

Abstract
The 14-position of natural opiates (e.g. morphine) are unsubstituted, however synthetic approaches have uncovered that functionalizing position 14 gives rise to a wide range of diverse activities. This review focuses on SAR of the position, with the aim of aiding in the search for opioid analgesics with improved clinical profiles.
Keywords: Opioid agonist; opioid antagonist; 14-alkoxymorphinans; 14-amino-4,5-epoxymorphinan derivatives; 14-O-heterocyclic naltrexones
I. Introduction
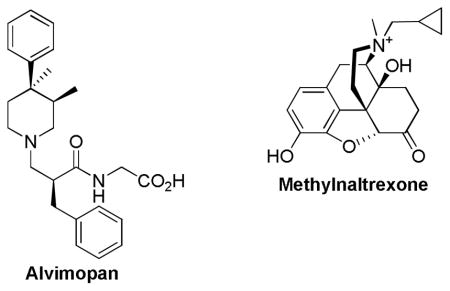
The provision of effective pain management is essential in a clinical setting where pain is common in individuals treated for cancer, post-operative patients, or in cases of severe trauma. There are two major classes of drugs that are commonly used in treating moderate to severe clinical pain; opioids and nonsteroidal anti-inflammatory agents [1]. Even though opioids are known to be most problematic [2], they are the mainstay of treatment of severe clinical pain [3, 4]. Undesirable side effects such as tolerance, dependence [5], respiratory depression, constipation and nausea [6] have been the leading cause of under-medication and inadequate pain management [7, 8]. Patients that receive opioid treatment often receive additional medications to treat or prevent some of the undesirable side effects. For example, constipation can be managed with stool softeners and laxatives, but not chronically [9]. More recently, alvimopan and methylnaltrexone have been approved as selective antagonists of gastrointestinal opioid receptors to treat constipation [10]. While additional medication may lessen or even prevent some of the adverse effects, in some cases it may dramatically decrease the effectiveness of the opioid itself due to drug-drug interaction [11]. Another problem associated with taking additional medication is that it adds to the regimen of drugs already taken by the patients.
Opioid receptors are G-protein coupled receptors that contain seven transmembrane domains and are primarily located in the brain and the spinal cord as well as the gastrointestinal tract (GIT) [12]. The three types of opioid receptors that have been cloned and pharmacologically characterized are κ [13], δ [14, 15], and μ [16], and each exhibits unique pharmacological response upon stimulation. μ Agonists produce analgesia, euphoria, respiratory depression, tolerance, and constipation [17]. Agonists of the κ receptor have been shown to produce dysphoria, by interacting though central nervous system (CNS) mechanisms, tremendously limiting the use of κ agonists in a clinical setting [18]. δ Agonists are not effective against severe pain and are known to produce convulsions [19, 20]. The growing body of evidence concerning the physiological relevance of homo- and heterodimers of opioid receptors [21, 22], leads to the potential of designing ligands that target the dimers and give rise to different effects. However, at present, μ opioid receptors remain the preferred target for more severe pain therapeutics.
Tremendous effort has been put towards the development of novel opioids lacking side effects that are commonly seen in opioid treatment [23]. For example, orvinols (such as bupronorphine) developed by Bentley, exhibit extreme potency but are unsuccessful in elimination of the frequently seen side effects [24]. More recently, several μ-receptor antagonists have been approved for treatment of opioid induced constipation: alvimopan [25], and methylnaltrexone bromide [26]. Alvimopan’s large molecular weight, zwitterionic form, and polarity reduce its CNS penetration, thereby allowing the agent to selectively antagonize the effect of opioids on μ receptors in the GI tract. Another significant limitation to prolonged use is the risk of a heart attack. Consequently, alvimopan is only available as a short-term treatment, in hospitals approved by the Entereg Access Support and Education (E.A.S.E.) program, and cannot be dispensed to patients after discharge [25, 27]. Methylnaltrexone bromide is a derivative of naltrexone which has a high peripheral selectivity resulting from the low lipid solubility due to its quaternary salt form. Moreover, methylnaltrexone must be administered subcutaneously as it exhibits poor oral bioavailability [26].
Recent modifications at position 14 have opened a new realm of possibilities. Though natural opiates are unsubstituted at position 14, introduction of 14-OH and 14NH2 has been achieved starting from thebaine [28, 29]. Substituents in position 14 have shown to not only improve potency but also selectivity for certain receptor types. For example, Schmidhammer et. al., showed that extremely high potency can be achieved at all three opioid receptors with 14-alkoxymorphinan derivatives [30]. While, Husbands’ group presented modest selectivity with 14-aminodihydromorphinones and 14-aminodihydrocodeinones, clocinnamox analogs [31]. Most recently, studies by Zhang et. al., showed that high binding affinity for the μ opioid receptor with high selectivity over the δ and the κ receptors can be achieved with 14-O-heterocyclic substituted naltrexone [32]. This review will present the most recent developments and modifications in the 14 position of the morphine analogs as potential therapeutic opportunities.
