

Abstract
Hydroxamic acid is a potent moiety not only in the field of cancer therapy but also as a mutagenic agent. Among the various derivatives of hydroxamic acid, SAHA (Suberoylanilide Hydroxamic Acid) is considered as a potent anticancer agent. Scientists from the different corner synthesized different hydroxamic acid moieties with some straight chain oxazole, thiadiazole, biphenyl moieties in the terminal position. Acetylation and deacetylation of histones of the core proteins of nucleosomes in chromatin play an important role in the regulation of gene expression. The level of acetylation of histones is established and maintained by two classes of enzymes, histone acetyltransferase and histone deacetylases, which have been identified as transcriptional coactivators and transcriptional corepressors, respectively. There is increasing evidence that aberrant histone acetylation has been linked to various malignant diseases. Great efforts are currently underway for the design of more potent and less toxic candidates for the treatment of cancer. In recent years, hydroxamic acid derivatives have attracted increasing attention for their potential as highly efficacious in combating various etiological factors associated with cancer. Our main intention to draw an attention is that this single functional moiety has not only fit in the receptor but also create a diversified activity.
Keywords: Cancer, histone deacetylase, hydroxamic acid, MMP inhibitor
INTRODUCTION
For the last four decades, a number of potential approaches have been proposed for the treatment of cancer. Several targets have already been identified in the process of novel anticancer therapy. One of the recent targets in which nowadays the scientists are showing their maximum interest is Histone Deacetylase (HDAC) enzyme. Great efforts are currently underway for the design of more potent and less toxic candidates for the treatment of cancer. In recent years, hydroxamic acid derivatives have attracted increasing attention for their potential as highly efficacious in combating various etiological factors associated with cancer. Several attempts have already been made prior to synthesizing novel compounds irrespective of the targets; they are molecular modeling, molecular docking, QSAR studies, etc. Among them, molecular docking study was found to be a promising tool as compared with High Throughput Screening (HTS) in overcoming the limitations of unprecedented number of novel leads.
HISTONE DEACETYLASE STRUCTURAL FEATURES
HDAC enzyme was found to be one of the leading targets in the process of anticancer drug development. HDAC have been divided into three distinct structural classes as class (i/ii) zinc dependent and class (iii) as NAD (Nicotinamide adenine dinucleotide) dependent. These enzymes are a part of multiprotein complexes, catalyzing the removal of acetyl group from lysine residue on protein, including histones. HDAC inhibitors have shown to bind the active site and block the substrate access, causing a resultant accumulation of acetylated histones. HDACIs (Histone deacetylase inhibitors) induce tumor growth inhibition, cell differentiation, and programmed cell death. HDACIs induce cancer cell cycle arrest, growth inhibition, differentiation, and programmed cell death. HDACI-induced cell cycle arrest and growth inhibition is usually correlated with transcriptional activation of p21WAF1/CIP1 (cyclin-dependent kinase inhibitor 1 or CDK-interacting protein), p27KIP1 (a cell-cycle regulatory protein that Interacts with cyclin-CDK2 and -CDK4, inhibiting cell cycle progression at G1), GADD45 (Growth Arrest and DNA Damage), inhibition of cyclin A, cyclin D, and thymidylate synthetase.[1]
HYDROXAMIC ACID AS ANTICANCER
Ragno et al. synthesized a new class of synthetic HDACIs, 3-(4-Aroyl-1-methyl- 1H-pyrrol-2-yl)-N-hydroxy-2-propenamides[2] which show minimal inhibitory concentration against HDAC in restricting the over expression of HDAC.
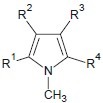
Kim et al. developed a new class of HDACIs, 3-(4-Substituted-phenyl)-N-hydroxy-2-propenamides by incorporating 1, 4-phenylene carboxamide as linker shows greater HDAC inhibition.[3]
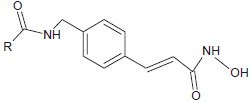
Lu et al. developed N-hydroxy-4-(4-phenylbutyryl-amino)benzamide,[4] a hydroxamate-tethered phenylbutyrate derivative with sub-micromolar potency in inhibiting HDAC activity and cancer cell proliferation.
