

Abstract
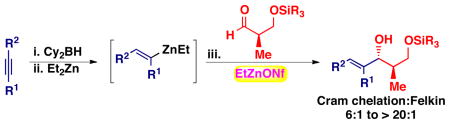
A general diastereoselective method for the addition of dialkylzincs and (E)-di and (E)-trisubstituted vinylzinc reagents to β-silyloxy aldehydes is presented. This method employs alkyl zinc triflate and nonaflate Lewis acids and affords chelation-controlled products (6:1 to > 20:1 dr).
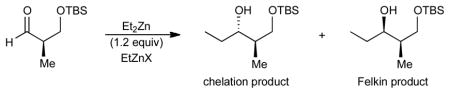
The traditional approach to complex molecule synthesis is to utilize existing substrate stereogenic centers to influence the introduction of new stereocenters.1 In particular, this strategy has proven useful in the addition of organometallic reagents to protected α- and β-hydroxy aldehydes and ketones to afford diol moieties. For quite some time, the paradigm that rationalizes the stereochemical outcomes of these additions has encompassed the Felkin-Anh,2 Cornforth-Evans,3 and Cram-chelation4 models.5 According to these models, stereoinduction in such additions is dependent on the size of the protecting group.6 Sterically undemanding protecting groups such as Me, Bn, and MOM promote chelation (Figure 1, Cram-chelation model).7 In contrast, bulky silyl protecting groups disfavor chelation and give Felkin addition products (Figure 1). One major shortcoming in the application of this paradigm is that the protecting group is chosen to afford the desired stereochemistry during the carbonyl addition step and may not be best suited for the global protecting group strategy. To override substrate control, chemists have utilized chiral catalysts,8 enantioenriched stoichiometric auxiliaries, and optically active stoichiometric additives.9 Our approach is to develop methods to reverse the diastereoselectivity predicted by the aforementioned paradigm through introduction of Lewis acids capable of chelating substrates that normally undergo Felkin addition.
Figure 1.

Models for 1,2-Asymmetric Induction for Protected α-Methyl β-Hydroxy Aldehydes.
To date, few exceptions to the current stereoinduction models have been reported.6f,7c,10 In particular, there are limited examples of methods to promote chelation in additions to β-silyloxy aldehydes and ketones. Evans and coworkers have demonstrated the remarkable ability of Me2AlCl and MeAlCl2 to chelate β-silyloxy aldehydes in Mukaiyama aldol reactions.7c As shown in Scheme 1a, the active Lewis acid results from the disproportionation of MeAlCl2. Somfai and coworkers have disclosed interesting studies to elucidate the origin of reversed diastereoselectivity of Mukaiyama aldol additions to α-silyloxy and chloro aldehydes.11 En route to developing methods for the in situ generation of (Z)-vinylzinc reagents, our group observed unexpected chelation-controlled additions to β-silyloxy aldehydes in the presence of ZnBr2, albeit in modest selectivity (Scheme 1b).12 In efforts toward the synthesis of mycolactone polyketides, Burkart observed high selectivity for the chelation-controlled product in the allylation of aldehyde ent-1 using allyltrichlorostannane (Scheme 1c).13
Scheme 1.

Previous Examples of β-Chelation to β-Silyloxy Aldehydes
More recently, we have shown that alkyl zinc halides and triflates are viable Lewis acids to chelate α-silyloxy aldehydes and ketones to enable chelation-controlled additions to these substrates.14 Given the high levels of diastereoselectvity of these reactions and their generality, we set out to determine whether highly diastereoselective additions of organozinc reagents to β-silyloxy aldehydes could be achieved. We perceived this to be a challenge considering that α-chelation is more favorable than β-chelation and usually furnishes product of higher dr.15 Furthermore, general chelation-controlled additions of organometallic reagents to β-silyloxy aldehydes are unknown.
