in a more physiological context. To this end, we used ERα+ breast cancer cells which were treated with different concentrations of the P-TEFb inhibitory drugs DRB or flavopiridol for 4 h. Drug concentrations (2.5–25.0 nM flavopiridol and 2.5–25.0 µM DRB) and treatment time were chosen so that drug-mediated effects on MYB transcription could be observed without compromising cell viability. Expression of MYB was analyzed by qRT-PCR and compared to that of the ‘housekeeping’ gene PPIA (cyclophilin A). In the experiments shown in Figure 2A and B, we observed a substantial dose-dependent down-regulation of cellular MYB expression in MCF-7 cells treated with drugs which varied from 6-fold to 10-fold with DRB and flavopiridol, respectively. In contrast, no effect was observed on cyclophilin A expression. It is also important to note that the effective concentrations of flavopiridol required to down-regulate MYB expression are about 1000-fold lower than those of DRB. This observation correlates well with the minimal P-TEFb inhibitory concentrations for those two drugs determined earlier by Chao and Price (30). To further confirm the role of P-TEFb in relieving transcription attenuation, we over-expressed DNCDK9 in MCF-7 cells together with GFP. GFP-positive cells were isolated 48 h after transfection and used to determine the effect of functionally inactive P-TEFb on endogenous MYB gene expression. In Figure 2C, ectopic expression of DNCDK9 down-regulated the expression of MYB in comparison to the untransfected control while having little effect on cyclophilin A expression. In contrast, a substantial activation, more than 5-fold, was observed upon over-expression of WTCDK9, suggesting that this factor may normally be rate-limiting. Collectively, from the above data, we conclude that CDK9 activity is required for ERα-mediated relief of the block to MYB transcriptional elongation.
Figure 2.

Functionally active P-TEFb complex is selectively required for MYB expression. CDK9 inhibitors DRB and flavopiridol down-regulate the endogenous MYB gene expression. MCF-7 cells grown in regular growth medium (RGM) were treated with (A) DRB and (B) flavopiridol for 4 h. Cells were harvested and expression of MYB and cyclophilin A (Cyclo) was measured by qRT-PCR. (C) Over-expression of DN CDK9 inhibits endogenous MYB gene expression. MCF-7 cells were transfected with WT CDK9 or DN-CDK9 along with pCMV-GFP and treated with 10 nM estrogen. Cells were harvested 16 h after the hormone treatment and GFP-positive cells were isolated by FACS. RNA samples were isolated from the GFP-positive cells and used in qRT-PCR. In all experiments, relative expression of each gene was normalized by 18s Ribosomal RNA expression values as a control. Each experiment was done in duplicate and repeated at least twice.
Estrogen depletion and drug-mediated inhibition of the P-TEFb complex blocks transcription beyond the SL-dT region
According to the general model of transcription pausing, RNA Pol II initiates transcription and pauses within 30–50 nt downstream from the transcription start site (16,31). It has been demonstrated that the generation of the resultant short transcripts is not inhibited by P-TEFb inhibitory drugs, such as DRB or flavopiridol, but that elongation leading to the synthesis of longer transcripts is inhibited (32,33). In the case of the MYB gene, however, transcriptional pausing occurs at/near the SL-dT motif which is about 1.7 kb downstream of the initiation site, as shown by nuclear run-on assays (9,17). If P-TEFb is essential for the synthesis of the complete MYB transcript, we would predict that inhibition of this complex would significantly reduce the synthesis of pre-mRNA that extends beyond this SL-dT region, without significant change in the level of the shorter transcripts that are blocked at/before this region.
To measure transcription upstream and downstream of the SL-dT motif, we designed several RT-PCR primers based on DNA sequence before and after the SL region, as depicted in Figure 3A. The qRT-PCR data of Figure 3C show that addition of estrogen not only significantly enhanced the synthesis of mature MYB mRNA (detected by exon8/9-based primers), but also increased the level of intronic pre-mRNA transcripts beyond the SL-dT region several-fold, as detected by Post-III and Post-IV primers (Post-PCR product). In contrast, there was little change in the levels of transcripts upstream of the SL-dT motif (detected by Pre-I primers,) or of exonic or intronic cyclophilin A transcripts. These data are completely consistent with our previous data obtained using nuclear run-on assays (9) and thus validate the use of this qRT-PCR assay.
Figure 3.