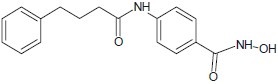
Belvedere et al. synthesized vorinostat-derived series of substrate-based HDAC inhibitors and evaluated their inhibitory potential.[5]
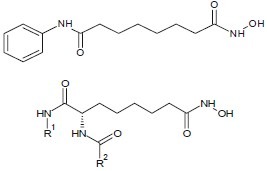
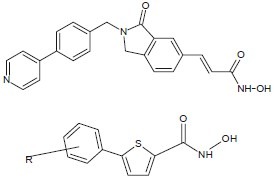
Lee et al. synthesized and evaluated hydroxamic acid derivatives, bearing a 4-(3-pyridyl)phenyl group as a cap structure which exhibited potent HDAC inhibitory activity.[6]
Price et al.[7] investigated a series of thienyl-based hydroxamic acids[7] that include ADS100380 and ADS102550, which led to the identification of the 5-pyridin-2-yl-thiophene-2-hydroxamic acid, possessed modest HDAC inhibitory activity.
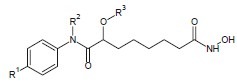
Hanessian et al. developed a series of ω-alkoxy ether by altering the chain length from six to eight of Suberoylanilide Hydroxamic Acid (SAHA).[8]
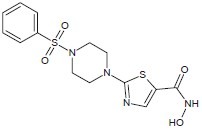
A series of structurally novel hydroxamic acid-based HDACIs were synthesized by Anandan et al., where a zinc chelating group attached to a thiazole ring connecting the piperazine spacer and sulfonamide cap.[9]
Dallavalle et al. designed, synthesized, and evaluated a series of hydroxamic acid-based histone deacetylase (HDAC) inhibitors characterized by a cinnamic spacer, capped with a substituted phenyl group.[10]
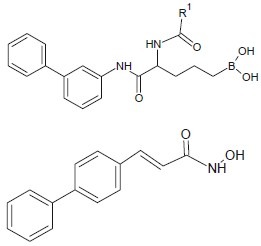
Suzuki et al. synthesized a series of boronic acid-based HDAC inhibitors, bearing an R-amino acid moiety which show some potent HDAC inhibitory activity.[11]
Charrier et al. synthesized and modeled new benzofuranone HDACIs that stimulate tumor suppressor gene expression as well as very effective in case of cancerous cell.[12]
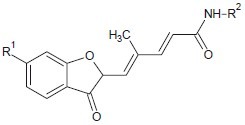
Koncic et al. examined few hydroxamic acid derivatives of nonsteroidal anti-inflammatory drugs and evaluated their antioxidant, radical scavenging activity with reference to BHA (Butylated Hydroxyanisole).[13]
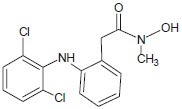
Lu et al. developed N-hydroxy-4-(4-phenylbutyryl-amino)benzamide, a hydroxamate-tethered phenylbutyrate derivative with sub-micromolar potency in inhibiting HDAC activity and cancer cell proliferation.[14]
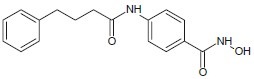
Zhou et al. designed and synthesized a N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide[15] which blocked cancer cell proliferation and induces histone acetylation.