II. 14-Alkoxymorphinans

One of the most promising subclass of opioids with the potential for reduced undesired effects is the 14-alkoxymorphinans, which was developed by Schmidhammer et. al. [30] During the initial structure-activity relationship (SAR) studies, Schmidhammer’s group showed that introduction of a 14-methoxy in oxymorphone (1) result in increased binding affinities at all three opioid receptors (0.10 nM at μ receptor; 4.80 nM at δ receptor; and 10.2 nM at κ receptor) [33]. The 14-O-methoxymorphone was reported to possess agonist properties with 400-fold greater potency than morphine and 800-fold greater potency than the parent compound oxymorphone by hot-plate test in mice [34]. Like the parent compound, 14-O-oxymorphone induced respiratory depression, physical dependence, and constipation [34].
Further studies revealed that introduction of a 14-benzyloxy group (2) compared to 14-methoxy group produced similar μ binding affinities (0.12 nM and 0.10 nM respectively), but lower selectivity over δ opioid receptors (2.14 nM and 4.80 nM, respectively) and κ opioid receptors (1.18 nM and 10.2 nM, respectively). Moreover, 14-O-benzyloxymorphone was reported to have 4-fold greater potency than the 14-methoxy analog and 700-fold greater potency than morphine. Most interestingly, 14-O-benzyloxymorphone displayed 2.5-fold less constipative activity as compared to morphine and 7.0-fold less constipation effects than 14-O- methoxymorphone in mice after s.c. administration [33].
Subsequently, the same group showed that introduction of a 14-methoxy in an N-methylmorphinan-6-one series (3), produced similar μ binding affinity as 14-O-methoxymorphone (0.15 nM and 0.10 nM, respectively) with a slightly better selectivity over δ opioid receptors (13.3 nM and 4.80 nM, respectively) and κ opioid receptor (25.2 nM and 10.2 nM, respectively) [35]. Remarkably high antinociceptive activity was reported for 14-methoxymetopon, which exhibited approximately 20,000-fold greater potency than morphine and 1500-fold greater potency than oxymorphone by the acetylcholine-writhing test in rats and mice [36]. Upon supraspinal administration, 14-methoxymetopon can elicit potency of up to one million-fold greater than morphine [37]. Perhaps the most exciting finding was that 14-methoxymetopon lacked tolerance and physical dependence after repeated treatment [38]. Studies also showed that 14-methoxymetopon has reduced constipation [37] and respiratory depression [38] commonly associated with highly potent opioids. These results indicate that a more favorable interaction is possible with the receptor via position 14 in the N-methylmorphinan-6-one series.
Furthermore, the 14-alkoxymorphinan series shows that potency can be further magnified by C14 arylalkyl substituents as seen with 14-benzyloxy (4) [33] and 14-phenylpropyloxymetopon (5) [39] derivatives. These 14-arylalkyloxymetapon derivatives displayed enhanced δ and κ affinities while maintaining high μ affinities. Though the 14-phenylpropyloxymetopon derivative exhibited complete loss in μ-selectivity with 0.20 nM at μ receptor, 0.14 nM at δ receptor, and 0.40 nM at κ receptor, it was reported to have extreme potency (24,000-fold higher in the tail flick assay and 8,500-fold higher in the hot plate assay as compared to morphine) [39]. This analog is even more potent than etorphine which makes 14-phenylpropoloxymetapon unsuitable for clinical use due to its extreme potency [39].
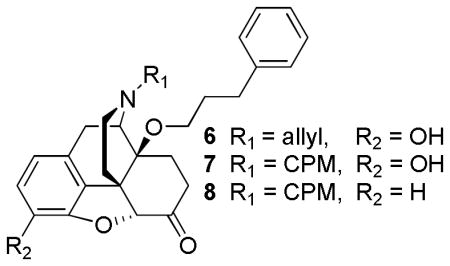
While developing novel μ agonists for the treatment of pain is beneficial, their reinforcing properties make for strong abuse potential [40]. Thus, there has been a growing interest in the development of μ antagonists to block the actions of the abused μ agonists [41]. For many years, it has been generally acknowledged that the introduction of either cyclopropylmethyl or allyl groups on the nitrogen position 17 typically results in antagonism [23]. However, in contrast to the generally accepted antagonist SAR models, 14-O-phenylpropyl derivatives containing N-cyclopropylmethyl and N-allyl groups (6–8) displayed full agonist activity [28]. These results indicate that the nature of the N-substituent does not determine the efficacy, but rather the position of the N-substituent is important for efficacy [28]. In addition, both analogs 6 and 7 displayed enhanced potency, about 100–400-fold more potent in the hot plate assay than morphine [28]. Moreover, 14-alkoxymorphinans such as 14-O-phenylpropyloxy-3-desoxy naltrexone (8) was capable of maintaining subnanomolar affinity for μ (0.84 nM) even when there is no C3 oxygen function [42]. Here it is evident that the substituents in position 3 that were previously considered essential for μ activity are not required in the 14-alkoxymorphinone subclass [42].