Synthesis of transcripts beyond the SL-region strongly correlates with the estrogen mediated stimulation or drug mediated down-regulation of endogenous MYB gene. Diagrammatic representation of human MYB (A) and cyclophilin A (B) genes. In the case of MYB, the position of the SL and poly dT sites are shown. Positions of intron based primers are indicated as Pre-I, Post-III and Post-IV. Full-length MYB transcripts are detected by MYB exon primer recognizes the sequence of exon8/9 region. Cyclophilin A full-length transcripts are detected by Cyclo Exon primers which recognize exon 5. (C) Ligand bound ERα relieves transcriptional attenuation of MYB gene expression. MCF-7 cells were kept 48 h in DMEM medium supplemented with CSS and incubated 2 h with 10 nM estrogen. The difference in gene expression in the presence of estrogen is shown as a fold change in comparison to the non-estrogen-treated cells. (D) DRB-mediated down-regulation of MYB transcription is associated with significant reduction in the synthesis of transcripts beyond the SL-dT region. MCF-7 cells were treated with 25 µM DRB for 4 h and synthesis of transcripts was estimated by qRT-PCR. Gene expression in the presence of drug was compared with the untreated control cells.
Next, MCF-7 cells were treated with DRB for 4 h and RNA was isolated to determine the expression of the various transcripts by qRT-PCR, which was then normalized to the levels in untreated cells. This revealed a substantial down-regulation of the synthesis of mature MYB mRNA (5-fold) and of post-SL-dT transcripts detected by primers Post-III and Post-IV (5- to 7-fold) in the drug-treated cells with respect to the untreated control (Figure 3D). However, little difference was seen in the levels of PCR products generated by the Pre-I primer set, located upstream of the SL-dT region or in the level of cyclophilin transcript synthesis, detected by intron based primers. A difference in the levels of Pre- and Post-PCR products was also observed when we used another set of primers (EV-I for Pre and EV-III and EV-IV for Post) for analysis by non-quantitative PCR. A clear down-regulation was observed in the level post-PCR products detected by EV-III and EV-IV in the presence of drug whereas a only minor change was observed in the Pre-product identified by EV-I (Supplementary Figure S1). Therefore, we conclude that transcription pausing either induced by inhibiting P-TEFb complex or relieved by addition of estrogen strongly correlates with the absence or presence, respectively, of intronic transcription immediately downstream of the regulatory SL-dT region, but not with apparently constitutive transcription upstream of this region.
ERα forms a complex with P-TEFb
We have shown that estrogen is required for MYB transcriptional elongation (Figure 3B) and that ERα is recruited to chromatin near the SL-dT motif (9), while the data shown in Figures 1 and 2 have now established that functionally active CDK9, and thus probably P-TEFb, is required to relieve transcriptional attenuation. Taken together, these observations suggested that ERα and P-TEFb act in concert to overcome the block to MYB transcriptional elongation, and raise the possibility that ERα may actually recruit P-TEFb. Indeed, an association between endogenous ERα and CyclinT1 has previously been shown in MCF-7 cells (34). We therefore wished to determine whether ERα associated with the P-TEFb complex. First, HEK293T cells were co-transfected either with CDK9 and ERα or CyclinT1 and ERα, and extracts were used for co-immunoprecipitation experiments. The data shown in Figure 4A indicate that anti-ERα antibody can pull down both CDK9 and CyclinT1. Complex formation of ERα with CDK9 and CyclinT1 was confirmed by using anti-CDK9 and anti-CyclinT1 antibody to co-precipitate ERα. In order to confirm the existence of this complex with endogenous proteins, whole cell extracts prepared from MCF-7 cells were subjected to immunoprecipitation with the same antibodies. Additionally, to examine the role of estrogen in complex formation, the cells were grown either in a regular medium (RGM control in Figure 4B), in a hormone-free serum supplemented medium (CSS) for 48 h, or overnight in CSS medium supplemented with estrogen. Using anti-CyclinT1 and anti-ERα antibody, we detected CDK9 in immunoprecipitates from all three treatments, and moreover, the levels of co-precipitated proteins were independent of the presence or absence of hormone (Figure 4B, left panel and Figure 4C, right panel). These observations were confirmed by performing additional immunoprecipitations with anti-CyclinT1 and anti-ERα antibodies. We could detect the presence of CyclinT1 (Figure 4C, left panel) and ERα (Figure 4B, right panel) in the immunocomplexes formed by the anti-ERα and anti-CyclinT1 antibodies, respectively, and moreover, no significant difference in the levels of co-precipitated proteins were detected with or without estrogen. These data strongly suggest that at least a fraction of ERα forms a complex with P-TEFb as all three proteins (ERα, CyclinT1 and CDK9) can be co-immunoprecipitated in pair-wise combination, and that complex formation is independent of the presence of estrogen. The latter observation differs to some extent from that of Wittmann et al. (34) who detected enhanced CyclinT1–ERα interaction in the presence of estrogen using a cell-free GST pull-down assay.