Initially, we investigated the reaction of diethylzinc with aldehyde 1. Interestingly, in the absence of EtZnX Lewis acids, the reaction of diethylzinc (1.2 equiv) with (R)-3-TBS-2-methylpropanal 1 provided the chelation controlled product with 3.5:1 dr in < 10% yield (Table 1, entry 1). The absolute stereochemistry of the major diastereomer was ascertained through comparison to literature data16 and confirmed by modified Mosher ester analysis.17 To achieve synthetically useful diastereoselectivities and yields, Lewis acids that could chelate the substrates were utilized. Additions employing 25–150 mol % of EtZnCl led to chelation-controlled products with slighly improved dr (entries 2–5). Lowering the temperature did not improve the diastereoselectivity of the reaction (entries 6 and 7). Furthermore, increasing the concentration of the reaction afforded the addition product in 89% yield with 8.9:1 dr (entry 8). Encouraged by these results, we next surveyed other zinc Lewis acids (entries 9–12). Employing either EtZnOTf or EtZnONf gave the expected addition product with 15.5:1 and 10:1 dr, respectively (entries 10 and 11). These Lewis acids can be prepared simply by reaction of the dialkylzinc with the sulfonic acid at low temperature.18
Table 1.
Optimization of Diethylzinc Addition to 1
| |||||
|---|---|---|---|---|---|
| entry | temp (°C) | concentration (M)a | LA (mol %)b | drc | yield (%)d |
| 1 | 0 | 0.2 | 0 | 3.5:1 | <10 |
| 2 | 0 | 0.2 | EtZnCl (25) | 5.7:1 | 50 |
| 3 | 0 | 0.2 | EtZnCl (50) | 5.7:1 | 61 |
| 4 | 0 | 0.2 | EtZnCl (100) | 7.4:1 | 77 |
| 5 | 0 | 0.2 | EtZnCl (150) | 6.5:1 | 83 |
| 6 | −15 | 0.2 | EtZnCl (150) | 6.9:1 | 83 |
| 7 | −30 | 0.2 | EtZnCl (150) | 5.2:1 | 61 |
| 8 | 0 | 0.5 | EtZnCl (150) | 8.9:1 | 89 |
| 9 | 0 | 0.5 | EtZnBr (150) | 7.9:1 | 85 |
| 10 | 0 | 0.5 | EtZnOTf (150) | 15.5:1 | 78 |
| 11 | 0 | 0.5 | EtZnONf (150) | 10:1 | 87 |
| 12 | 0 | 0.5 | EtZnN(Tf)2 (150) | 6:1 | 72 |
Concentration is with respect to the aldehyde.
mol % of Lewis acid is with respect to the aldehyde.
dr determined by 1H NMR of the unpurified product or by GC analysis of the TMS-protected product derivatives and refers to the ratio of chelation:Felkin addition products.
Refers to yield of isolated, purified product.
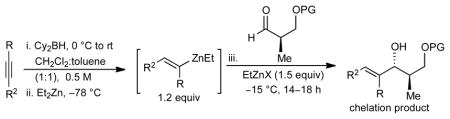
The optimized reaction conditions in Table 1 (entry 10) were applied to the addition of other dialkylzincs to aldehyde 1. It is important to note that R groups on the dialkylzinc and Lewis acid must be the same due to expeditious alkyl exchange. As shown in Scheme 2, the addition of dimethylzinc to afford 3 was less selective (dr = 5:1) and can be attributed to smaller size of the nucleophile, resulting in a lower energy difference between the transition states leading to the chelation or Felkin addition products. Additionally, MeZnOTf has limited solubility in dichloromethane. Addition of di-n-butylzinc to aldehyde 1 occurred to give 4 with comparable dr to diethylzinc (16:1). An excess of di-n-butylzinc (3 equiv) was required due to the facile formation of a reduction side product via a β-hydride reduction mechanism.19
Scheme 2.