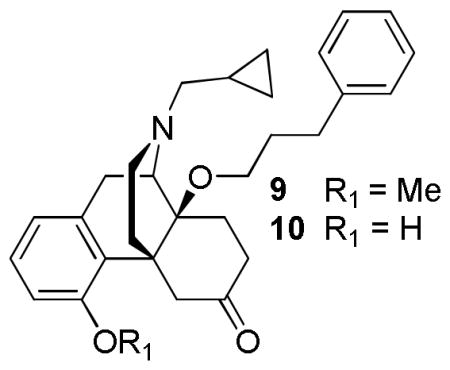
Further SAR studies revealed that partial agonism at μ and δ can be attained by introducing a 14-phenylpropyl group into cyprodime [42], a selective μ-antagonist. Although antagonism was observed at κ opioid receptors by GTPγS functional assays, the cyprodime derivatives, 9 and 10 showed no antagonist activity against morphine in the mouse tail flick assay. These results further imply that the overall conformation of the N-substituent in relation to its skeleton, rather than the substituent itself, dictates the efficacy [42]. The presence of 14-alkoxy showed an increase in binding affinity at all three opioid receptors and acted as a potent antinociceptive agent in vivo with potency similar to that of 14-metoxymetopon [42].
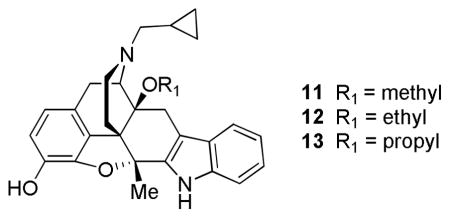
Schmidhammer’s group also showed that conversion of a hydroxyl to alkoxy in naltrindole with a methyl moiety located at position 5 produced lower affinity for δ while increasing δ selectivity when compared to naltrindole. Further studies showed that the nature of the substituent in position 14 determines the binding strength. The 14-ethoxy substituent (12) showed increased interaction with the δ receptor (Ki = 0.78 nM) when compared to the 14-methoxy (11: Ki = 1.15 nM) and 14-propoxy (13: Ki = 5.3 nM) naltrindole derivatives. All 14-alkoxy derivatives possessed antagonist activity in the GTPγS functional assay. Some loss in δ affinity and selectivity was seen with the 14-arylalkoxy naltrindole derivatives (8–30 nM) [43].
Evidence that δ antagonists such as naltrindole and 7-benzylspiroindanylnaltrexone may be involved in allograft survival [44] persuaded Schmidhammer’s group to investigate such a phenomena with analog 12, which was previously shown to be superior to naltrindole [43]. The results showed that 12 inhibited rat lymphocyte proliferation in vitro (IC50 = 0.54 μM) [45]. Additionally, compound 11 showed immunosuppressive activity in vitro and reduced interleukin-2 (IL-2) production in mouse and human lymphocytes [46]. In contrast to the previous finding, these naltrindole derivatives did not exhibit immunosupression via δ opioid receptors as seen in the mixed lymphocyte reaction assay that uses μ/δ/κ receptor knock-out mice. Furthermore, it has been suggested that the indolo moiety is involved in immunosuppressive activity [47].
III. 14-Aminomorphinones and codeinones
Another important subclass of opioids contains 14-aminomorphinones and codeinones. Clocinnamox (14, C-CAM) and methoclocinnamox (15, MC-CAM) were the first analogs developed in their structural class by Lewis et al. [48]. MC-CAM and its parent compound C-CAM had very similar affinities (μ = 0.46 nM and 7.2 nM; δ = 29 nM and 7.2 nM; and κ = 4.5 nM and 1.6 nM respectively) [49]. While C-CAM displayed μ antagonism with no agonist activity [50], MC-CAM was reported to have higher efficacy, displaying partial agonism at the μ receptor after peripheral administration in vivo [51]. Potentially, the most exciting finding was that MC-CAM had pseudo-irreversible effects with its extremely long duration of antagonist action similar to that of buprenorphine [52]. Initially, MC-CAM was believed to exhibit its delayed long-term antagonist effect via its de-methylated metabolite C-CAM [48]. However, it was later shown that MC-CAM was capable of producing μ-antagonist effects after i.c.v. administration [53]. Although long duration of action μ-antagonists can be used to treat drug abuse by blocking the effects of the drug upon subsequent administration [54], MC-CAM does not possess a superior profile to buprenorphine.
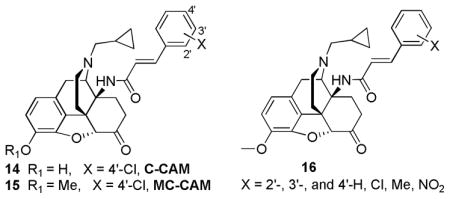
Other studies presented by the groups of Husbands and Lewis looked at the effect of the aryl ring substituent orientation (16) [55]. In these studies, the μ efficacy decreases in the order: ortho- > meta- > para- for the methyl and chloro substituents while no effect was seen with the fluoro substituent [55]. In contrast, a reduction in μ agonist efficacy and potency was seen when the nitro orientation was changed from the para- to the ortho- position possibly due to the lipophilicity rather than steric or electronic effects [55]. Conclusions drawn from these studies showed that 2′-chloro, 2′-methyl, 4′-fluoro and 4′-nitro substituted cinnamylaminomorphinone analogs possessed potent agonist effects, with ED50 of 0.003 mg/kg to 0.014 mg/kg compared to morphine’s 0.66 mg/kg in the rat tail pressure in vivo assay [31]. Interestingly, the 4′-nitro analog acted as a short-term agonist in the mouse hot water tail-withdrawal (TW) assay[53]. However, when pretreated for 24 hours, the 4′-nitro analog had morphine antagonist activity with a long duration of action [55].
