

Abstract
Background
Endotoxemia from lipopolysaccharide (LPS) induces systemic cytokine production, whereas traumatic brain injury (TBI) increases intracerebral cytokine production. In anesthetic doses, ketamine has potent anti-inflammatory properties. However, its anti-inflammatory effects at sub-anesthetic doses and its effects upon TBI induced inflammation have not been fully investigated. We hypothesized that ketamine would attenuate both LPS and TBI induced inflammatory responses.
Methods
Male rats received intraperitoneal ketamine (70, 7, or 1 mg/kg IP) or saline one hour before LPS (20 mg/kg IP) or saline. Five hours after LPS, rats were sacrificed. Serum was collected for cytokine analysis. In other experiments, male rats were given ketamine (7 mg/kg IP) or saline one hour prior to induction of TBI with controlled cortical impact (or sham). One and six hours following injury, brain was extracted for analysis of cerebral edema and cytokine production.
Results
LPS increased the serum concentrations of IL-1α, IL-1β, IL-6, IL-10, TNF-α, and IFN-γ. Ketamine dose dependently attenuated these changes. TBI caused cerebral edema and increased concentrations of cerebral IL-1α, IL-1β, IL-6, IL-10, and TNF-α. However, ketamine had minimal effect on TBI induced inflammation.
Conclusions
While ketamine did not appear to exert any beneficial effects against TBI in the rat, it did not exacerbate cytokine production or enhance cerebral edema as some studies have suggested.
Keywords: cytokine, cerebral edema, ketamine, lipopolysaccharide, traumatic brain injury
Introduction
Ketamine, an anesthetic agent with sedative and analgesic properties, has been shown to have potent anti-inflammatory effects in a variety of models of systemic inflammation, including endotoxemia, sepsis, ischemia, and burns (1-6). These effects have been found in multiple organ systems and involve modulation of the molecular mediators of the inflammatory response, including transcription factors such as nuclear factor-κB (NF-κB) and peroxisome proliferator-activated receptor-γ (PPAR-γ), and proteins such as heme oxygenase-1 (HO-1), inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) (2, 7, 8). Additionally, when given either before or after various pro-inflammatory insults, ketamine has been shown to diminish systemic production of cytokines, such as interleukins -1α, -1β, and -6 (IL-1α, IL-1β, and IL-6), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ), and improve survival (3, 5, 9). Furthermore, recent evidence suggests that ketamine improves end-organ dysfunction and has anti-inflammatory effects that appear to be present even at sub-anesthetic (sedative) doses (10-12).
The role of inflammation in traumatic brain injury (TBI) has also recently become better understood and a focus of investigation. TBI elicits a local inflammatory response involving the activation of astrocytes and microglia, local cytokine production, and recruitment and infiltration of immuno-inflammatory cells (13, 14). This inflammatory response has been shown to play a reparative role in response to the injury. However, in the acute phase, this response may contribute to neuronal injury and cell death (13-15). In particular, IL-1α, IL-1β, IL-6, and TNF-α have been found to be involved in the acute inflammatory response that results from TBI and may significantly contribute to the secondary damage that results from the TBI induced inflammatory response (16-18). In rodent models of TBI, pharmacologic interventions capable of attenuating the intracerebral cytokine response have been shown to improve histologic and functional outcomes (19-21). The effects of ketamine on TBI are largely unknown. Because some studies suggested that ketamine increases cerebral metabolic oxygen requirements and cerebral perfusion pressure, which could aggravate neuronal damage after TBI, its use in patients with TBI was considered contraindicated (22-24). However, recent investigations have shown that such concerns may not be warranted in certain conditions (25, 26). Ketamine, at anesthetic doses, has been found to be neuroprotective in models of cortical injury and ischemia (27, 28). In addition, ketamine has been shown to inhibit microglial activation in response to LPS in vitro (29). Moreover, recent evidence suggests that the use of ketamine has shown promise in patients with TBI (26, 30-32). Because the effects of ketamine on TBI induced inflammatory responses in the brain have not been fully investigated, and because the effects of sub-anesthetic doses of ketamine on serum cytokine production in response to LPS remain to be fully elucidated, this study was done to better examine these effects. We hypothesized that sub-anesthetic doses of ketamine would attenuate both LPS induced changes in serum cytokine production and TBI induced changes in cortical cytokine production.
Materials and Methods
Animals
The University of Texas at Houston Animal Welfare Committee approved all experiments prior to investigation and all studies were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals. Male Sprague-Dawley rats weighing approximately 200 – 230 g (mean 220 g) were used in this study. Rats were housed at constant room temperature with a 12-h-light:12-h-dark cycle and fasted for 18 hours prior to and during the experiments, but allowed unrestricted access to water.
Lipopolysaccharide model
In this model, rats were administered a single intraperitoneal (IP) injection of ketamine (70 mg/kg, 7 mg/kg, or 1 mg/kg) or an equivalent volume of saline. In previous studies, the 70 mg/kg dose of ketamine resulted in complete general anesthesia. One hour after ketamine or saline, rats were given a single injection of lipopolysaccharide (LPS, 20 mg/kg IP) or saline. Because this dose of LPS consistently incites a systemic inflammatory response with reproducible organ dysfunction within five hours, rats were sacrificed under anesthesia five hours after LPS (9, 11). At sacrifice, the rats were anesthetized with intraperitoneal ketamine and euthanized by ventricular puncture and exsanguination. Blood was collected and centrifuged at 1000×g for fifteen minutes at 4°C. Serum was collected as the supernatant and each sample was divided in 2 – 4 microtubes and stored at -80°C until cytokine analysis as previously described (9, 33).