Chelation-Controlled Addition of Dialkylzincs to 1
Chiral allylic alcohols are important structural motifs that are commonly used as key intermediates in synthesis and are found in many natural products.1c,20 To broaden the substrate scope of our method, we studied the addition of (E)-vinylzinc reagents to silyl-protected β-hydroxy aldehydes. The (E)-vinylzinc reagents were generated in situ using the Srebnik/Oppolzer procedure.21 Hydroboration of alkynes and subsequent B to Zn transmetallation with Et2Zn gave the (E)-vinylzinc intermediates. These vinylzinc reagents were then added to β-silyloxy aldehydes in the presence of EtZnX Lewis acids. It is well precedented that vinyl- and arylzinc reagents add to aldehdyes significantly faster that alkylzinc reagents.22
After extensive screening, we found that optimal yields and diastereoselectivities were achieved at −15 °C in 1:1 toluene to dichloromethane solvent. Similar to the addition of dialkylzincs, intially EtZnOTf proved to be the most effective Lewis acid, providing the chelation-controlled addition product with moderate to good dr (Table 2). However, employing EtZnONf further improved the diastereoselectivity of the reaction. For example, a twofold increase in the dr of product 5 is seen in entry 1 when EtZnONf is used compared to the same reaction using EtZnOTf (dr = 10:1 vs. 4.8:1). It is conceivable that this improved diastereoselectivity is due to the greater solubility of EtZnONf in dichloromethane at low temperature. As shown in Table 2, a variety of terminal alkynes can be employed in the reaction with 1.5 equiv EtZnONf and both TBS or TES-protected 3-hydroxy-2-methylproponal. The (E)-disubstituted allylic alcohol products were furnished with ≥ 5.8:1 dr and 61–80% yield (entries 1–3 and 6–8). Lower yields were observed in some cases owing to the formation of ethyl addition byproducts (entries 2 and 8 using EtZnOTf). Furthermore, when internal alkynes were utilized in the reaction, (E)-trisubstituted allylic alcohols were generated with ≥ 17:1 dr (entries 4, 5, and 9). Reactions with less sterically hindered TES-protected 3-hydroxy-2-methylproponal generally yielded products with higher dr, most likely due to more favorable chelate formation.
Table 2.
Generation of (E)-Di and Trisubstituted Allylic Alcohols
| |||||
|---|---|---|---|---|---|
| entry | PG | alkyne | dra, EtZnONf (yield) | dra, EtZnOTf (yield) | major product |
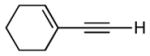
| 1 | TBS |
|
10:1 (61%) | 4.8:1 (83%) |
5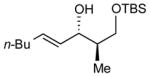
|
| 2 |
|
5.8:1 (66%) | 2.3:1 (50%) |
6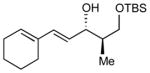
|
|
| 3 |
|
10:1 (70%) | 3.6:1 (78%) |
7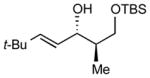
|
|
| 4 |
|
20:1 (80%) | 11:1 (77%) |
8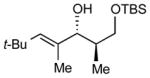
|
|
| 5b |
|
17:1 (78%) | 7.8:1 (77%) |
9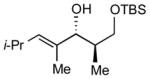
|
|
| 6 | TES |
|
11:1 (65%) | 7.8:1 (84%) |
10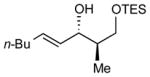
|
| 7 |
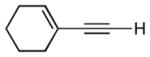
|
9:1 (78%) | 4.6:1 (67%) |
11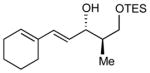
|
|
| 8 |
|
18:1 (71%) | 10:1 (45%) |
12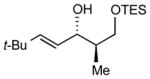
|
|
| 9 |
|
> 20:1 (76%) | 17.5:1 (76%) |
13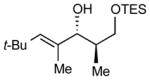
|
|
dr determined by 1H NMR of the unpurified product and refers to the ratio of chelation:Felkin addition products.
The relative stereochemistry was determined by modified Mosher ester analysis (see Supporting Information).