Figure 4.

Complex formation between CyclinT1, CDK9 and ERα. (A) Expression plasmids encoding CyclinT1, CDK9 and ERα were co-transfected into HEK293 cells and immunoprecipitated (IP) with the indicated primary antibodies and the resultant complexes were analyzed by western blotting (WB) using the indicated secondary antibodies. (B) and (C) Identification of complex formation by endogenous proteins. MCF-7 cells were grown in estrogen free (CSS), 10 nM estrogen added and in regular growth medium (control). Extracts, prepared from cells grown in the three different conditions, were used for immunoprecipitation (IP) with anti-CyclinT1 in (B) and anti-ERα in (C). Complexes were analyzed by WB using the antibodies indicated under each panel. In each case, 10% of the total cell extract was loaded for comparison and is labeled ‘In’ (input).
The P-TEFb complex is specifically recruited to the regulatory region of the MYB gene through ERα
Recruitment of the P-TEFb complex at the pausing site is one of the most critical steps in resuming transcription. In the case of HIV, the virus-encoded Tat protein recruits this complex to the TAR RNA sequence to allow transcription of full-length viral RNA (35). The P-TEFb complex is also recruited to cellular target genes by transcription factors, such as NFκB, MyoD, c-Myc or chromatin remodeling proteins such as Brd4, in part, by protein–protein interactions (25,36). In MCF-7 cells, published data from our laboratory (9) and global ChIP-on-chip or ChIP-seq analyses (37,38) have shown in vivo ERα binding in a region close to the SL-dT motif in the presence of estrogen. We were interested to know whether the P-TEFb complex is also recruited to this region and whether, given that P-TEFb forms a complex with ERα, the latter is required for its recruitment. To do this, we used ChIP followed by qPCR with primers that amplify 419 bp of MYB intron1 sequence including 181 bp upstream and 166 bp downstream from the 72 bp SL-dT region, as described previously (9). As a control for an estrogen-regulated gene, we used primers designed to amplify the promoter segment of the pS2 gene, which contains a well-defined ERα-binding site (39). As expected, qPCR analysis of ChIP samples generated using ERα-specific antibody detected binding (several fold above the control IgG) to the regulatory region of the MYB gene and to the pS2 promoter element (Figure 5A and B). Significantly, we also detected the presence of both CyclinT1 and CDK9 when antibodies to each of these proteins and the same MYB intron1 primers were used in ChIP assays. Somewhat surprisingly, we did not detect CDK9 or CyclinT1 association with the pS2 promoter, indicating a selective recruitment of these P-TEFb components to particular sites (Figure 5C). Since co-immunoprecipitation suggested complex formation of P-TEFb with ERα, we next wished to determine the role of ERα in the recruitment of the components of this complex to the MYB SL-dT region. Therefore, ChIP assays using antibodies against ERα, CyclinT1 and CDK9 were carried out on MCF-7 cells that had been incubated in estrogen-free medium for 48 h. Results presented in Figure 6A show negligible binding of ERα to the Myb-regulatory region and no significant accumulation of CDK9 or CyclinT1 under these conditions, but subsequent exposure to 10 nM estrogen for 16 h showed a significant recovery of the ERα binding to the MYB regulatory region. In parallel to ERα binding, a significant increase in the binding of CyclinT1 and CDK9 was observed following estrogen addition (Figure 6B). The response to estrogen in each case is actually quite rapid, as strong binding was also seen after 1 h (Supplementary Figure S2). To establish further the role of ERα in the recruitment of P-TEFb complex, we used the drug Fulvestrant/ICI182780 which not only blocks nuclear localization of this receptor, but also expedites its proteasome-mediated degradation (21). ChIP assays were performed using extracts prepared from MCF-7 treated with 100 nM Fulvestrant for 16 h (Figure 6C). A significant down-regulation (about 3- to 4-fold) of both ERα and P-TEFb binding was observed in the drug-treated sample. Note that Fulvestrant did not affect the total amounts of CDK9 and cyclinT1 in these cells (Supplementary Figure S3). We also performed a recovery experiment where cells were returned to Fulvestrant-free medium for 24 h after treatment as above. ChIP assay of this extract showed a significant re-accumulation of both ERα and P-TEFb complex binding (Figure 6D). Collectively, from the above results, we conclude that P-TEFb is recruited to the MYB-regulatory region as part of the complex formed with ligand-bound ERα.