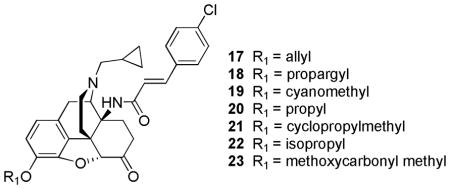
Subsequently, the groups of Lewis and Husbands studied the effect of a variety of 3-alkyl ether to further investigate the possibility of the MC-CAM’s delayed long duration of action antagonism to be a result of the C-CAM metabolite. Interestingly, higher efficacy was achieved with 3-alkyl ether C-CAM analogs [41, 56]. Specifically, 3-allyl (17), 3-propargyl (18), cyanomethyl (19), and propyl (20) ethers displayed higher efficacy than MC-CAM, with 3-propargyl ether analog having the greatest activity by TW assay [56]. The 3-propargyl ether analog was reported to have similar potency to morphine with higher efficacy than buprenorphine in mice, meanwhile a lack of change in efficacy was seen in rhesus monkeys [56]. Other substituents like cyclopropylmethyl, isopropyl and methoxycarbonyl methyl ether were reported to have antagonist activity by the TW assay in mice. All the ether analogs were reported to have long-term antagonism effects in the TW assay when administered 24 hours prior to morphine administration [56]. In this series, the propagyl ether analog had the preferred long-lived μ-antagonist effects in mice and rhesus monkeys in addition to the increased efficacy when compared to buprenorphine [56]. These results further indicate that the delayed antagonist activity of MC-CAM is not related to its metabolism [56].
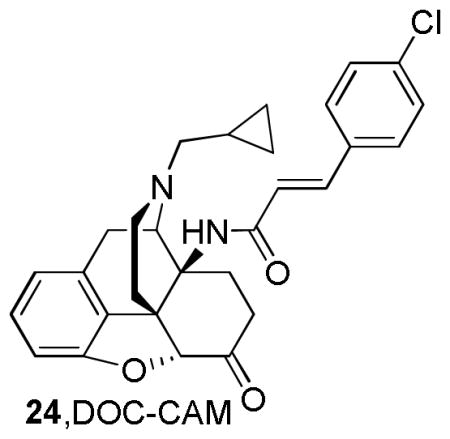
Similar to Schmidhammer’s compounds, [42] the removal of the 3-hydroxy group from C-CAM to give DOC-CAM, 24 resulted in similar μ-affinity as its parent compounds MC-CAM and C-CAM (Ki= 0.54 nM, 0.46 nM, and 0.25 nM, respectively). Although DOC-CAM was reported to qbe an antagonist, it did not exhibit irreversible effects as its parent compound in vivo. Therefore, even though it is evident that the 3-hydroxyl substituent is not required for μ-opioid activity, it is essential for the irreversible μ antagonist activity in the 14-cinnamoylamino series [31, 57].

Studied by Archer et al. were the 14-aminomorphinone derivatives containing sulfhydryl group, which can form covalent disulfide bonds with the receptor via oxidative coupling [58–60]. One such compound is TAMO which displays moderate selectivity for the μ-receptor over δ and κ [58]. This compound was shown to have antinociceptive effects immediately after i.p. and i.c.v. administration for up to 5 h and 2.5 h, respectively [59, 60]. These effects appeared to be mediated via the μ opioid receptor as they were antagonized by the β-funaltrexamine[60]. Additionally, TAMO was found to act as a selective μ opioid receptor antagonist with long duration of action (from 8h to 48h after administration) against morphine [60]. Interestingly, N-cyclopropylmethyl analog of TAMO (N-CPM-TAMO) had no antinociceptive activity but acted as a selective μ antagonist with long duration of action [61].
IV. 14-O-heterocyclic naltrexones
Antagonists such as naloxone and naltrexone are the approved drugs used for treatment of opiate overdose [62]. Since there is no crystal structure of the μ receptor in existence to date, these μ antagonists play an important role in the study of opioid receptors [32]. More recently, studies showed that μ antagonists can be used to treat obesity, psychosis, and Parkinson’s disease [63], making the development of novel μ antagonists a valuable tool not only for studying the structure of opioid receptors, but also for the development of the much needed therapeutics. 14-O-heterocyclic substituted naltrexone derivatives were most recently developed by Guo et. al. [32], using a constructed homology model based on bovine rhodopsin. This model contained transmembrane helical domains with extracellular and intracellular loops, and was further optimized in a membrane-aqueous system using molecular dynamic simulations. The model revealed that the non-conserved residues, Tyr212 and Trp320, may interact with the receptor via hydrogen bonding interactions with the ligand [32]. Thus, a new series of compounds were developed to incorporate a hetero-aromatic moiety on position 14 of naltrexone enabling hydrogen bonding and/or aromatic stacking interactions with Tyr212 and Trp320 [32].