Controlled cortical impact model
In this model, and based on our dose response studies with ketamine and LPS, rats were administered ketamine (7 mg/kg IP) or an equivalent volume of saline. One hour later rats underwent craniectomy and controlled cortical impact (CCI) or sham injury. A CCI device (eCCI Model 6.3; Custom Design) was used to cause unilateral brain injury. Rats were anesthetized with 4% isoflurane and a 1:1 mixture of N2O:O2 and then mounted on a stereotaxic frame. The head was held in a horizontal plane, a midline incision used for exposure, and a 7 – 8 mm craniectomy was performed on the right cranial vault. The center of the craniectomy was placed at the midpoint between bregma and lambda, approximately 3 mm lateral to the midline, overlying the right temporoparietal cortex. A 6 mm diameter impactor tip was positioned at an angle of 10° from the vertical plane, to produce an impact orthogonal to the surface of the cortex. Animals received a single impact with a depth of deformation measuring 3.1 mm, an impact velocity of 6 m/s, and a dwell of 150 ms. In our model this impact produces a moderate to severe injury (34, 35). An audible baseline monitor was used to ensure that the location of the tip, relative to the surface of the brain, was consistent prior to each impact. The impact was delivered onto the parietal association cortex. Sham injury was performed by conducting all procedures except the impact injury, including anesthesia, craniectomy, and stereotaxic mounting. Body temperature was maintained at 37°C using a heating pad. After CCI or sham injury, the skin was closed with staples and the rats were placed in isolation with a heating pad and observed. Once recovered from anesthesia, the animals were returned to their cages for observation until sacrifice. Because this model of TBI produces significant changes in the measured data points of interest (cerebral cytokine concentrations and edema) by 6 hours, rats were sacrificed at 1 and 6 hours after CCI (34, 35). At sacrifice, the rats were anesthetized with isoflurane and euthanized by ventricular puncture and exsanguination followed by immediate decapitation. The brain was extracted and two regions were isolated, the site of direct injury and the penumbral region (Figure 1). The sections were weighed to ensure each section was 120 mg, minced with a pellet pestle, diluted in low glucose Dulbecco's modified eagle medium (GIBCO®), vortexed for 30 seconds, and centrifuged for 2 minutes at 1000×g. The supernatant (intracerebral interstitial fluid) was collected and stored at -80°C until cytokine analysis.
Figure 1.
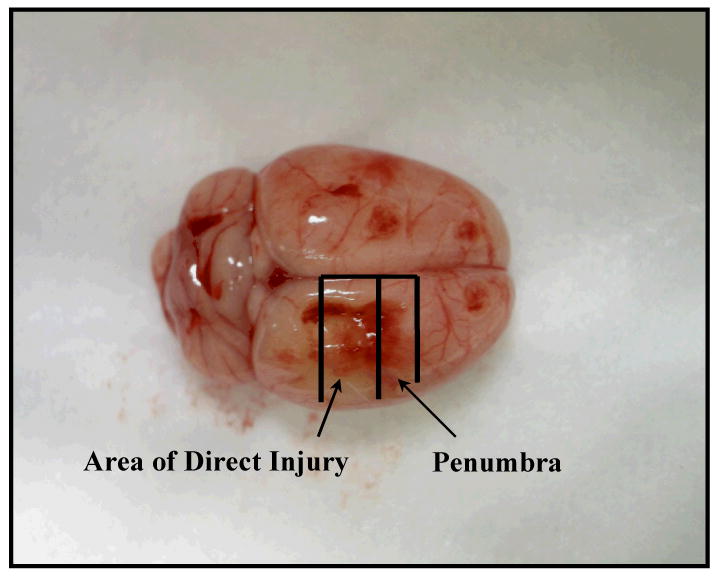
Brain of the rat. The site of direct injury and penumbra were isolated after sacrifice for measurement of tissue percent water and tissue cytokine concentrations.
Assessment of cerebral edema
Because the regions of direct injury and penumbra removed were processed for analysis of cytokines and both cytokine concentration and edema formation cannot be measured from the same section, an additional section of penumbra from each extracted brain was obtained. These sections were weighed and then placed in a desiccator at 60°C. After 72 h, the duration determined to provide appropriate dehydration, the sections were weighed, and tissue weights after 72 h remain stable (35). Edema was measured as tissue percent water:
Analysis of cytokine concentrations
Cytokine concentrations of the serum (LPS model) and intracerebral interstitial fluid (CCI model) were quantitatively assessed using a multiplex bead-based suspension immunoassay (Bio-Rad Laboratories) following the manufacturer's protocol. This assay measures nine cytokines, IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ, and granulocyte-monocyte colony stimulating factor (GM-CSF). Briefly, fluorescently dyed microspheres (beads) are conjugated with antibodies for target cytokines. The antibody-coupled beads are washed with serum containing an unknown amount of cytokine. A series of washes are performed to remove unbound protein, and the beads exposed to a detection antibody specific for a separate epitope on the cytokine, resulting in a sandwich immunoassay format. Streptavidin-phycoerythrin is added, to bind the detection antibody. Beads are drawn into a flow cytometer that identifies and quantitates each reaction based on bead fluorescence. Unknown concentrations are calculated using a standard curve derived from recombinant cytokine standard (Bioplex manual). This method of cytokine analysis has been reported by our group and we have shown that measured cytokine concentrations do not significantly change over time with freezing or thawing (33). Because serum concentrations of IL-2, IL-4, and GM-CSF are undetectable or have minimal change in response to LPS, they are not reported here. The same is true for the intracerebral production of these three cytokines and IFN-γ.