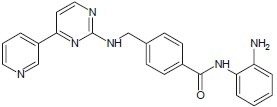
Pabba et al. developed a novel hydroxamic acid derivatives with aryl ether and aryl sulfone residues at the terminus of a substituted with unsaturated 5-carbon spacer moiety which showed greater HDAC inhibitory activity.[16]
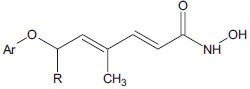
Angibaud et al. developed a series of pyrimidyl-5-hydroxamic acids which was prepared for evaluation as inhibitors of HDAC. Amino-2-pyrimidinyl can be used as a linker to provide HDAC inhibitors of good enzymatic potency.[17]
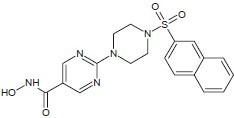
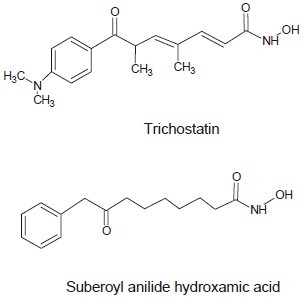
Codd et al. discovered SAHA (vorinostat, Zolinza®) and trichostatin A are inhibitors of the Zn(II)-dependent class I and class II HDACs resulting inhibition of Zn(II)-dependent HDACs that relaxes the chromatin structure and upregulates transcription.[18]
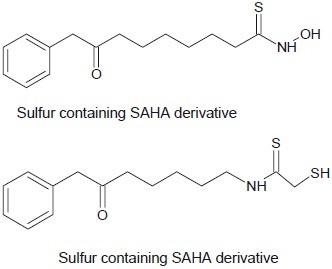
Gu et al. developed a new series of carbonyl and sulfur-containing analogs of SAHA as a potent inhibition of HDACIs.[19]
Rajak et al. developed two novel series of 2-[5-(4-substitutedphenyl)-[1,3,4]-oxadiazol/thiadiazol-2-ylamino]-pyrimidine-5-carboxylic acid[20] (tetrahydro-pyran-2-yloxy)-amides which were designed and synthesized as novel hydroxamic acid-based HDACIs.
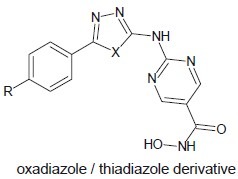
Jiao et al. developed a novel series of N-hydroxy-4-(3-phenylpropanamido)benzamide derivatives comprising N-hydroxybenzamide group as zinc-chelating moiety which showed greater HDAC inhibitory activity.[21]
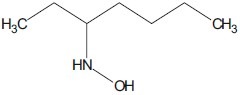
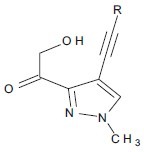
Mshvidobadze et al. synthesized various pyrazolo hydroxamic acid molecules which showed greater efficacy against cancer.[22]
Shen et al. suggested that both the resistance of tumor cells to cisplatin and dose-related toxicity remain two of the most important problems in the chemotherapy of clinical oral squamous cell carcinoma. Cell viability and apoptotic assay were examined.[23]
Hrubec et al. described new linker-less HDAC inhibitors designed to exploit a unique sub-pocket in the HDAC8 active site which showed minimal concentration to inhibit apoptotic cells.[24]
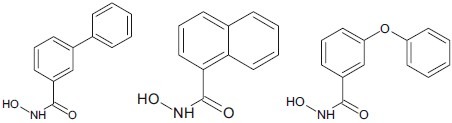
Various derivatives of hydroxamic acid molecule such as pyridyl hydroxamic acid, biphenyl hydroxamic acid, or the other substituted heterocyclic ring associated with hydroxamic acid has not only a great impact against HDAC but also human ovarian cancer, non-small cell lung cancer, and breast cancer cell line [Table 1].
Table 1.