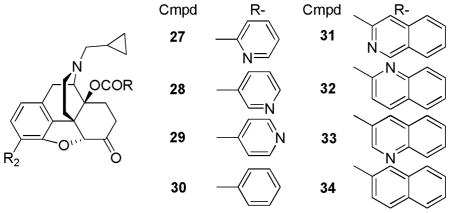
Zhang’s group further investigated the effect of the pyridyl nitrogen position and bulkiness via additional aromatic moieties on the 14-O-heterocyclic naltrexone derivatives. Almost all compounds were reported to have antagonist activity in GTPγS assays except for compound 33 [32]. When compared to previously reported compounds by Schmidhammer and Husbands [30, 31] this series had similar binding affinities; however, compound 27 had higher selectivity, approximately 800-fold selectivity for the μ over δ and 200-fold selectivity for the μ over κ [32]. Introduction of an additional aromatic moiety (compounds 31–34) did not improve the interaction with the μ receptor but rather lowered their selectivity [32].
V. Conclusion
Advances in the development of highly potent and selective opioid agonists and antagonists via position 14 in 14-alkoxymorphinan, 14-aminomorphinone, and 14-O-heterocyclic naltrexone series provide valuable insights into opioid ligand-receptor interaction. It is evident that the nature of the substituent on position 14 and its orientation has a strong influence on receptor binding and post-receptor mechanisms. The advances in SAR illustrated in this review serve as a valuable tool for designing novel molecules with optimal configuration that may aid in identification of ideal opioid medications.
Acknowledgments
The authors wish to thank the National Institute on Drug Abuse, National Institutes of Health (NIDA, NIH) (DA-13583) for the financial support of ongoing research.
References
- 1.Block JH, Beale JM. Organic Medicinal and Pharmceutical Chemistry. Vol. 11. Lippincott Williams and Wilkins; Philadelphia: 2004. [Google Scholar]
- 2.Fries DS. Principles of Medicinal Chemistry. Vol. 4. Williams and Wilkins; Philadelphia: 1995. Analgesics. [Google Scholar]
- 3.Stein C, Schafer M, Machelska H. Attacking pain at its source: new perspectives on opioids. Nat Med. 2003;9(8):1003–1008. doi: 10.1038/nm908. [DOI] [PubMed] [Google Scholar]
- 4.Zieglgansberger W, Tolle TR, Zimprich A, Hollt V, Spanagel R. Endomorphins, Pain Relief, and Euphoria. Pain and the Brain. 1995;22:439–457. [Google Scholar]
- 5.Kieffer BL, Evans CJ. Opioid tolerance-in search of the holy grail. Cell. 2002;108(5):587–590. doi: 10.1016/s0092-8674(02)00666-9. [DOI] [PubMed] [Google Scholar]
- 6.McNicol E, Horowicz-Mehler N, Fisk RA, Bennett K, Gialeli-Goudas M, Chew PW, Lau J, Carr D. Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J Pain. 2003;4(5):231–256. doi: 10.1016/s1526-5900(03)00556-x. [DOI] [PubMed] [Google Scholar]
- 7.Hill CS., Jr The barriers to adequate pain management with opioid analgesics. Semin Oncol. 1993;20(2 Suppl 1):1–5. [PubMed] [Google Scholar]
- 8.Cherny N, Ripamonti C, Pereira J, Davis C, Fallon M, McQuay H, Mercadante S, Pasternak G, Ventafridda V. Strategies to manage the adverse effects of oral morphine: an evidence-based report. J Clin Oncol. 2001;19(9):2542–2554. doi: 10.1200/JCO.2001.19.9.2542. [DOI] [PubMed] [Google Scholar]
- 9.Klaschik E, Nauck F, Ostgathe C. Constipation--modern laxative therapy. Support Care Cancer. 2003;11(11):679–685. doi: 10.1007/s00520-003-0525-x. [DOI] [PubMed] [Google Scholar]
- 10.Hipkin RW, Dolle RE, John EM. Annual Reports in Medicinal Chemistry. Vol. 45. Academic Press; pp. 142–155. [Google Scholar]
- 11.Armstrong SC, Cozza KL. Pharmacokinetic Drug Interactions of Morphine, Codeine, and Their Derivatives: Theory and Clinical Reality, Part II. Psychosomatics. 2003;44:515–520. doi: 10.1176/appi.psy.44.6.515. [DOI] [PubMed] [Google Scholar]
- 12.Ossipov MH, Lai J, King T, Vanderah TW, Malan TP, Jr, Hruby VJ, Porreca F. Antinociceptive nociceptive actions of opioids. J Neurobiol. 2004;61(1):126–148. doi: 10.1002/neu.20091. [DOI] [PubMed] [Google Scholar]
- 13.Mansson E, Bare L, Yang D. Isolation of a human kappa opioid receptor cDNA from placenta. Biochem Biophys Res Commun. 1994;202(3):1431–1437. doi: 10.1006/bbrc.1994.2091. [DOI] [PubMed] [Google Scholar]