Statistics
All values are expressed as mean ± standard error of the mean (SEM). For all experimental groups a sample size of five or more rats per group was used. Statistical significance was determined using analysis of variance followed by Tukey's post hoc test. A p value less than 0.05 was considered statistically significant.
Results
Ketamine dose dependently attenuates the LPS induced increases in serum cytokines
In these dose response studies with ketamine, the 70 mg/kg dose conferred complete anesthesia. In contrast, when the 1 mg/kg or 7 mg/kg dose of ketamine was administered, minimal, if any, differences in rat arousability or behavior were observed when compared to rats receiving saline (not shown). Subjectively, no differences in the 1 mg/kg and 7 mg/kg doses of ketamine were noted. Different results were seen with respect to their effects on LPS induced cytokine production. The effects of ketamine in doses ranging from 1 – 70 mg/kg on LPS induced increases in serum cytokines are shown in Figures 2-4. As shown, LPS significantly increased serum levels of IL-1α, IL-1β, IL-6, IL-10, TNF-α, and IFN-γ when compared to saline treated controls. When given in doses of 7 and 70 mg/kg, ketamine attenuated LPS induced increases in serum IL-1α, IL-1β, and IL-6. LPS induced increases in TNF-α were attenuated by 1, 7, and 70 mg/kg ketamine. However, production of IFN-γ was attenuated by 70 mg/kg ketamine only. Additionally, for IL-1α, IL-1β, and IL-6, both 7 and 70 mg/kg of ketamine produced significantly different results when compared to rats receiving 1 mg/kg ketamine. When compared to LPS treated rats not receiving ketamine, serum concentrations of IL-10 were not affected by 1 or 70 mg/kg of ketamine, while 7 mg/kg produced a moderate, but statistically significant, attenuation of the LPS induced increase in this cytokine.
Figures 2A-B.

Effects of intraperitoneal ketamine (K; 70, 7, and 1 mg/kg), administered 1 hour prior to intraperitoneal lipopolysaccharide (LPS, 20 mg/kg), on serum concentrations of IL-1α (2A) and IL-1β (2B) 5 hours after LPS. Data are mean serum cytokine concentration (pg/mL) ± SEM. * p < 0.05 versus Saline; ** p < 0.05 vs Saline-LPS; *** p < 0.05 vs Ketamine (1 mg/kg)-LPS; n = 8 for Saline-Saline, n = 10 for all other groups.
Figures 4A-B.

Effects of intraperitoneal ketamine (K; 70, 7, and 1 mg/kg), administered 1 hour prior to intraperitoneal lipopolysaccharide (LPS, 20 mg/kg), on serum concentrations of TNF-α (4A) and IFN-γ (4B) 5 hours after LPS. Data are mean serum cytokine concentration (pg/mL) ± SEM. * p < 0.05 versus Saline; ** p < 0.05 vs Saline-LPS; *** p < 0.05 vs Ketamine (1 mg/kg)-LPS; **** p < 0.05 vs Ketamine (1 mg/kg)-LPS; n = 8 for Saline-Saline, n = 10 for all other groups.
Ketamine does not prevent TBI induced cortical edema
The macroscopic appearance of the rat brain 6 hours after injury is shown in Figure 1. Because there is a significant hemorrhagic component in the area of direct injury, cortical edema was only measured in the penumbra. As shown in Table 1, TBI caused a significant increase in tissue water percentage at 6 hours in the penumbra, indicative of cerebral edema formation (p = 0.05). Increased tissue percent water was not present 1 hour after CCI. Ketamine did not prevent cerebral edema formation when compared to saline or sham controls.