Different hydroxamic acid derivatives as HDAC inhibitor
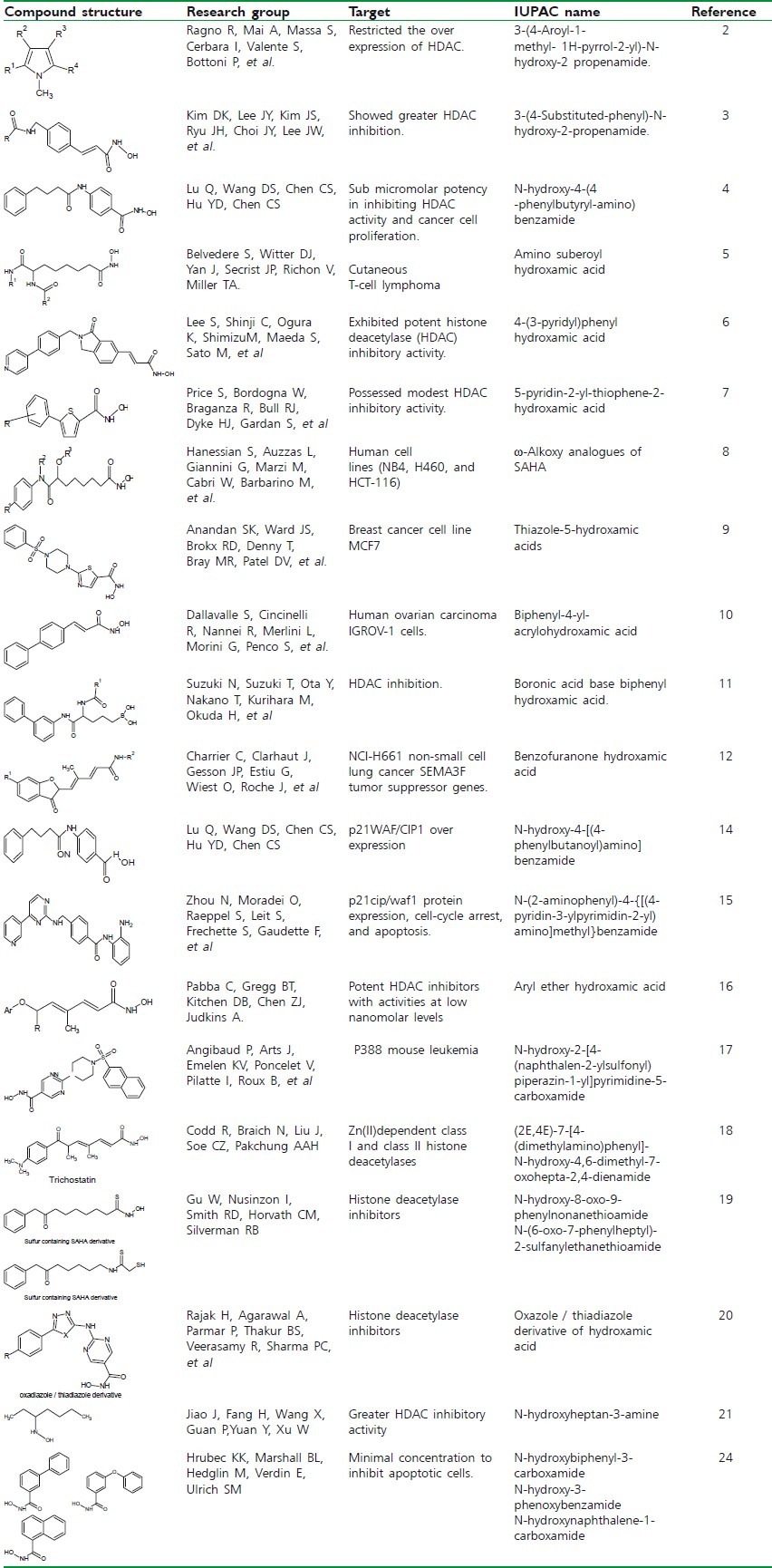
HYDROXAMIC ACID AS MUTAGENIC AND MMP (MATRIX METALLOPROTEINASE) INHIBITOR
Wang et al. evaluated the mutagenicity and antibacterial activity of a series of hydroxamic acids over a huge diversified range of bacterial strains. Among them, Salicylhydroxamic acid has shown antitubercular activity both in vivo and in vitro.[25]
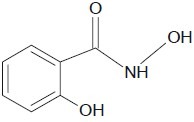
An in vitro antimicrobial evaluation was carried out by Davis et al. for cyclic hydroxamic acid and related lactams which showed the maximal antibacterial effects against E. aerogenes 13048, S. marcescens 13880, K. pneumoniae 10031, P. aeruginosa 10145, E. coli 9723, etc.[26]
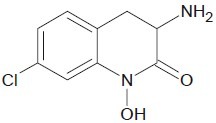
Brien et al. developed a complex with iron(III), copper(II), nickel(II), and zinc(II) complexes of salicylhydroxamic acid, anthranilic hydroxamic acid, and benzohydroxamic acid which showed some hydroxamate-peroxidase inhibitor interactions.[27]
Kokare et al. successfully used a novel reagent, phosphoric acid diethyl ester 2-phenyl-benzimidazol-1-yl ester,[28] that was designed and synthesized and its applicability was demonstrated for the preparation of O-alkyl hydroxamic acids. The O-alkyl hydroxamic acids of N-protected amino acids were also synthesized.