- 14.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258(5090):1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 15.Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci U S A. 1992;89(24):12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JB, Johnson PS, Persico AM, Hawkins AL, Griffin CA, Uhl GR. Human mu opiate receptor. cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 1994;338(2):217–222. doi: 10.1016/0014-5793(94)80368-4. [DOI] [PubMed] [Google Scholar]
- 17.Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol Sci. 1999;20(1):19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- 18.Hasebe K, Kawai K, Suzuki T, Kawamura K, Tanaka T, Narita M, Nagase H. Possible pharmacotherapy of the opioid kappa receptor agonist for drug dependence. Ann N Y Acad Sci. 2004;1025:404–413. doi: 10.1196/annals.1316.050. [DOI] [PubMed] [Google Scholar]
- 19.Comer SD, Hoenicke EM, Sable AI, McNutt RW, Chang KJ, De Costa BR, Mosberg HI, Woods JH. Convulsive effects of systemic administration of the delta opioid agonist BW373U86 in mice. J Pharmacol Exp Ther. 1993;267(2):888–895. [PubMed] [Google Scholar]
- 20.Broom DC, Nitsche JF, Pintar JE, Rice KC, Woods JH, Traynor JR. Comparison of receptor mechanisms and efficacy requirements for delta-agonist-induced convulsive activity and antinociception in mice. J Pharmacol Exp Ther. 2002;303(2):723–729. doi: 10.1124/jpet.102.036525. [DOI] [PubMed] [Google Scholar]
- 21.George SR, O’Dowd BF, Lee SP. G-Protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1(10):808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 22.Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2(4):274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- 23.Casy AF, Parfitt RT. Opioid Analgesics. Plenum Press; New York and London: 1986. [Google Scholar]
- 24.Lewis JW, Bentley KW, Cowan A. Narcotic analgesics and antagonists. Annu Rev Pharmacol. 1971;11:241–270. doi: 10.1146/annurev.pa.11.040171.001325. [DOI] [PubMed] [Google Scholar]
- 25.Lavine G. New drug to restore bowel function approved under new FDA rules. Am J Health Syst Pharm. 2008;65(13):1204. doi: 10.2146/news080052. [DOI] [PubMed] [Google Scholar]
- 26.Yuan CS, Doshan H, Charney MR, O’Connor M, Karrison T, Maleckar SA, Israel RJ, Moss J. Tolerability, gut effects, and pharmacokinetics of methylnaltrexone following repeated intravenous administration in humans. J Clin Pharmacol. 2005;45(5):538–546. doi: 10.1177/0091270004273491. [DOI] [PubMed] [Google Scholar]
- 27.Chappelle R. US Food and Drug Administration. 2008. p. 1. [Google Scholar]
- 28.Greiner E, Spetea M, Krassnig R, Schullner F, Aceto M, Harris LS, Traynor JR, Woods JH, Coop A, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 18. N-substituted 14-phenylpropyloxymorphinan-6-ones with unanticipated agonist properties: extending the scope of common structure-activity relationships. J Med Chem. 2003;46(9):1758–1763. doi: 10.1021/jm021118o. [DOI] [PubMed] [Google Scholar]
- 29.Bentley KW, Horsewood P, Kirby GW, Singh S. Diels-Alder adducts from thebaine and nitroso-arenes. Journal of the Chemical Society D: Chemical Communications. 1969;(23):1411a–1411a. [Google Scholar]
- 30.Nagase H, Schmidhammer H, Spetea M. Chemistry of Opioids. Vol. 299. Springer; Berlin/Heidelberg: pp. 63–91. [DOI] [PubMed] [Google Scholar]
- 31.Nagase H, Lewis J, Husbands S. Chemistry of Opioids. Vol. 299. Springer; Berlin/Heidelberg: pp. 93–119. [Google Scholar]
- 32.Li G, Aschenbach LC, He H, Selley DE, Zhang Y. 14-O-Heterocyclic-substituted naltrexone derivatives as non-peptide mu opioid receptor selective antagonists: design, synthesis, and biological studies. Bioorg Med Chem Lett. 2009;19(6):1825–1829. doi: 10.1016/j.bmcl.2008.12.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lattanzi R, Spetea M, Schullner F, Rief SB, Krassnig R, Negri L, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 22. (1) Influence of the 14-alkoxy group and the substitution in position 5 in 14-alkoxymorphinan-6-ones on in vitro and in vivo activities. J Med Chem. 2005;48(9):3372–3378. doi: 10.1021/jm040894o. [DOI] [PubMed] [Google Scholar]