In summary, chelation-controlled addition of organozinc reagents to TBS and TES-protected β-hydroxy aldehydes can be accomplished in the presence of EtZnOTf or EtZnONf. Our method represents an alternative approach to the use of stoichiometric amounts of chiral auxiliaries to reverse the diastereoselectivity in additions of organometallic reagents to α-chiral β-silyloxy aldehydes.
Supplementary Material
Acknowledgments
We thank the NSF (CHE-0848467 and 1152488). We are also grateful to Prof. Marisa Kozlowski (University of Pennsylvania) for helpful discussions. G.R.S. thanks UNCF–Merck and NOBCChE/GlaxoSmithKline programs, ACS Organic Division, and Amgen for graduate scholarships. We would also like to thank the NIH for the purchase of a Waters LCTOF-Xe Premier ESI mass spectrometer (Grant No. 1S10RR23444-1).
Footnotes
Supporting Information Available. Experimental procedures and full characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley; New York: 1989. [Google Scholar]; (b) Koskinen A. Asymmetric Synthesis of Natural Products. Wiley; Chichester, NY: 1993. [Google Scholar]; (c) Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307–1370. [Google Scholar]; (d) Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. VCH; Weinheim, Germany: 1996. [Google Scholar]; (e) Nicolaou KC, Snyder SA. Classics in Total Synthesis II : More Targets, Strategies, Methods. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]; (f) Carreira EM, Kvaerno L. Classics in Stereoselective Synthesis. Wiley-VCH; Weinheim, Germany: 2009. [Google Scholar]
- 2.(a) Chérest M, Felkin H, Prudent N. Tetrahedron Lett. 1968;9:2199–2204. [Google Scholar]; (b) Chérest M, Felkin H. Tetrahedron Lett. 1968;9:2205–2208. [Google Scholar]; (c) Anh NT, Eisenstein O. Tetrahedron Lett. 1976;17:155–158. [Google Scholar]; (d) Anh N, Eisenstein O. Nouv J Chim. 1977;1:61–70. [Google Scholar]; (e) Anh NT. Top Curr Chem. 1980;88:145–162. [Google Scholar]
- 3.(a) Cornforth JW, Cornforth RH, Mathew KK. J Chem Soc. 1959:112–127. [Google Scholar]; (b) Evans DA, Siska SJ, Cee VJ. Angew Chem, Int Ed. 2003;42:1761–1765. doi: 10.1002/anie.200350979. [DOI] [PubMed] [Google Scholar]; (c) Cee VJ, Cramer CJ, Evans DA. J Am Chem Soc. 2006;128:2920–2930. doi: 10.1021/ja0555670. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cram DJ, Elhafez FAA. J Am Chem Soc. 1952;74:5828–5835. [Google Scholar]; (b) Cram DJ, Kopecky KR. J Am Chem Soc. 1959;81:2748–2755. [Google Scholar]; (c) Leitereg TJ, Cram DJ. J Am Chem Soc. 1968;90:4019–4026. [Google Scholar]
- 5.Mengel A, Reiser O. Chem Rev. 1999;99:1191–1224. doi: 10.1021/cr980379w. [DOI] [PubMed] [Google Scholar]