Reiter et al. used a series of sterically hindered sulfonamide hydroxamic acids which are the potent inhibitors of MMP-13. The metabolically more stable compounds in the series contain either a monocyclic or bicyclic pyran ring adjacent to the hydroxamate group.[29]
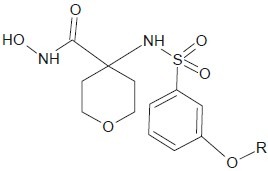
Tommasi et al. discovered a series of hydroxamic acid inhibitors of MMP-13 that do not significantly inhibit MMP-2 (gelatinase-1). MMP-2 has been implicated in the musculoskeletal side effects resulting from pan-MMP inhibition due to findings from spontaneously occurring human MMP-2 deletions.[30]
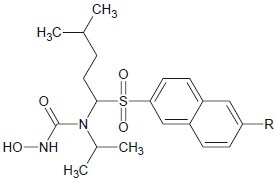
Sulfonamide hydroxamic acid molecule has a great influence against metalloproteinase enzyme and if that molecule was further substituted with naphthyl molecule, it also shows a greater affinity to that enzyme [Table 2].
Table 2.
Different hydroxamic acid derivatives as mutagenic and MMP inhibitor
CONCLUSION
So, hydroxamic acid molecule has a greater impact in the field of pharmaceutical chemistry, though it has not only used as an anticancer but also for other mutagenic problems. From the total review, we can conclude that there was a diversified activity profile that occurred due to the changes in the various position of a molecule with the same hydroxamic acid group on the terminal. So, this single molecule can work further more and more with various other compatible groups and not only in the field of cancer therapy but also other associated diseases.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
REFERENCES
- 1.Mai A, Massa S, Lavu S, Pezzi R, Simeoni S, Ragno R. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J Med Chem. 2005;48:7789–95. doi: 10.1021/jm050100l. [DOI] [PubMed] [Google Scholar]
- 2.Ragno R, Mai A, Massa S, Cerbara I, Valente S, Bottoni P, et al. 3-(4-Aroyl-1-methyl-1H-pyrrol-2-yl)-N-hydroxy-2-propenamides as a New Class of Synthetic Histone Deacetylase Inhibitors. 3. Discovery of Novel Lead Compounds through Structure-Based Drug Design and Docking Studies. J Med Chem. 2004;47:1351–9. doi: 10.1021/jm031036f. [DOI] [PubMed] [Google Scholar]
- 3.Kim DK, Lee JY, Kim JS, Ryu JH, Choi JY, Lee JW, et al. Synthesis and Biological Evaluation of 3-(4-Substituted-phenyl)-N-hydroxy-2-propenamides, a New Class of HistoneDeacetylase Inhibitors. J Med Chem. 2003;46:5745–51. doi: 10.1021/jm030377q. [DOI] [PubMed] [Google Scholar]
- 4.Lu Q, Wang DS, Chen CS, Hu YD, Chen CS. Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J Med Chem. 2005;48:5530–5. doi: 10.1021/jm0503749. [DOI] [PubMed] [Google Scholar]
- 5.Belvedere S, Witter DJ, Yan J, Secrist JP, Richon V, Miller TA. Aminosuberoylhydroxamic acids (ASHAs): A potent new class of HDAC inhibitors Bioorg. Med Chem Lett. 2007;17:3969–71. doi: 10.1016/j.bmcl.2007.04.089. [DOI] [PubMed] [Google Scholar]
- 6.Lee S, Shinji C, Ogura K, Shimizu M, Maeda S, Sato M, et al. Design, synthesis, and evaluation of isoindolinone-hydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Bioorg Med Chem Lett. 2007;17:4895–900. doi: 10.1016/j.bmcl.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 7.Price S, Bordogna W, Braganza R, Bull RJ, Dyke HJ, Gardan S, et al. Identification and optimisation of a series of substituted 5-pyridin-2-yl-thiophene-2-hydroxamic acids as potenthistone deacetylase (HDAC) inhibitors. Bioorg Med Chem Lett. 2007;17:363–9. doi: 10.1016/j.bmcl.2006.10.045. [DOI] [PubMed] [Google Scholar]