- 34.Schmidhammer H, Aeppli L, Atwell L, Fritsch F, Jacobson AE, Nebuchla M, Sperk G. Synthesis and biological evaluation of 14-alkoxymorphinans. 1. Highly potent opioid agonists in the series of (−)-14-methoxy-N-methylmorphinan-6-ones. J Med Chem. 1984;27(12):1575–1579. doi: 10.1021/jm00378a009. [DOI] [PubMed] [Google Scholar]
- 35.Spetea M, Toth F, Schutz J, Otvos F, Toth G, Benyhe S, Borsodi A, Schmidhammer H. Binding characteristics of [3H]14-methoxymetopon, a high affinity mu-opioid receptor agonist. Eur J Neurosci. 2003;18(2):290–295. doi: 10.1046/j.1460-9568.2003.02744.x. [DOI] [PubMed] [Google Scholar]
- 36.Furst Z, Buzas B, Friedmann T, Schmidhammer H, Borsodi A. Highly potent novel opioid receptor agonist in the 14-alkoxymetopon series. Eur J Pharmacol. 1993;236(2):209–215. doi: 10.1016/0014-2999(93)90591-5. [DOI] [PubMed] [Google Scholar]
- 37.King MA, Su W, Nielan CL, Chang AH, Schutz J, Schmidhammer H, Pasternak GW. 14-Methoxymetopon, a very potent mu-opioid receptor-selective analgesic with an unusual pharmacological profile. Eur J Pharmacol. 2003;459(2–3):203–209. doi: 10.1016/s0014-2999(02)02821-2. [DOI] [PubMed] [Google Scholar]
- 38.Furst Z, Buzas B, Friedmann T, Schmidhammer H, Borsodi A. Highly potent novel opioid receptor agonist in the 14-alkoxymetopon series. Eur J Pharmacol. 1993;236(2):209–215. doi: 10.1016/0014-2999(93)90591-5. [DOI] [PubMed] [Google Scholar]
- 39.Schutz J, Spetea M, Koch M, Aceto MD, Harris LS, Coop A, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 20. 14-phenylpropoxymetopon: an extremely powerful analgesic. J Med Chem. 2003;46(19):4182–4187. doi: 10.1021/jm030878b. [DOI] [PubMed] [Google Scholar]
- 40.Compton WM, Volkow ND. Major increases in opioid analgesic abuse in the United States: concerns and strategies. Drug Alcohol Depend. 2006;81(2):103–107. doi: 10.1016/j.drugalcdep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 41.Husbands SM, Lewis JW. Opioid ligands having delayed long-term antagonist activity: potential pharmacotherapies for opioid abuse. Mini Rev Med Chem. 2003;3(2):137–144. doi: 10.2174/1389557033405395. [DOI] [PubMed] [Google Scholar]
- 42.Spetea M, Schullner F, Moisa RC, Berzetei-Gurske IP, Schraml B, Dorfler C, Aceto MD, Harris LS, Coop A, Schmidhammer H. Synthesis and biological evaluation of 14-alkoxymorphinans. 21. Novel 4-alkoxy and 14-phenylpropoxy derivatives of the mu opioid receptor antagonist cyprodime. J Med Chem. 2004;47(12):3242–3247. doi: 10.1021/jm031126k. [DOI] [PubMed] [Google Scholar]
- 43.Biyashev D, Monory K, Benyhe S, Schütz J, Koch M, Schmidhammer H, Borsodi A. Novel delta-opioid-receptor-selective ligands in the14-alkoxy-substituted indolo- and benzofuromorphinan series. Helv Chim Acta. 2001;84:2015–2021. [Google Scholar]
- 44.Linner KM, Stickney BJ, Quist HE, Sharp BM, Portoghese PS. The delta1-opioid receptor antagonist, 7-(benzospiroindanyl)naltrexone [correction of 7-benzylspiroindanylnaltrexone], prolongs renal allograft survival in a rat model. Eur J Pharmacol. 1998;354(1):R3–5. doi: 10.1016/s0014-2999(98)00477-4. [DOI] [PubMed] [Google Scholar]
- 45.Spetea M, Harris HE, Berzetei-Gurske IP, Klareskog L, Schmidhammer H. Binding, pharmacological and immunological profiles of the delta-selective opioid receptor antagonist HS 378. Life Sci. 2001;69(15):1775–1782. doi: 10.1016/s0024-3205(01)01271-1. [DOI] [PubMed] [Google Scholar]
- 46.D’Ambrosio A, Noviello L, Negri L, Schmidhammer H, Quintieri F. Effect of novel non-peptidic delta opioid receptor antagonists on human T and B cell activation. Life Sci. 2004;75(1):63–75. doi: 10.1016/j.lfs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Gaveriaux-Ruff C, Filliol D, Simonin F, Matthes HW, Kieffer BL. Immunosuppression by delta-opioid antagonist naltrindole: delta- and triple mu/delta/kappa-opioid receptor knockout mice reveal a nonopioid activity. J Pharmacol Exp Ther. 2001;298(3):1193–1198. [PubMed] [Google Scholar]
