

Abstract
Adrenocortical carcinomas are rare tumors with an incidence of one to two cases per million population and are still more rarer in the pediatric age group. Adrenocortical carcinomas can be functional or may be unassociated with syndromes of hormone overproduction. It is very important to differentiate an adrenocortical adenoma from a carcinoma, as both share a large number of phenotypic features, and assess their prognosis, as adrenocortical carcinoma may need an adjuvant therapy. In this communication, we describe the case of a two-year-old boy, who presented with iso-sexual precocious puberty, having features of virilization, which included growth of facial and pubic hair, deepening of voice, and penile growth.
Keywords: Adrenocortical tumor, virilization, Weiss criteria
INTRODUCTION
Adrenocortical carcinoma (ACC) is a very rare entity in the pediatric population, and is rarely associated with virilizing symptoms. The gland is grossly enlarged and the phenotypic features overlap with an adrenocortical adenoma, which need to be evaluated carefully, so as to distinguish it from the latter, as complete surgical excision is curative in an adenoma unlike in ACC which may need adjuvant therapy and follow-up.
CASE REPORT
A two-year-old male child presented with features of virilization, which included growth of facial and pubic hair, deepening of voice, and penile growth for five months [Figure 1]. A palpable abdominal lump was also present. Bilateral testes were normal. The features of Cushing syndrome (moon face, pot belly, skin stria) were not present. Blood pressure was 128 / 74 mmHg. Ultrasonography showed a complex echogenic mass in the left paravertebral region. The child's testosterone and dehydroepiandrostenidione levels were elevated and the serum cortisol level was within normal limits [Table 1]. A provisional diagnosis of adrenocortical carcinoma was made, an adrenalectomy was done, and a histopathological diagnosis of adrenocortical carcinoma was made.
Figure 1.

Two-year-old male child with features of virilization (pubic hair and penile growth)
Table 1.
Preoperative and postoperative (after two months) hormonal profile
Pathology
Grossly, the adrenalectomy specimen measured 11 × 9 × 7 cm and the weighed 240 g. The external surface was unremarkable. The cut section was reddish brown with areas of hemorrhage. A narrow strip of normal adrenal tissue was seen compressed to the periphery. [Figure 2]
Figure 2.