- 6.(a) Overman LE, McCready RJ. Tetrahedron Lett. 1982;23:2355–2358. [Google Scholar]; (b) Keck GE, Castellino S, Wiley MR. J Org Chem. 1986;51:5478–5480. [Google Scholar]; (c) Kahn SD, Keck GE, Hehre WJ. Tetrahedron Lett. 1987;28:279–280. [Google Scholar]; (d) Keck GE, Castellino S. Tetrahedron Lett. 1987;28:281–284. [Google Scholar]; (e) Keck GE, Andrus MB, Castellino S. J Am Chem Soc. 1989;111:8136–8141. [Google Scholar]; (f) Reetz MT, Hüellmann MJ. Chem Soc, Chem Commun. 1986:1600–1602. [Google Scholar]; (g) Frye SV, Eliel EL. Tetrahedron Lett. 1986;27:3223–3226. [Google Scholar]; (h) Frye SV, Eliel EL. J Am Chem Soc. 1988;110:484–489. [Google Scholar]
- 7.(a) Keck GE, Castellino S. J Am Chem Soc. 1986;108:3847–3849. [Google Scholar]; (b) Reetz MT. Acc Chem Res. 1993;26:462–468. [Google Scholar]; (c) Evans DA, Allison BD, Yang MG, Masse CE. J Am Chem Soc. 2001;123:10840–10852. doi: 10.1021/ja011337j. [DOI] [PubMed] [Google Scholar]
- 8.(a) Soai K, Hatanaka T, Yamashita T. J Chem Soc, Chem Commun. 1992:927–929. [Google Scholar]; (b) Watanabe M, Komota M, Nishimura M, Araki S, Butsugan YJ. Chem Soc, Perkin Trans 1. 1993:2193–2196. [Google Scholar]; (c) Jeon S-J, Chen YK, Walsh PJ. Org Lett. 2005;7:1729–1732. doi: 10.1021/ol050255n. [DOI] [PubMed] [Google Scholar]; (d) Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito: 2008. [Google Scholar]
- 9.(a) Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348–6359. [Google Scholar]; (b) El-Sayed E, Anand NK, Carreira EM. Org Lett. 2001;3:3017–3020. doi: 10.1021/ol016431j. [DOI] [PubMed] [Google Scholar]; (c) Marshall JA, Bourbeau MP. Org Lett. 2003;5:3197–3199. doi: 10.1021/ol034918h. [DOI] [PubMed] [Google Scholar]; (d) Marshall JA, Eidam P. Org Lett. 2004;6:445–448. doi: 10.1021/ol0363568. [DOI] [PubMed] [Google Scholar]; (e) Guillarme S, Plé K, Banchet A, Liard A, Haudrechy A. Chem Rev. 2006;106:2355–2403. doi: 10.1021/cr0509915. [DOI] [PubMed] [Google Scholar]; (f) Williams DR, Kiryanov AA, Emde U, Clark MP, Berliner MA, Reeves JT. Angew Chem, Int Ed. 2003;42:1258–1262. doi: 10.1002/anie.200390322. [DOI] [PubMed] [Google Scholar]; (g) Rodriguez-Escrich C, Olivella A, Urpi F, Vilarrasa J. Org Lett. 2007;9:989–992. doi: 10.1021/ol063035y. [DOI] [PubMed] [Google Scholar]; (h) Kolakowski RV, Williams LJ. Tetrahedron Lett. 2007;48:4761–4764. [Google Scholar]; (i) Sharon O, Monti C, Gennari C. Tetrahedron. 2007;63:5873–5878. [Google Scholar]; (j) Georges Y, Ariza X, Garcia J. J Org Chem. 2009;74:2008–2012. doi: 10.1021/jo8025753. [DOI] [PubMed] [Google Scholar]
- 10.(a) Frye SV, Eliel EL, Cloux R. J Am Chem Soc. 1987;109:1862–1863. [Google Scholar]; (b) Chen X, Hortelano ER, Eliel EL, Frye SV. J Am Chem Soc. 1990;112:6130–6131. [Google Scholar]; (c) Chen X, Hortelano ER, Eliel EL, Frye SV. J Am Chem Soc. 1992;114:1778–1784. [Google Scholar]; (d) Evans DA, Allison BD, Yang MG. Tetrahedron Lett. 1999;40:4457–4460. [Google Scholar]; (e) Mohanta PK, Davis TA, Gooch JR, Flowers RA., II J Am Chem Soc. 2005;127:11896–11897. doi: 10.1021/ja052546x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Borg T, Danielsson J, Somfai P. Chem Commun. 2010;46:1281–1283. doi: 10.1039/b922954j. [DOI] [PubMed] [Google Scholar]; (b) Borg T, Danielsson J, Mohiti M, Restorp P, Somfai P. Adv Synth Catal. 2011;353:2022–2036. [Google Scholar]