Table 1. Intracerebral edema after CCI.
| Treatment | 1 hour | 6 hours | |
|---|---|---|---|
| Saline | Sham | 79.13±0.20 | 78.72±0.33 |
| Saline | CCI | 79.19±0.23 | 79.76±0.34* |
| Ketamine | Sham | 79.09±0.27 | 78.80±0.28 |
| Ketamine | CCI | 79.42±0.53 | 80.00±0.65 |
Mean tissue percent water ± SEM 1 and 6 hours after CCI
CCI: controlled cortical impact, 1 hour after intraperitoneal Ketamine (7mg/kg) or saline; n = 6 for all groups
p = 0.05 vs Saline-Sham
Ketamine does not prevent TBI induced intracerebral cytokine production
The effects of TBI on intracerebral production of IL-1α, IL-1β, IL-6, IL-10, and TNF-α in the areas of direct injury and penumbra at 1 hour and 6 hours after injury are shown in Tables 2 and 3. As shown, CCI induced increased tissue concentrations of IL-1α, IL-1β, and TNF-α at 1 hour after injury, and IL-1α, IL-1β, IL-6, IL-10, and TNF-α at 6 hours after injury, in the area of cortical injury in the absence and presence of ketamine (p < 0.05). In contrast to the LPS model, ketamine did not significantly affect tissue cytokine concentrations in the area of cortical injury when compared to saline or sham controls. In the penumbra, CCI alone significantly increased tissue concentrations of IL-1α, IL-1β, and TNF-α at 1 hour (p < 0.05). Ketamine did not affect tissue concentrations of IL-1α and IL-1β in the penumbra at 1 hour, but did attenuate the CCI induced increase in TNF-α (p < 0.05). At 6 hours, CCI increased tissue concentrations of IL-1α, IL-1β, IL-6, IL-10, and TNF-α in the penumbra, both in the absence and presence of ketamine (p < 0.05). Ketamine did not significantly influence tissue cytokine concentrations in the penumbra at 6 hours.
Table 2. Intracerebral cytokine concentration: Area of direct injury.
| Treatment | IL-1α | IL-1β | IL-6 | IL-10 | TNF-α | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| 1 hour | 6 hours | 1 hour | 6 hours | 1 hour | 6 hours | 1 hour | 6 hours | 1 hour | 6 hours | ||
| Saline | Sham | 6.3±0.8 | 8.8±1.2 | 25.6±1.9 | 33.2±2.3 | 118.3±10.8 | 113.0±9.8 | 8.2±1.2 | 8.3±2.2 | 20.8±4.8 | 16.2±2.7 |
| CCI | 66.2±12.6* | 174.2±20.8* | 484.5±136.0* | 2987.8±726.2* | 130.8±13.6 | 463.1±34.0* | 16.6±3.0 | 28.5±3.7* | 39.6±4.4* | 35.3±2.2* | |
| Ketamine | Sham | 8.0±0.7 | 8.5±0.7 | 28.2±2.4 | 37.5±3.6 | 155.9±17.6 | 133.2±10.1 | 10.2±1.0 | 9.3±0.6 | 17.3±4.1 | 21.9±3.7 |
| CCI | 71.8±14.1** | 260.5±35.4** | 454.6±124.4** | 4081.0±531.2** | 150.0±22.0 | 501.0±63.0** | 18.6±2.0 | 32.5±2.5** | 36.7±3.8** | 36.8±4.8** | |
Mean cytokine concentration (pg/mL) ± SEM;
p < 0.05 vs Saline-Sham;
p < 0.05 vs Ketamine-Sham
1 hr data: n = 6 for all groups
6 hr data: n = 5 for Saline-Sham and Saline-CCI, n = 6 for Ketamine-Sham and Ketamine-CCI
CCI: controlled cortical impact, 1 hour after Ketamine or Saline; Sham: craniectomy without CCI; Ketamine: 7 mg/kg, intraperitoneal
Table 3. Intracerebral cytokine concentration: penumbra.
| Treatment | IL-1α | IL-1β | IL-6 | IL-10 | TNF-α | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| 1 hour | 6 hours | 1 hour | 6 hours | 1 hour | 6 hours | 1 hour | 6 hours | 1 hour | 6 hours | ||
| Saline | Sham | 10.5±1.7 | 9.8±1.4 | 28.7±1.5 | 38.1±3.3 | 152.9±22.7 | 130.0±6.4 | 10.4±1.2 | 10.3±1.9 | 35.3±3.7 | 39.1±4.7 |
| CCI | 29.2±3.7* | 62.3±11.2* | 187.6±38.0* | 1109.0±295.7* | 147.8±6.1 | 346.7±56.6* | 19.1±4.2* | 19.1±2.9* | 53.7±4.5* | 45.9±3.0 | |
| Ketamine | Sham | 9.8±1.6 | 8.2±1.5 | 30.6±3.2 | 31.6±3.2 | 177.3±11.2 | 144.7±18.7 | 10.4±1.0 | 9.7±1.5 | 33.9±2.9 | 34.0±3.9 |
| CCI | 26.1±1.6** | 92.9±15.8** | 178.3±47.0** | 1286.1±145.4** | 135.4±16.5 | 292.0±29.8** | 12.1±1.7 | 23.3±5.3** | 39.4±4.0ˆ | 45.9±3.0** | |
Mean cytokine concentration (pg/mL) ± SEM;
p < 0.05 vs Saline-Sham;
p < 0.05 vs Ketamine-Sham;
p < 0.05 vs Saline-CCI
n = 6 for all groups
CCI: controlled cortical impact, 1 hour after Ketamine or Saline; Sham: craniectomy without CCI; Ketamine: 7 mg/kg, intraperitoneal
Discussion
Trauma and septic shock initiate simultaneous pro- and anti-inflammatory responses. Based on ongoing laboratory studies, we believe that clinical outcomes can be favorably altered by differential modulation of anti-inflammation over pro-inflammation. While other groups continue to define the complexities involved in the pathophysiologic responses to traumatic and septic shock, we have focused on examining how commonly used ICU interventions such as anesthetics and sedatives can be used to beneficially modulate inflammation. Critically ill patients often have a prolonged hospital course with management in the intensive care unit and may require multiple surgical procedures. As a result, they receive a variety of anesthetic, sedative, and analgesic interventions. Due to its anti-inflammatory properties, ketamine may be a useful therapeutic adjunct in this setting. Moreover, ketamine has been shown to have anti-inflammatory effects in some patient populations, including cardiac surgery and liver transplant, and may be a safe and beneficial adjunct in the brain-injured patient (31, 32, 36, 37). However, others have suggested that its use in patients with TBI is contraindicated as it could potentially exacerbate TBI due to its effects on cerebral perfusion pressure. While perfusion pressures were not measured in this study, we clearly demonstrated that a 7 mg/kg dose of ketamine does not exacerbate TBI-induced inflammation.