Figure 5.

ChIP assays detect ERα and P-TEFb components in the attenuation region of the MYB gene. (A,B) ERα binding to the MYB intron-I attenuation region and the pS2 promoter. In total, 0.25, 0.5 or 1.0 µg of anti-ERα antibody, no antibody (mock) or control IgG (1.0 µg) was added to MCF-7 cell extracts and ChIP assays were performed as described in ‘Materials and Methods’ section. (C) P-TEFb complex components are selectively recruited to the MYB intron-1 region. Anti-CyclinT1 and anti-CDK9 antibody (0.25 or 0.5 µg) or 0.5 µg of IgG was added to the MCF-7 extract for ChIP assays. The amount of DNA bound to ERα, CyclinT1 and CDK9 was detected by qPCR using MYB intron- and pS2 promoter-specific primers and the results are plotted as percentage of input.
Figure 6.

Recruitment of the P-TEFb complex to the MYB regulatory region is dependent on ligand-bound ERα. (A,B) MCF-7 cells were cultured in estrogen-free medium for 48 h, following which 10 nM estrogen or vehicle was added for a further 16 h. Extracts prepared from cells cultured in both conditions were used to perform ChIP assays using (A) anti-ERα antibody (1.0 µg), (B) anti-CyclinT1 (0.5 µg) and anti-CDK9 (0.5 µg) antibodies and corresponding control antibody (IgG) or no antibody (mock). The amount of binding was estimated by qPCR and expressed as percent input. (C,D) MCF-7 cells were incubated with 10 µM ICI 182780 for 16 h (ICI). Half of these cells were harvested and the remaining cells were incubated in fresh drug-free growth medium for 24 h (Recovery). An extract prepared from MCF-7 cells grown in regular growth medium (RGM) was used as control. Extracts of harvested cells were used for ChIP assays using (C) anti-ERα (1.0 µg), (D) anti-CyclinT1 (0.5 µg) or anti-CDK9 (0.5 µg) antibodies as indicated.
P-TEFb recruitment to the MYB attenuation region results in enhanced Pol II phosphorylation
Phosphorylation of the C-terminal domain (CTD) of RNA Pol II on its serine 2 residue is required order to resume transcriptional elongation after promoter-proximal pausing. Of the two serine residues at positions 2 and 5 of the CTD, CDK9 preferentially phosphorylates Ser-2 (40). It was also reported that estrogen-dependent regulation of transcription is dominated by the changing phosphorylation of RNA Pol II rather than recruitment of Pol II holoenzyme (41). We therefore wanted to determine the CTD phosphorylation status in the presence or absence of estrogen by ChIP assay, using an antibody that can detect the Ser-2 phosphorylated RNA Pol II. To detect the level of phosphorylated Pol II at the transcription attenuation region of the MYB gene, samples from ChIP assays were analyzed by RT-PCR using the primer set described earlier. As observed in Figure 7A, Ser-2 phosphorylation of Pol II was significantly up-regulated (5- to 6-fold) after addition of estrogen, which is consistent with phosphorylation by CDK9. At the same time, the amount of total polymerase bound to that site was at least 2-fold down-regulated which was estimated by an antibody recognizing total Pol II. We suggest that the decrease in total polymerase at the attenuation site after estrogen addition is most likely because of the mobilization of stalled polymerase after estrogen addition and P-TEFb recruitment. Most importantly, the large increase in Ser-2 phosphorylation correlates well with the recruitment of P-TEFb complex to the attenuation region of the MYB gene (Figures 5C and 6B). As a control, we used the pS2 gene to measure total and Ser-2 phosphorylated Pol II in proximity of promoter and transcription start site. In the presence of estrogen, we found an enrichment of RNA Pol II at the proximal promoter region of pS2. Although we noticed a significant basal level of Ser-2 phosphorylated Pol-ll, no further enrichment of Ser-2-phosphorylated Pol II was observed after estrogen stimulation (Figure 7B). This observation matches well with the data in Figure 5 showing a very low basal level of P-TEFb complex at the actively transcribing pS2 gene. This is reinforced by comparison of the amount of Ser-2 phosphorylation of Pol II in both genes with respect to the total amount of polymerase. We observed a 10-fold increase in the proportion of Ser-2-phosphorylated Pol II in the presence of estrogen at the MYB attenuation site, whereas a small decrease was apparent at the pS2 promoter (Figure 7C).
Figure 7.