- 12.Jeon SJ, Fisher EL, Carroll PJ, Walsh PJ. J Am Chem Soc. 2006;128:9618–9619. doi: 10.1021/ja061973n. [DOI] [PubMed] [Google Scholar]
- 13.(a) Ko KS, Alexander MD, Fontaine SD, Biggs-Houck JE, La Clair JJ, Burkart MD. Org Biomol Chem. 2010;8:5159–5165. doi: 10.1039/c0ob00540a. [DOI] [PubMed] [Google Scholar]; (b) Keck GE, Abbott DE. Tetrahedron Lett. 1984;25:1883–1886. [Google Scholar]
- 14.(a) Kerrigan MH, Jeon SJ, Chen YK, Salvi L, Carroll PJ, Walsh PJ. J Am Chem Soc. 2009;131:8434–8445. doi: 10.1021/ja809821x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stanton GR, Johnson CN, Walsh PJ. J Am Chem Soc. 2010;132:4399–4408. doi: 10.1021/ja910717p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stanton GR, Koz G, Walsh PJ. J Am Chem Soc. 2011;133:7969–7976. doi: 10.1021/ja201629d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carey FA, Sundberg RJ. Advanced Organic Chemistry: Part A: Structure and Mechanisms. 5. Springer; New York: 2007. p. 179. [Google Scholar]
- 16.Pilli RA, Riatto VB. J Braz Chem Soc. 1999;10:363–368. [Google Scholar]
- 17.(a) Dale JA, Mosher HS. J Am Chem Soc. 1968;90:3732–3738. [Google Scholar]; (b) Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]; (c) Hoye TR, Jeffrey CS, Shao F. Nat Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 18.Oppolzer W, Schroder F, Kahl S. Helv Chim Acta. 1997;80:2047–2057. [Google Scholar]
- 19.(a) DiMauro EF, Kozlowski MC. Org Lett. 2002;4:3781–3784. doi: 10.1021/ol026315w. [DOI] [PubMed] [Google Scholar]; (b) DiMauro EF, Kozlowski MC. J Am Chem Soc. 2002;124:12668–12669. doi: 10.1021/ja026498h. [DOI] [PubMed] [Google Scholar]; (c) Fennie MW, DiMauro EF, O’Brien EM, Annamalai V, Kozlowski MC. Tetrahedron. 2005;61:6249–6265. [Google Scholar]
- 20.Katsuki T, Martin VS. In: Org React. Paquette LA, editor. Vol. 48. Wiley; New York: 1996. pp. 1–300. [Google Scholar]
- 21.(a) Srebnik M. Tetrahedron Lett. 1991;32:2449–2452. [Google Scholar]; (b) Oppolzer W, Radinov RN. Helv Chim Acta. 1992;75:170–173. [Google Scholar]; (c) Oppolzer W, Radinov RN, Brabander JD. Tetrahedron Lett. 1995;36:2607–2610. [Google Scholar]; (d) Oppolzer W, Radinov RN, El-Sayed E. J Org Chem. 2001;66:4766–4770. doi: 10.1021/jo000463n. [DOI] [PubMed] [Google Scholar]
- 22.(a) Bolm C, Hildebrand JP, Muniz K, Hermanns N. Angew Chem, Int Ed. 2001;40:3285–3308. [Google Scholar]; (b) Rudolph J, Rasmussen T, Bolm C, Norrby PO. Angew Chem, Int Ed. 2003;42:3002–3005. doi: 10.1002/anie.200351150. [DOI] [PubMed] [Google Scholar]; (c) Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chemical Society Reviews. 2006;35:454–470. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]; (d) Rudolph J, Bolm C, Norrby PO. J Am Chem Soc. 2005;127:1548–1552. doi: 10.1021/ja046254s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.