In contrast, ketamine has potent anti-inflammatory effects on LPS induced changes in serum concentrations of IL-1α, IL-1β, IL-6, TNF-α, and IFN-γ and these effects were dose dependent. Indeed the anti-inflammatory effects were found at both anesthetic (70 mg/kg) and sub-anesthetic (7 mg/kg) doses, but were limited at lower doses (1 mg/kg). Ketamine may be most useful as an adjunct in the critical care setting at subanesthetic doses, exploiting its sedative and analgesic properties without producing total intravenous anesthesia. Because the subanesthetic dose of ketamine effectively attenuated LPS induced changes in serum cytokine concentrations and did not anesthetize the rat, this dose of ketamine was examined in a model of TBI induced intracerebral inflammation. However, this dose of ketamine had little effect upon TBI induced cerebral edema formation or changes in cerebral cytokine concentrations.
This study extends our previously published data regarding the effects of ketamine at 70 mg/kg on LPS induced changes in serum cytokines (9). In that study, we reported that ketamine (70 mg/kg) blunted the LPS induced increases in serum concentrations of IL-1β, IL-6, TNF-α, and IFN-γ. Here we report its effects on IL-1α as well as the effects of ketamine at doses of 7 and 1 mg/kg on LPS induced changes in these cytokines. Taken together, these data indicate that the anti-inflammatory effects of ketamine, with regards to LPS induced changes in serum cytokines, are dose dependent and exist at doses considered sub-anesthetic.
Our results are consistent with the literature. However, these other investigations have been limited to IL-6 and TNF-α (10, 12). In a cecal ligation and puncture model of rodent sepsis, Yu et al reported that ketamine, given intravenously in a dose of 2.5 mg/kg attenuated the increase in serum concentration of TNF-α, but not IL-6 (12). Similarly, we found that the lowest dose of ketamine investigated in our study (1 mg/kg IP) was able to significantly blunt the increase in systemic production of TNF-α in response to LPS, but had no significant effects on the other cytokines studied. Thus, these data suggest that the most potent anti-inflammatory effect of ketamine, with regards to LPS induced changes in serum cytokine production, is its ability to attenuate the production of TNF-α. TNF-α is produced by macrophages as a direct response to LPS through a toll-like receptor 4 mediated pathway. That ketamine, at a dose of 1 mg/kg, is able to attenuate the LPS induced production of TNF-α, in the absence of effects upon the LPS induced changes in other cytokines, indicates that ketamine acts, in part, upon the macrophage mediated response to LPS in a manner independent from its effects on other cytokines. Indeed, ketamine has been shown to inhibit macrophage production of TNF-α in vitro (38). This effect alone, however, was not sufficient for ketamine to modulate the global cytokine response, as evidenced by the marked reduction of TNF-α production in response to 1 mg/kg of ketamine and the lack of response in the other measured cytokines at this dose. This could reflect an insufficient dose of ketamine to affect other aspects of macrophage function, such as IL-1 production, an insufficient dose of ketamine to affect other cells involved in the inflammatory response, or both. Conversely, ketamine appears to be much less potent with regards to its ability to modulate production of IFN-γ, as neither the 1 mg/kg nor the 7 mg/kg doses of ketamine were able to significantly diminish the LPS induced increases in this cytokine. This finding suggests that the modulation of the inflammatory response by ketamine is not mediated through IFN-γ. Additionally, ketamine had little effect upon the LPS production of IL-10, an anti-inflammatory cytokine. Although ketamine produced a statistically significant attenuation of LPS induced IL-10 production at a dose of 7 mg/kg, this effect is relatively modest and production of IL-10 is largely preserved. That ketamine attenuates the LPS induced production of pro-inflammatory cytokines, while having minimal effect upon the anti-inflammatory cytokine IL-10, suggests that ketamine exerts its effect through selective modulation of the inflammatory response, rather than through a broad non-discriminatory mechanism. Thus, ketamine may modulate the inflammatory response in such a way that the pro-inflammatory response is suppressed, while the anti-inflammatory response is relatively preserved. In addition to the elucidation of the exact mechanism of its action, future studies should also evaluate the effects of ketamine upon LPS induced inflammation and injury when administered in conjunction with and after LPS.