Estrogen stimulation selectively induces P-TEFb complex-driven Ser-2 phosphorylation of RNA Pol II in the MYB attenuation site. ChIP assays were performed with MCF-7 extract from cells grown either in the absence or presence of estrogen using anti-RNA Pol II antibody or anti-RNA Pol II CTD phospho Ser-2 antibody. The amount of total RNA Pol II and Ser-2 phospho-Pol II bound was detected by qPCR using MYB intron-1-specific primers (A) and pS2 promoter-specific primers (B), and expressed with respect to the input. These data are also plotted (C) to show the proportion of Ser-2 phosphorylated RNA Pol II compared with the total RNA Pol II present at the pS2 promoter and MYB intron-I pausing site in the presence or absence of estrogen.
DISCUSSION
Previous studies from our laboratory have shown that MYB is required for the proliferation of breast cancer cells. In these and other cells, MYB expression is regulated by transcriptional pausing in the first intron, which in turn is relieved by binding of the estrogen–ERα complex. We wished to investigate the mechanisms involved and, in particular, to know whether the P-TEFb complex, known to be involved in stimulating promoter-proximal transcriptional elongation, is also required here. Using CDK9 inhibitory drugs and dominant negative CDK9, we have demonstrated here that functionally active P-TEFb complex is indeed required to overcome the transcriptional block imposed by the previously described SL-dT motif in intron 1. Our data also strongly suggest that a tripartite complex is formed between ERα, CyclinT1 and CDK9. In the presence of hormone, the ligand-bound complex is recruited to the transcriptional pausing site, where its ability to phosphorylate RNA Pol II is essential for the resumption of transcriptional elongation as illustrated in the model shown in Figure 8. We will consider each of these key findings below.
Figure 8.
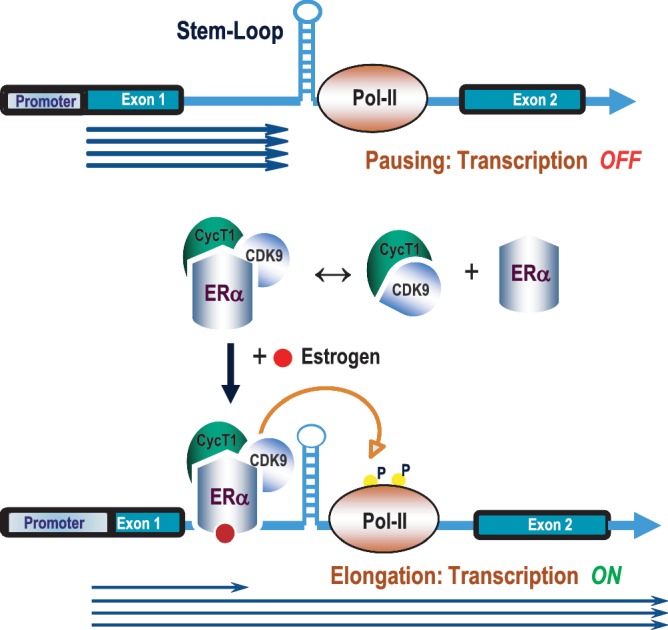
Model for estrogen-mediated relief of MYB transcriptional attenuation. Transcription is paused at the SL-dT region in the absence of estrogen in MCF-7 cells where Pol II is hypophosphorylated at Ser-2. Upon estrogen stimulation, the ERα–CyclinT1–CDK9 tripartite complex is recruited to the attenuation (SL-dT) region; this, in turn, phosphorylates the Ser-2 residue of RNA Pol II to allow resumption of elongation.
We have shown that transcription of MYB in estrogen-dependent breast cancer cells is directly regulated by the P-TEFb complex. First generation Cdk9 inhibitors, such as DRB and flavopiridol, as well as ectopic expression of a dominant-negative CDK9, result in significant down-regulation of MYB transcription when cells are exposed to these agents. Critically, RT-PCR showed that this is a result of re-imposition of a transcriptional block imposed by the SL-dT attenuation motif, which is normally overcome by ERα. Moreover, in Figure 3A, we observed a very low IC50 for inhibition of MYB expression in MCF-7 cells of ∼3–4 nM when exposed for 4 h. However, concentrations of flavopiridol of up to 300 nM are required for substantial inhibition of total Pol II transcription as determined by nuclear run-on assays (30). Thus, we suggest that because of the stringent requirement for transcriptional elongation, and hence for P-TEFb activity, imposed by the SL-dT attenuation motif, MYB expression is particularly sensitive to inhibition of transcriptional elongation and CDK9 activity. This parallels the heightened sensitivity of HIV to flavopiridol (30), which is likely to reflect the requirement for Tat recruitment of P-TEFb for elongation of HIV transcripts through the TAR motif (42).