- 8.Hanessian S, Auzzas L, Giannini G, Marzi M, Cabri W, Barbarino M, et al. ω-Alkoxy analogues of SAHA (vorinostat) as inhibitors of HDAC: A study of chain-length and stereochemical dependence. Bioorg Med ChemLett. 2007;17:6261–5. doi: 10.1016/j.bmcl.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 9.Anandan SK, Ward JS, Brokx RD, Denny T, Bray MR, Patel DV, et al. Design and synthesis of thiazole-5-hydroxamic acids as novel histone deacetylase inhibitors. Bioorg Med ChemLett. 2007;17:5995–9. doi: 10.1016/j.bmcl.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 10.Dallavalle S, Cincinelli R, Nannei R, Merlini L, Morini G, Penco S, et al. Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur. J Med Chem. 2009;44:1900–12. doi: 10.1016/j.ejmech.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki N, Suzuki T, Ota Y, Nakano T, Kurihara M, Okuda H, et al. Design, synthesis, and biological activity of boronic acid-based histone deacetylase inhibitors. J Med Chem. 2009;52:2909–22. doi: 10.1021/jm900125m. [DOI] [PubMed] [Google Scholar]
- 12.Charrier C, Clarhaut J, Gesson JP, Estiu G, Wiest O, Roche J, et al. Synthesis and modeling of new benzofuranone histone deacetylase inhibitors that stimulate tumor suppressor gene expression. J Med Chem. 2009;52:3112–5. doi: 10.1021/jm9002439. [DOI] [PubMed] [Google Scholar]
- 13.Koncic MZ, Rajic Z, Petric N, Zorc B. Antioxidant activity of NSAID hydroxamic acids. Acta Pharm. 2009;59:235–42. doi: 10.2478/v10007-009-0017-8. [DOI] [PubMed] [Google Scholar]
- 14.Lu Q, Wang DS, Chen CS, Hu YD, Chen CS. Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J Med Chem. 2005;48:5530–5. doi: 10.1021/jm0503749. [DOI] [PubMed] [Google Scholar]
- 15.Zhou N, Moradei O, Raeppel S, Leit S, Frechette S, Gaudette F, et al. Discovery of N-(2-Aminophenyl)- 4- [(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide (MGCD0103),an Orally Active Histone Deacetylase Inhibitor. J Med Chem. 2008;51:4072–5. doi: 10.1021/jm800251w. [DOI] [PubMed] [Google Scholar]
- 16.Pabba C, Gregg BT, Kitchen DB, Chen ZJ, Judkins A. Design and synthesis of aryl ether and sulfonehydroxamic acids as potent histone deacetylase (HDAC) inhibitors. Bioorg Med Chem Lett. 2011;21:324–8. doi: 10.1016/j.bmcl.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Angibaud P, Arts J, Emelen KV, Poncelet V, Pilatte I, Roux B, et al. Discovery of pyrimidyl-5-hydroxamic acids as new potent histone deacetylase inhibitors. Eur J Med Chem. 2005;40:597–606. doi: 10.1016/j.ejmech.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Codd R, Braich N, Liu J, Soe CZ, Pakchung AA. Zn(II)-dependent histone deacetylase inhibitors: Suberoylanilidehydroxamic acid and trichostatin A. Int J Biochem Cell Biol. 2009;41:736–9. doi: 10.1016/j.biocel.