Our current study also extends our knowledge with respect to TBI and confirms that TBI results in a local inflammatory response as measured by formation of edema and interstitial cytokine concentrations in cortical tissues. The presence of edema at 6 hours after injury, but not at 1 hour after injury, is consistent with previous work as edema formation is expected to be present by 6 hours and to peak at 48 hours (35, 39). Treatment with ketamine prior to TBI was not able to attenuate or delay the formation of post-TBI cortical edema. The changes in cerebral cytokine concentrations as a result of TBI in this study are expected and consistent with previous results from our laboratory and the work of others (34, 40-42). At the site of injury, pro-inflammatory cytokines IL-1α, IL-1β, and TNF-α were found to be elevated at 1 and 6 hours after CCI, while the production of IL-6 was not observed until 6 hours after injury. IL-10, an anti-inflammatory cytokine, was found to be increased at 1 and 6 hours after injury. Despite the dramatic effects seen with ketamine on LPS induced production of serum cytokines, ketamine only blunted the TBI induced increase in cerebral concentrations of TNF-α, and even then only at 1 hour after CCI and only in the penumbra (Table 3). With the lone exception of TNF-α, ketamine had no impact on the cytokine responses in our model of TBI. Taken together, the results of our investigations utilizing a model of TBI indicate that ketamine, at the subanesthetic dose of 7 mg/kg, has minimal effects on the early local inflammatory state in the brain that develops in response to TBI.
In our LPS model, the anti-inflammatory effects of ketamine were very potent, especially in regards to its ability to abrogate production of TNF-α. Consequently, we originally hypothesized that the anti-inflammatory effects of ketamine would be present in another model of inflammation, in this case, TBI. However, this was not the case. Nevertheless, it remains possible that higher doses of ketamine may be required to observe any anti-inflammatory effects when using other models of inflammation. That ketamine lacked efficacy during TBI may be secondary to differing mechanisms of the inflammatory insults. Endotoxemia from LPS produces a profound systemic inflammatory response with subsequent organ damage and dysfunction, whereas CCI results in local tissue injury, a localized inflammatory response, and, in more severe cases, a subsequent systemic inflammatory response. Although these two responses, to LPS and to TBI, share many cellular responses, signaling molecules, and molecular pathways, they are clearly the result of two different initial insults and progression. The underlying molecular mechanisms likely differ as well, but remain to be fully elucidated.
The major limitations of this study, specifically in regard to TBI, include the examination of early time-points and the choice of a relatively low dose of ketamine. Previous work in our laboratory indicates that edema can be detected by 6 hours, and persists up to 60 hours (35). Additionally, local cytokine production varies depending upon the cytokine measured, with increased tissue concentrations observed between 1 to 24 hours. Changes in IL-1α, IL-1β, and TNF-α are detectable up to 12 hours, while changes in IL-6 are found from 6 to 24 hours (34). In the present study, measurements at the early time-points after CCI evaluate early local inflammatory response. The prolonged course of the local inflammatory response was not evaluated. Thus the progression of edema, the resolution of early cytokine production (IL-1α, IL-1β, and TNF-α), and the sustained production of the pro-inflammatory cytokine IL-6 were not examined. Furthermore, given the differing mechanism responsible for the inflammatory response in TBI when compared to endotoxemia, it is possible that a relatively low dose of ketamine is not sufficient to produce effects. Finally, we have previously shown that pre-treatment with the anesthetic isoflurane is also capable of attenuating the systemic production of IL-1α and IL-1β in response to LPS (9). In our model of TBI, isoflurane anesthesia is used at the time of CCI, and potential anti-inflammatory effects from the administration of this agent could make any effect that ketamine might produce difficult to detect. Future investigations involving the anti-inflammatory effects of ketamine in TBI are warranted and should incorporate higher doses of ketamine and later time-points.
In conclusion, this study found that ketamine attenuates LPS induced systemic cytokine production, but has little effect on the early local inflammatory response that occurs as a result of CCI induced TBI. However, it is noteworthy that ketamine did not exacerbate this response either. Further studies with this agent to confirm its safety and efficacy in the presence of TBI are warranted. Moreover, the data suggest that ketamine may have utility as a sedative adjunct in septic patients.
Figures 3A-B.

Effects of intraperitoneal ketamine (K; 70, 7, and 1 mg/kg), administered 1 hour prior to intraperitoneal lipopolysaccharide (LPS, 20 mg/kg), on serum concentrations of IL-6 (3A) and IL-10 (3B) 5 hours after LPS. Data are mean serum cytokine concentration (pg/mL) ± SEM. * p < 0.05 versus Saline; ** p < 0.05 vs Saline-LPS; *** p < 0.05 vs Ketamine (1 mg/kg)-LPS; n = 8 for Saline-Saline, n = 10 for all other groups.
Acknowledgments
The authors would like to thank Lily Chang, Yan Cui, Raymond Kwan, and Daniel MacDougall for their technical assistance in performing the animal studies.
Supported by NIGMS GM-38529 and 08782
Contributor Information
Matthew T. Harting, Email: mharting@med.umich.edu.
Charles S. Cox, Jr, Email: charles.s.cox@uth.tmc.edu.