The ChIP assay data obtained in the absence or presence of estrogen or Fulvestrant treatment indicated that recruitment of the P-TEFb complex by ERα to the attenuation region in MYB intron 1 depends upon the binding of estrogen to the receptor. Since our co-immunoprecipitation data show that formation of the ERα–CyclinT1–CDK9 complex is independent of the presence of hormone, this implies the hormone-dependent step is DNA binding by ERα itself. Analysis of ChIP-on-chip and ChIP-seq data (37,38) shows a receptor-binding region within a span of ∼298 bp located ∼200 bp upstream from the beginning of the SL-dT attenuation motif. Indeed, we have confirmed ERα binding in the vicinity of this motif previously (9) and in the present report (Figures 5–7). While there are no consensus full-length ERα-binding elements (EREs) within that region, we have noticed two half EREs. One (GGTCA) is located very close to the polydT sequence and another (TGACC) is within the ∼298 nt in vivo-binding region. However, introducing mutations into each of these sites individually within our WT-Myb elongation reporter construct had no effect on the estrogen response of this construct (our unpublished results). Taken together, these observations suggest that ERα binds either to a highly divergent motif or via tethering by another transcription factor (43). Distinguishing between these alternatives will require further study.
It is worth commenting on the fact that while we observed selective recruitment of Cyclin T1 and CDK upon ER binding to the MYB intronic region, we did not observe recruitment of this P-TEFb complex to the pS2 promoter. In contrast, Wittmann et al. (34) reported the estrogen/ERα-stimulated recruitment of CyclinT1 to the pS2 promoter. However, in our experiments, MCF-7 cells were treated with estrogen for 12 h rather than the shorter times (45 and 90 min) used by Wittmann et al. One possibility is that in the initial stage of estrogen/ERα-stimulated pS2 gene expression, CyclinT1 is recruited to the promoter as a part of an ERα–CyclinT1 complex, but that it may rapidly dissociate as transcription continues, resulting in a low/undetectable steady-state level. In the case of the MYB intronic pausing region, high levels of P-TEFb may be continuously required to maintain elongation, by promoting Ser-2 phosphorylation of the Pol II CTD, in the presence of the attenuating effect of the SL-dT motif. Our data showing (i) accumulation of Pol II near the attenuation region and (ii) the strong stimulation by estrogen of Ser-2 CTD phosphorylation of Pol II bound to this region, compared to in each case to the pS2 promoter, are entirely consistent with such a notion.
Our recent work has shown an essential role for MYB in the proliferation of human breast cancer cells in vitro and in vivo (9,11), and that it is required for mammary tumorigenesis in (at least) two murine transgenic breast cancer models (11). Given these observations, and that MYB knockdown potentiates differentiation and apoptosis (10), targeting of MYB in breast cancer is a potentially attractive therapeutic approach. Moreover, MYB’s widespread involvement in other major cancers, such as colon carcinoma and leukemia, would further enhance the utility of a general strategy for targeting MYB. The fact that MYB expression is largely regulated at the level of transcriptional elongation suggests inhibition of this process may be an effective and possibly selective approach (1).
The data reported here show a stringent requirement for CDK9 activity to maintain estrogen/ERα-dependent MYB expression and identify inhibition of this kinase with existing compounds as to some extent selectively down-modulating MYB expression. We might therefore predict that MYB-expressing ERα+ breast cancer cells might be more sensitive to CDK9 inhibitors than ERα−/MYB−ve cells. Indeed, preliminary studies have shown that the IC50 for flavopiridol in an MCF-7 cell survival assay is ∼15 nM, while viability of ERα−/MYB− MDA-MB-231 cells is unaffected by that concentration (Z. Talaash, our unpublished observations). The extent to which such effects are, in fact, due to MYB inhibition and whether MYB expression can be selectively suppressed by CDK9 inhibition in other MYB-dependent cancer types will require further investigation.
While this manuscript was under revision, Stadhouders et al. (44) reported that P-TEFb was recruited in mouse erythroid cells to a site in Myb intron 1 that probably corresponds to the SL-dT attenuation region mentioned here. They also reported that transcriptional elongation was strongly inhibited by Cdk9 inhibitors as we have found here.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–3.