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 19.Gu W, Nusinzon I, Smith RD, Horvath CM, Silverman RB. Carbonyl- and sulfur-containing analogs of suberoylanilidehydroxamic acid: Potent inhibition of histone deacetylases. Bioorg Med Chem. 2006;14:3320–9. doi: 10.1016/j.bmc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 20.Rajak H, Agarawal A, Parmar P, Thakur BS, Veerasamy R, Sharma PC, et al. 2,5-Disubstituted-1,3,4-oxadiazoles/thiadiazole as surface recognition moiety: Design and synthesis of novel hydroxamic acid based histone deacetylase inhibitors. Bioorg Med Chem Lett. 2011;21:5735–8. doi: 10.1016/j.bmcl.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 21.Jiao J, Fang H, Wang X, Guan P, Yuan Y, Xu W. Design, synthesis and preliminary biological evaluation of N-hydroxy-4-(3-phenylpropanamido)benzamide (HPPB) derivatives as novel histonedeacetylase inhibitors. Eur J Med Chem. 2009;30:1–7. doi: 10.1016/j.ejmech.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 22.Mshvidobadze EV, Vasilevskya SF, Elguero J. A new route to pyrazolo[3,4-c] and [4,3-c]pyridinones via heterocyclization of vic-substituted hydroxamic acids of acetylenylpyrazoles. Tetrahedron. 2004;60:11875–8. [Google Scholar]
- 23.Shen J, Huang C, Jiang L, Gao F, Wang Z, Zhang Y, et al. Enhancement of cisplatin induced apoptosis by suberoylanilidehydroxamic acid in human oral squamous cell carcinoma cell lines. Bio Pharmaco. 2007;73:1901–9. doi: 10.1016/j.bcp.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Hrubec KK, Marshall BL, Hedglin M, Verdin E, Ulrich SM. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg Med Chem Lett. 2007:2874–8. doi: 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 25.Wang CY, Lee LH. A. Mutagenicity and antibacterial activity of hydroxamic acids. AmSocMicrobiol. 1977;11:753–5. doi: 10.1128/aac.11.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis A, Hulme KL, Wilson GT, Mccord TJ. In vitro antimicrobial activity of some cyclic hydroxamic acids and related lactams. AmSocMicrobiol. 1978;13:542–4. doi: 10.1128/aac.13.3.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brien EC, Farkas E, Gil MJ, Fitzgerald D, Castineras A, Nolan KB. Metal complexes of salicylhydroxamic acid (H2Sha), anthranilichydroxamic acid and benzohydroxamic acid. Crystal and molecular structure of [Cu(phen)2(Cl)]ClPH2Sha, a model for a peroxidase inhibitor complex. J Inorg Biochem. 2000;79:47–51. doi: 10.1016/s0162-0134(99)00245-7. [DOI] [PubMed] [Google Scholar]
- 28.Kokare ND, Nagawade RR, Ranea VP, Shinde DB. Design, synthesis and utilization of a novel coupling reagent for the preparation of O-alkyl hydroxamic acids. Tetrahedron Lett. 2007;48:4437–40. [Google Scholar]
- 29.Reiter LA, Robinson RP, McClure KF, Jones CS, Reese MR, Mitchell PG. Pyran-containing sulfonamide hydroxamic acids: potent MMP inhibitors that spare MMP-1. Bioorg Med Chem Lett. 2004;14:3389–95. doi: 10.1016/j.bmcl.2004.04.083. [DOI] [PubMed] [Google Scholar]
- 30.Tommasi RA, Weiler S, McQuire LW, Rogel O, Chambers M, Clark K, et al. Potent and selective 2-naphthylsulfonamide substituted hydroxamic acid inhibitors of matrix metalloproteinase-13. Bioorg Med Chem Lett. 2011;21:6440–5. doi: 10.1016/j.bmcl.2011.08.087. [DOI] [PubMed] [Google Scholar]