David W. Mercer, Email: dwmercer@unmc.edu.
References
- 1.Suliburk JW, Gonzalez EA, Moore-Olufemi SD, et al. Ketamine inhibits lipopolysacharide (LPS) induced gastric luminal fluid accumulation. J Surg Res. 2005;127:203–207. doi: 10.1016/j.jss.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Suliburk JW, Helmer KS, Gonzalez EA, et al. Ketamine attenuates liver injury attributed to endotoxemia: role of cyclooxygenase-2. Surgery. 2005;138:134–140. doi: 10.1016/j.surg.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 3.Shaked G, Czeiger D, Dukhno O, et al. Ketamine improves survival and suppresses IL-6 and TNFalpha production in a model of Gram-negative bacterial sepsis in rats. Resuscitation. 2004;62:237–242. doi: 10.1016/j.resuscitation.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Camara CR, Guzman FJ, Barrera EA, et al. Ketamine anesthesia reduces intestinal ischemia/reperfusion injury in rats. World J Gastroenterol. 2008;14:5192–5196. doi: 10.3748/wjg.14.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gurfinkel R, Czeiger D, Douvdevani A, et al. Ketamine improves survival in burn injury followed by sepsis in rats. Anesth Analg. 2006;103:396–402. doi: 10.1213/01.ane.0000226140.84281.3e. [DOI] [PubMed] [Google Scholar]
- 6.Yu M, Shao D, Yang R, et al. Effects of ketamine on pulmonary inflammatory responses and survival in rats exposed to polymicrobial sepsis. J Pharm Pharm Sci. 2007;10:434–442. doi: 10.18433/j3rp46. [DOI] [PubMed] [Google Scholar]
- 7.Helmer KS, Cui Y, Dewan A, et al. Ketamine/xylazine attenuates LPS-induced iNOS expression in various rat tissues. J Surg Res. 2003;112:70–78. doi: 10.1016/s0022-4804(03)00138-0. [DOI] [PubMed] [Google Scholar]
- 8.Helmer KS, Suliburk JW, Mercer DW. Ketamine-induced gastroprotection during endotoxemia: role of heme-oxygenase-1. Dig Dis Sci. 2006;51:1571–1581. doi: 10.1007/s10620-005-9013-0. [DOI] [PubMed] [Google Scholar]
- 9.Adams SD, Radhakrishnan RS, Helmer KS, et al. Effects of anesthesia on lipopolysaccharide-induced changes in serum cytokines. J Trauma. 2008;65:170–174. doi: 10.1097/TA.0b013e31805824ca. [DOI] [PubMed] [Google Scholar]
- 10.DeClue AE, Cohn LA, Lechner ES, et al. Effects of subanesthetic doses of ketamine on hemodynamic and immunologic variables in dogs with experimentally induced endotoxemia. Am J Vet Res. 2008;69:228–232. doi: 10.2460/ajvr.69.2.228. [DOI] [PubMed] [Google Scholar]
- 11.Ward JL, Adams SD, Delano BA, et al. Ketamine Suppresses LPS-induced Bile Reflux and Gastric Bleeding in the Rat. Journal of Trauma. 2010;68:69–75. doi: 10.1097/TA.0b013e3181a8b3a7. [DOI] [PubMed] [Google Scholar]
- 12.Yu M, Shao D, Liu J, et al. Effects of ketamine on levels of cytokines, NF-kappaB and TLRs in rat intestine during CLP-induced sepsis. Int Immunopharmacol. 2007;7:1076–1082. doi: 10.1016/j.intimp.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt OI, Heyde CE, Ertel W, et al. Closed head injury--an inflammatory disease? Brain Res Brain Res Rev. 2005;48:388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 14.Morganti-Kossmann MC, Rancan M, Otto VI, et al. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165–177. doi: 10.1097/00024382-200116030-00001. [DOI] [PubMed] [Google Scholar]
- 15.Morganti-Kossmann MC, Rancan M, Stahel PF, et al. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Lu KT, Wang YW, Yang JT, et al. Effect of interleukin-1 on traumatic brain injury-induced damage to hippocampal neurons. J Neurotrauma. 2005;22:885–895. doi: 10.1089/neu.2005.22.885. [DOI] [PubMed] [Google Scholar]
- 17.Stahel PF, Shohami E, Younis FM, et al. Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab. 2000;20:369–380. doi: 10.1097/00004647-200002000-00019. [DOI] [PubMed] [Google Scholar]
- 18.Tehranian R, Andell-Jonsson S, Beni SM, et al. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19:939–951. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- 19.Chen SF, Hsu CW, Huang WH, et al. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br J Pharmacol. 2008;155:1279–1296. doi: 10.1038/bjp.2008.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd E, Somera-Molina K, Van Eldik LJ, et al. Suppression of acute proinflammatory cytokine and chemokine upregulation by post-injury administration of a novel small molecule improves long-term neurologic outcome in a mouse model of traumatic brain injury. J Neuroinflammation. 2008;5:28. doi: 10.1186/1742-2094-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch JR, Wang H, Mace B, et al. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192:109–116. doi: 10.1016/j.expneurol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Gardner AE, Dannemiller FJ, Dean D. Intracranial cerebrospinal fluid pressure in man during ketamine anesthesia. Anesth Analg. 1972;51:741–745. [PubMed] [Google Scholar]