FUNDING
Australian Research Council and the Association for International Cancer Research (to T.J.G. and R.G.R.); University of Queensland Sister Janet Mylonas Memorial scholarship (to Y.D.); and a Research Fellowship and Program Grant from the National Health and Medical Research Council (to R.G.R.). Funding for open access charge: University of Queensland Diamantina Institute.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Zia Talaash and Dr Pamela Mukhopadhyay for contributions to preliminary data and assistance with bioinformatics, respectively.
REFERENCES
- 1.Gonda TJ, Leo P, Ramsay RG. Estrogen and MYB in breast cancer: potential for new therapies. Expert Opin. Biol. Ther. 2008;8:713–717. doi: 10.1517/14712598.8.6.713. [DOI] [PubMed] [Google Scholar]
- 2.Jin S, Zhao H, Yi Y, Nakata Y, Kalota A, Gewirtz AM. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J. Clin. Invest. 2010;120:593–606. doi: 10.1172/JCI38030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011;25:1628–1640. doi: 10.1101/gad.17269211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biroccio A, Benassi B, D'Agnano I, D'Angelo C, Buglioni S, Mottolese M, Ricciotti A, Citro G, Cosimelli M, Ramsay RG, et al. c-Myb and Bcl-x overexpression predicts poor prognosis in colorectal cancer: clinical and experimental findings. Am. J. Pathol. 2001;158:1289–1299. doi: 10.1016/S0002-9440(10)64080-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torelli G, Venturelli D, Colo A, Zanni C, Selleri L, Moretti L, Calabretta B, Torelli U. Expression of c-myb protooncogene and other cell cycle-related genes in normal and neoplastic human colonic mucosa. Cancer Res. 1987;47:5266–5269. [PubMed] [Google Scholar]
- 6.Trainer DL, Kline T, McCabe FL, Faucette LF, Feild J, Chaikin M, Anzano M, Rieman D, Hoffstein S, Li DJ, et al. Biological characterization and oncogene expression in human colorectal carcinoma cell lines. Int. J. Cancer. 1988;41:287–296. doi: 10.1002/ijc.2910410221. [DOI] [PubMed] [Google Scholar]
- 7.Anderson WF, Chatterjee N, Ershler WB, Brawley OW. Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results database. Breast Cancer Res. Treat. 2002;76:27–36. doi: 10.1023/a:1020299707510. [DOI] [PubMed] [Google Scholar]
- 8.Guerin M, Sheng ZM, Andrieu N, Riou G. Strong association between c-myb and oestrogen-receptor expression in human breast cancer. Oncogene. 1990;5:131–135. [PubMed] [Google Scholar]
- 9.Drabsch Y, Hugo H, Zhang R, Dowhan DH, Miao YR, Gewirtz AM, Barry SC, Ramsay RG, Gonda TJ. Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc. Natl Acad. Sci. USA. 2007;104:13762–13767. doi: 10.1073/pnas.0700104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drabsch Y, Ramsay RG, Gonda TJ. MYB suppresses differentiation and apoptosis of human breast cancer cells. Breast Cancer Res. 2010;12:R55. doi: 10.1186/bcr2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miao RY, Drabsch Y, Cross RS, Cheasley D, Carpinteri S, Pereira L, Malaterre J, Gonda TJ, Anderson RL, Ramsay RG. MYB is essential for mammary tumorigenesis. Cancer Res. 2011;71:7029–7037. doi: 10.1158/0008-5472.CAN-11-1015. [DOI] [PubMed] [Google Scholar]
- 12.Bender TP, Thompson CB, Kuehl WM. Differential expression of c-myb mRNA in murine B lymphomas by a block to transcription elongation. Science (New York, NY) 1987;237:1473–1476. doi: 10.1126/science.3498214. [DOI] [PubMed] [Google Scholar]
- 13.Thompson MA, Flegg R, Westin EH, Ramsay RG. Microsatellite deletions in the c-myb transcriptional attenuator region associated with over-expression in colon tumour cell lines. Oncogene. 1997;14:1715–1723. doi: 10.1038/sj.onc.1201007. [DOI] [PubMed] [Google Scholar]
- 14.Watson RJ. A transcriptional arrest mechanism involved in controlling constitutive levels of mouse c-myb mRNA. Oncogene. 1988;2:267–272. [PubMed] [Google Scholar]
- 15.Nechaev S, Adelman K. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta. 2011;1809:34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilmour DS. Promoter proximal pausing on genes in metazoans. Chromosoma. 2009;118:1–10. doi: 10.1007/s00412-008-0182-4. [DOI] [PubMed] [Google Scholar]
- 17.Hugo H, Cures A, Suraweera N, Drabsch Y, Purcell D, Mantamadiotis T, Phillips W, Dobrovic A, Zupi G, Gonda TJ, et al. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes Chromosomes Cancer. 2006;45:1143–1154. doi: 10.1002/gcc.20378. [DOI] [PubMed] [Google Scholar]