- 48.Lewis JW, Smith CFC, McCarthy PS, Walter D, Kobylecki RJ, Myers M, Haynes AS, Lewis CJ, Waltham K. New 14-aminomorphinones and codeinones. NIDA Res Monograph. 1988;80:136–143. [PubMed] [Google Scholar]
- 49.Zernig G, Burke T, Lewis JW, Woods JH. Mechanism of clocinnamox blockade of opioid receptors: evidence from in vitro and ex vivo binding and behavioral assays. J Pharmacol Exp Ther. 1996;279(1):23–31. [PubMed] [Google Scholar]
- 50.Comer SD, Burke TF, Lewis JW, Woods JH. Clocinnamox: a novel, systemically-active, irreversible opioid antagonist. J Pharmacol Exp Ther. 1992;262(3):1051–1056. [PubMed] [Google Scholar]
- 51.Woods JH, Lewis JW, Winger G, Butelman E, Broadbear J, Zernig G. Methoclocinnamox: a μ partial agonist with pharmacotherapeutic potential for heroin abuse. NIDA Res Monograph. 1995;147:195–219. [PubMed] [Google Scholar]
- 52.Aceto MD, Bowman ER, May EL, Harris LS, Woods JH, Smith CB, Medzihradsky F, Jacobson AE. Very long-acting narcotic antagonists: the 14 beta-p-substituted cinnamoylaminomorphinones and their partial mu agonist codeinone relatives. Arzneimittelforschung. 1989;39(5):570–575. [PubMed] [Google Scholar]
- 53.McLaughlin JP, Hill KP, Jiang Q, Sebastian A, Archer S, Bidlack JM. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of mu-selective agonist and antagonist activity. J Pharmacol Exp Ther. 1999;289(1):304–311. [PubMed] [Google Scholar]
- 54.Cowan A, Lewis JW. Buprenorphine. Whiley-Liss, Inc; New York: 1995. [Google Scholar]
- 55.Nieland NP, Moynihan HA, Carrington S, Broadbear J, Woods JH, Traynor JR, Husbands SM, Lewis JW. Structural determinants of opioid activity in derivatives of 14-aminomorphinones: effect of substitution in the aromatic ring of cinnamoylaminomorphinones and codeinones. J Med Chem. 2006;49(17):5333–5338. doi: 10.1021/jm0604777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Husbands SM, Sadd J, Broadbear JH, Woods JH, Martin J, Traynor JR, Aceto MD, Bowman ER, Harris LS, Lewis JW. 3-Alkyl ethers of clocinnamox: delayed long-term mu-antagonists with variable mu efficacy. J Med Chem. 1998;41(18):3493–3498. doi: 10.1021/jm9810248. [DOI] [PubMed] [Google Scholar]
- 57.Derrick I, Neilan CL, Andes J, Husbands SM, Woods JH, Traynor JR, Lewis JW. 3-Deoxyclocinnamox: the first high-affinity, nonpeptide mu-opioid antagonist lacking a phenolic hydroxyl group. J Med Chem. 2000;43(17):3348–3350. doi: 10.1021/jm0009641. [DOI] [PubMed] [Google Scholar]
- 58.Archer S, Seyed-Mozaffari A, Jiang Q, Bidlack JM. 14 alpha,14′ beta-[Dithiobis[(2-oxo-2,1-ethanediyl)imino]]bis (7,8-dihydromorphinone) and 14 alpha,14′ beta-[dithiobis[(2-oxo-2,1- ethanediyl)imino]]bis[7,8-dihydro-N-(cyclopropylmethyl)normorphinone]: chemistry and opioid binding properties. J Med Chem. 1994;37(11):1578–1585. doi: 10.1021/jm00037a008. [DOI] [PubMed] [Google Scholar]
- 59.Jiang Q, Seyed-Mozaffari A, Archer S, Bidlack JM. Antinociceptive properties of two alkylating derivatives of morphinone: 14 beta-(thioglycolamido)-7,8-dihydromorphinone (TAMO) and 14 beta-(bromoacetamido)-7,8-dihydromorphinone (H2BAMO) J Pharmacol Exp Ther. 1992;262(2):526–531. [PubMed] [Google Scholar]
- 60.Gatch MB, Negus SS, Mello NK, Archer S, Bidlack JM. Effects of the structurally novel opioid 14 alpha, 14′ beta-[dithiobis [(2-oxo-2,1-ethanediyl)imino]]bis(7,8-dihydromorphinone) on schedule-controlled behavior and thermal nociception in rhesus monkeys. J Pharmacol Exp Ther. 1996;278(3):1282–1289. [PubMed] [Google Scholar]
- 61.Jiang Q, Seyed-Mozaffari A, Archer S, Bidlack JM. Pharmacological study of 14 beta-(thioglycolamido)-7,8-dihydro-N(cyclopropylmethyl)-normor phinone (N-CPM-TAMO) J Pharmacol Exp Ther. 1993;264(3):1021–1027. [PubMed] [Google Scholar]
- 62.Ling W, Wesson DR. Drugs of abuse--opiates. West J Med. 1990;152(5):565–572. [PMC free article] [PubMed] [Google Scholar]
- 63.Goodman AJ, Le Bourdonnec B, Dolle RE. Mu opioid receptor antagonists: recent developments. ChemMedChem. 2007;2(11):1552–1570. doi: 10.1002/cmdc.200700143. [DOI] [PubMed] [Google Scholar]