- 23.Takeshita H, Okuda Y, Sari A. The effects of ketamine on cerebral circulation and metabolism in man. Anesthesiology. 1972;36:69–75. doi: 10.1097/00000542-197201000-00013. [DOI] [PubMed] [Google Scholar]
- 24.Shaprio HM, Wyte SR, Harris AB. Ketamine anaesthesia in patients with intracranial pathology. Br J Anaesth. 1972;44:1200–1204. doi: 10.1093/bja/44.11.1200. [DOI] [PubMed] [Google Scholar]
- 25.Langsjo JW, Maksimow A, Salmi E, et al. S-ketamine anesthesia increases cerebral blood flow in excess of the metabolic needs in humans. Anesthesiology. 2005;103:258–268. doi: 10.1097/00000542-200508000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Albanese J, Arnaud S, Rey M, et al. Ketamine decreases intracranial pressure and electroencephalographic activity in traumatic brain injury patients during propofol sedation. Anesthesiology. 1997;87:1328–1334. doi: 10.1097/00000542-199712000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Shapira Y, Artru AA, Lam AM. Ketamine decreases cerebral infarct volume and improves neurological outcome following experimental head trauma in rats. J Neurosurg Anesthesiol. 1992;4:231–240. doi: 10.1097/00008506-199210000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Shapira Y, Lam AM, Eng CC, et al. Therapeutic time window and dose response of the beneficial effects of ketamine in experimental head injury. Stroke. 1994;25:1637–1643. doi: 10.1161/01.str.25.8.1637. [DOI] [PubMed] [Google Scholar]
- 29.Chang Y, Lee JJ, Hsieh CY, et al. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediators Inflamm. 2009;2009:705379. doi: 10.1155/2009/705379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourgoin A, Albanese J, Leone M, et al. Effects of sufentanil or ketamine administered in target-controlled infusion on the cerebral hemodynamics of severely brain-injured patients. Crit Care Med. 2005;33:1109–1113. doi: 10.1097/01.ccm.0000162491.26292.98. [DOI] [PubMed] [Google Scholar]
- 31.Bourgoin A, Albanese J, Wereszczynski N, et al. Safety of sedation with ketamine in severe head injury patients: comparison with sufentanil. Crit Care Med. 2003;31:711–717. doi: 10.1097/01.CCM.0000044505.24727.16. [DOI] [PubMed] [Google Scholar]
- 32.Himmelseher S, Durieux ME. Revising a dogma: ketamine for patients with neurological injury? Anesth Analg. 2005;101:524–534. doi: 10.1213/01.ANE.0000160585.43587.5B. [DOI] [PubMed] [Google Scholar]
- 33.Ward JL, Delano BA, Adams SD, et al. Laparotomy attenuates lipopolysaccharide induced gastric bleeding in the rat. Dig Dis Sci. 2010;55:902–910. doi: 10.1007/s10620-009-0800-x. [DOI] [PubMed] [Google Scholar]
- 34.Harting MT, Jimenez F, Adams SD, et al. Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery. 2008;144:803–813. doi: 10.1016/j.surg.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harting MT, Smith CT, Radhakrishnan RS, et al. Regional Differences in Cerebral Edema After Traumatic Brain Injury Identified by Impedance Analysis. J Surg Res. 2010;159:557–564. doi: 10.1016/j.jss.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartoc C, Frumento RJ, Jalbout M, et al. A randomized, double-blind, placebo-controlled study assessing the anti-inflammatory effects of ketamine in cardiac surgical patients. J Cardiothorac Vasc Anesth. 2006;20:217–222. doi: 10.1053/j.jvca.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Zeyneloglu P, Pirat A, Sulemanji D, et al. Perioperative anesthetic management for recipients of orthotopic liver transplant undergoing nontransplant surgery. Exp Clin Transplant. 2007;5:690–692. [PubMed] [Google Scholar]
- 38.Wu GJ, Chen TL, Ueng YF, et al. Ketamine inhibits tumor necrosis factor-alpha and interleukin-6 gene expressions in lipopolysaccharide-stimulated macrophages through suppression of toll-like receptor 4-mediated c-Jun N-terminal kinase phosphorylation and activator protein-1 activation. Toxicol Appl Pharmacol. 2008;228:105–113. doi: 10.1016/j.taap.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 39.Markgraf CG, Clifton GL, Moody MR. Treatment window for hypothermia in brain injury. J Neurosurg. 2001;95:979–983. doi: 10.3171/jns.2001.95.6.0979. [DOI] [PubMed] [Google Scholar]
- 40.Kamm K, Vanderkolk W, Lawrence C, et al. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J Trauma. 2006;60:152–157. doi: 10.1097/01.ta.0000196345.81169.a1. [DOI] [PubMed] [Google Scholar]
- 41.Shohami E, Novikov M, Bass R, et al. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 42.Woodroofe MN, Sarna GS, Wadhwa M, et al. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. J Neuroimmunol. 1991;33:227–236. doi: 10.1016/0165-5728(91)90110-s. [DOI] [PubMed] [Google Scholar]