- 18.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 19.Lukasiak S, Schiller C, Oehlschlaeger P, Schmidtke G, Krause P, Legler DF, Autschbach F, Schirmacher P, Breuhahn K, Groettrup M. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene. 2008;27:6068–6074. doi: 10.1038/onc.2008.201. [DOI] [PubMed] [Google Scholar]
- 20.Bender TP, Catron KM, Kuehl WM, Thompson CB. Sense and anti-sense transcription in the murine c-myb attenuator region. Curr. Top. Microbiol. Immunol. 1988;141:324–329. doi: 10.1007/978-3-642-74006-0_43. [DOI] [PubMed] [Google Scholar]
- 21.Long X, Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J. Biol. Chem. 2006;281:9607–9615. doi: 10.1074/jbc.M510809200. [DOI] [PubMed] [Google Scholar]
- 22.Byers SA, Price JP, Cooper JJ, Li Q, Price DH. HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem. 2005;280:16360–16367. doi: 10.1074/jbc.M500424200. [DOI] [PubMed] [Google Scholar]
- 23.Krueger BJ, Jeronimo C, Roy BB, Bouchard A, Barrandon C, Byers SA, Searcey CE, Cooper JJ, Bensaude O, Cohen EA, et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008;36:2219–2229. doi: 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xue Y, Yang Z, Chen R, Zhou Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010;38:360–369. doi: 10.1093/nar/gkp977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 26.Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 27.Yik JH, Chen R, Pezda AC, Samford CS, Zhou Q. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol. Cell Biol. 2004;24:5094–5105. doi: 10.1128/MCB.24.12.5094-5105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yik JH, Chen R, Pezda AC, Zhou Q. Compensatory contributions of HEXIM1 and HEXIM2 in maintaining the balance of active and inactive positive transcription elongation factor b complexes for control of transcription. J. Biol. Chem. 2005;280:16368–16376. doi: 10.1074/jbc.M500912200. [DOI] [PubMed] [Google Scholar]
- 29.De Falco G, Bagella L, Claudio PP, De Luca A, Fu Y, Calabretta B, Sala A, Giordano A. Physical interaction between CDK9 and B-Myb results in suppression of B-Myb gene autoregulation. Oncogene. 2000;19:373–379. doi: 10.1038/sj.onc.1203305. [DOI] [PubMed] [Google Scholar]
- 30.Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 31.Margaritis T, Holstege FC. Poised RNA polymerase II gives pause for thought. Cell. 2008;133:581–584. doi: 10.1016/j.cell.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 32.Chodosh LA, Fire A, Samuels M, Sharp PA. 5,6-Dichloro-1-beta-d-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J. Biol. Chem. 1989;264:2250–2257. [PubMed] [Google Scholar]
- 33.Kephart DD, Marshall NF, Price DH. Stability of Drosophila RNA polymerase II elongation complexes in vitro. Mol. Cell Biol. 1992;12:2067–2077. doi: 10.1128/mcb.12.5.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wittmann BM, Fujinaga K, Deng H, Ogba N, Montano MM. The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor alpha and transcriptional elongation factor cyclin T1. Oncogene. 2005;24:5576–5588. doi: 10.1038/sj.onc.1208728. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998;17:3681–3691. doi: 10.1093/emboj/17.13.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 37.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 38.Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, Stunnenberg HG. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009;28:1418–1428. doi: 10.1038/emboj.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berry M, Nunez AM, Chambon P. Estrogen-responsive element of the human pS2 gene is an imperfectly palindromic sequence. Proc. Natl Acad. Sci. USA. 1989;86:1218–1222. doi: 10.1073/pnas.86.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol. Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kininis M, Isaacs GD, Core LJ, Hah N, Kraus WL. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol. Cell Biol. 2009;29:1123–1133. doi: 10.1128/MCB.00841-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Liu XY, De Clercq E. Role of the HIV-1 positive elongation factor P-TEFb and inhibitors thereof. Mini Rev. Med. Chem. 2009;9:379–385. doi: 10.2174/1389557510909030379. [DOI] [PubMed] [Google Scholar]
- 43.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 44.Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra R-J, Bryne JC, van den Heuvel A, Stevens M, de Boer E, Kockx C, van der Sloot A, et al. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. EMBO J. 2012;31:986–999. doi: 10.1038/emboj.2011.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.