Cut section of tumor showing reddish brown areas with areas of hemorrhage. A narrow strip of normal adrenal was seen compressed to the periphery
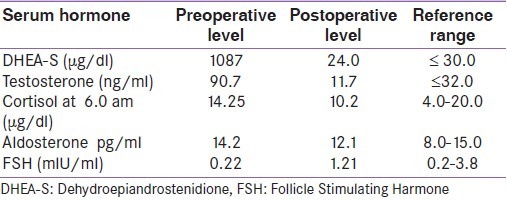
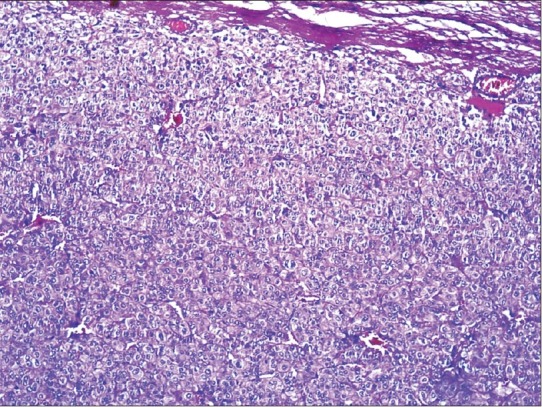
Microscopy revealed large cells with mild-to-moderate pleomorphism. The cells were arranged in sheets and trabaculae [Figure 3]; > 75% of the cells had abundant eosinophilic cytoplasm. The nucleus was mildly hyperchromatic with one to two nucleoli [Figure 4]. A majority of the tumor had a diffuse architecture, interspersed with areas of hemorrhage; there was no evidence of atypical mitosis or vascular and capsular invasion. A Weiss score of 3 was obtained.
Figure 3.
Photomicrograph showing cells arranged in sheets showing moderate pleomorphism. (H and E × 100)
Figure 4.
Photomicrograph showing cells having abundant eosinophilic cytoplasm and hyperchromatic nucleus with one-to-two nucleoli. (H and E × 400)
Based on the weight, size of the tumor, and microscopic features, and considering the criteria proposed by Weiss, a diagnosis of adrenocortical carcinoma was made. After the operation, the manifestations of virilization disappeared gradually, except for the lower voice. Serum androgens rapidly decreased to the normal level [Table 1]. The patient was asymptomatic after six months of follow-up.
DISCUSSION
Adrenocortical carcinoma is a rare pediatric neoplasm with poor prognosis. ACC and adrenocortical adenoma are the two ends of a spectrum that need to be differentiated. These carcinomas are also seen with increased frequency in children with the Li-Fraumeni syndrome, Beckwith-Wiedermann syndrome, and congenital adrenal hyperplasia.[1] A slight preponderance in the female population has been noted. ACC can be associated with Cushing's syndrome and minerlocorticoid production, although > 75% do not have any hormone overproduction. Virilizing adrenal carcinoma is a very rare entity.[2,3] Grossly, carcinomas, as compared to adenomas, are large and weigh more than 100 g. It has been stated that any adrenal tumor weighing more than 250 g, age > 3.5 years at diagnosis, and associated with symptoms of marked hormone overproduction, is to be considered malignant with poor prognosis. Pure virilizing carcinomas, in general, appear to have a better prognosis than other adrenal carcinomas.[4]
In ACC, the cut section of the gland is pink to yellow tan, with areas of hemorrhage and necrosis.[1] Microscopically, the tumor has different patterns of growth and as the distinction between benign and malignant neoplasms is difficult, the Weiss criteria, which require histopathological findings, are employed to distinguish the benign from the malignant tumors. This classification is based on the mitotic index, confluent necrosis, atypical mitosis, and nuclear grade, and a tumor is rendered as malignant when the score is three or more than three.[1,4,5] Our case has a score of 3, although the Weiss criteria is not useful in the pediatric population.[6] Hence, in our case, considering the weight and size of the gland, coupled with the microscopy, elevated hormonal levels, and the clinical symptoms, a diagnosis of ACC was made. In our case, good prognostic factors were, weight less than 250 g, age less than 3.5 years, and a pure virilizing form of carcinoma.
All adrenocortical carcinomas are highly malignant tumors, and hence, they need to be resected. Surgery is the single most important procedure for the successful treatment of ACC. The role of chemotherapy in the management of childhood ACC has not been established.[4]
Mitotane, an insecticide derivative that produces adrenocortical necrosis, has been extensively used in adults with ACC, but its efficacy in children who highlight a case of virilizing adrenocortical carcinoma, a very aggressive tumor, which is very rare in the pediatric age group and requires surgery, is not known.[4]
In this communication we highlight a rare case of paediatric virilising adrenocortical carcinoma.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Agrani P, Beckwith JB. Renal neoplasm of childhood. In: Mills SE, editor. Sternberg's Diagnostic surgical pathology. 4th ed. Philadelphia: Lippincott Williams and Wilkins; 2004. pp. 2016–9. [Google Scholar]
- 2.Pal DK, Kundu AK, Chakrabortty S, Das S. Virilizing adrenocortical carcinoma. J Indian Med Assoc. 2002;100:251–2. [PubMed] [Google Scholar]
- 3.Wooten MD, King DK. Adrenal cortical carcinoma: Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72:3145–52. doi: 10.1002/1097-0142(19931201)72:11<3145::aid-cncr2820721105>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 4.Riberio RC, Michalkiewicz EL, Figueiredo BC, DeLacerda L, Sandrini F, Pianovsky MD, et al. Adrenocortical tumors in children. Braz J Med Biol Res. 2000;33:1225–34. doi: 10.1590/s0100-879x2000001000013. [DOI] [PubMed] [Google Scholar]
- 5.Jain M, Kapoor S, Mishra A, Gupta S, Agarwal A. Weiss criteria in large adrenocortical tumors: A validation study. Indian J Pathol Microbiol. 2010;53:222–6. doi: 10.4103/0377-4929.64325. [DOI] [PubMed] [Google Scholar]
- 6.Budhwani KS, Ghritlaharey K, Debbarma MK. Adrenal cortex tumor in a six year old girl: A case report and review of literature. Indian J Med Paediatr Oncol. 2004;25:71–5. [Google Scholar]