

Abstract
Remodeling, a continuous physiological process maintains the strength of the bones, which maintains a delicate balance between bone formation and resorption process. This review gives an insight to the complex interaction and correlation between the bone remodeling and the corresponding changes in host immunological environment and also summarises the most recent developments occuring in the understanding of this complex field. T cells, both directly and indirectly increase the expression of receptor activator of nuclear factor kB ligand (RANKL); a vital step in the activation of osteoclasts, thus positively regulates the osteoclastogenesis. Though various cytokines, chemikines, transcription factors and co-stimulatory molecules are shared by both skeletal and immune systems, but researches are being conducted to establish and analyse their role and / or control on this complex but vital process. The understanding of this part of research may open new horizons in the management of inflammatory and autoimmune diseases, resulting into bone loss and that of osteoporosis also.
Keywords: Bone remodeling, immunoregulation, osteoblasts, osteoclasts, receptor activator of nuclear factor kB ligand
INTRODUCTION
Immune cells and bone cells have common site of origin i.e. bone marrow. Due to this approximity, they not only influence the genesis, maturation and activation but their functions also. It has been identified that various cytokines and signaling transducers participate in regulatory mechanism between immune and bone cells.
Bones are dynamic, viable, highly organised living tissue and is main constituent of musculoskeletal system. Though the main function of bones is to protect the internal structures and to provide support to various soft tissues, additionally they also provide haematopoiesis in bone marrow. Continuous adaption by the major and microarchitechture of the bone occurs in response to varying functional demands. This process of adaptation is known as ‘remodeling’. Bone modeling is a result of a balance between bone formation by osteoblasts and bone resorption by osteoclasts. It is a continuous process and at any given time approximately 5-25% of bone surface undergoes remodeling.[1] As normal physiological bone remodeling is imperative for the maintenance of bone strength and integrity, any imbalance will either lead to increase or decrease bone mass. By restoring the microdamages, remodeling ensures the mechanical integrity of the skeletal system, as well as regulates the release of calcium and phosphorus from the bones into the blood.
Remodeling involves four main processes: activation, resorption, reversal and formation.[2] It is initiated by the activation of the quiescent bone lining cells. Simultaneously the osteoclast precursor cells fuses to form mature osteoclasts. Osteoclasts initiate dissolution of calcified matrix with specific enzymes. As bone resorption subsides, osteoclasts disappear from resoption pits and mononuclear cells prepare the ground for new bone formation. Bone remodeling cycle ends with formation of bony canopy of these pits by the osteoblasts, keeping the material dormant until the next cycle.
BONE PHYSIOLOGY RELATED TO REMODELING
Bone is composed of extracellular matrix (ECM) and cells. ECM can be further subdivided into inorganic and organic part. The organic matrix is mainly constituted of the type I collagen (approximately 95%) and inorganic part predominantly containing calcium and phosphorus, appearing as hydroxyapatite crystals deposited in collagenous matrix. The major cells in bone are osteoclasts (responsible for resorption) and osteoblasts (responsible for bone tissue deposition). Osteoblasts do convert into osteocytes and bone lining cells. Fully differentiated osteoblasts forming bone lining cells are probably responsible for initiation of remodeling by degrading the matrix. The other fully differentiated osteoblasts are called osteocytes, which acts as mechanosensors (by communicating to each other by means of their dendritic processes) in bone tissue, thus regulating bone mass and bone structure.[3,4]
Osteoblasts synthesize collagen rich organic matrix and provided optimal conditions for mineralization by secreting various bone matrix proteins and matrix metalloproteinases (MMP).[5] Alkaline phosphatase, bone sialoprotein, osteocalcin and osteopontin expressed by mature osteoblasts while depositing matrix and its mineralization. Prominent golgi apparatus and endoplasmic retinaculum are universal features of osteoblasts primarily responsible for matrix and these are poor in osteocytes,[6] the bone cells.
Osteoclasts are tissue specific and derived from monocytes / macrophage hematopoietic lineage and are the only cells capable of breaking down mineralized bone, including calcified cartilage.[7,8] Osteoclasts attached tightly to matrix through integrins. They create an isolated lacuna, known as Howship's Lacuna with an acidic environment. This acidic environment is produced by release of acid – hydrolic acid formed by protons and chloride ions. This release occurs by the ruffled borders of osteoclasts formed by fusion of cytoplasmic acidophilic vacuoles. This results into resolution lacunae, responsible into rapid dissolution of hydroxyappetite crystals.[9]
REGULATION OF BONE REMODELING
Osteoblasts, Chondrocytes, Adipocytes, Stromal cells, Myoblasts and Tenocytes, all originate from a common progenitor cell, the mesenchymal stem cell (MSC). The development of bone cells has been supported by cells of immune system; such as macrophages encourage osteoblastogenesis by secreting interlukin-18 and T cells influences osteoclastogenesis by secreting interlukin-1, interlukin-6, interferon- γ and interlukin-4. Core binding factor 1 and Osterix are crucial transcription factors for osteoblast differentiation with lineage commitment.[10] It has been seen that core binding factor 1 deficient rats had only cartilaginous skeleton, indicating its importance in the development of osteoblasts development.[11] There are evidences that apoptotic osteocytes increase secretion of osteoclastic cytokines, increasing bone resorption.[12] The presence of Receptor Activator of Nuclear Factor kB Ligand (RANKL) and M-CSF (macrophage-colony – stimulating factor) are essential for formation and fusion of multinucleated cells, expressing osteoclasts specific markers such as tartrate-resistant acid phosphatase (TRAP), Cathepsin K, Calcitonin receptor (CTR) and Integrin receptors.[13,14]
It has been observed that the osteoclast-inducing capacity of activated T cells was mediated through the RANKL / Receptor activator of nuclear factor Kappa – B (RANK) / osteoprotegerin (OPG). RANKL is a key regulator of osteoclastogenesis and contributes significantly in bone remodeling.[15]
Bone remodeling is under influence of various local and systemic factors. Various prostaglandins, leukotrienes and hormones interact with bone cells. It has been established that Prostaglandin (PG) E2 and leukotrienes are metabolites of arachidonic acid in osteoblasts and have stimulatory as well as inhibitory effects on bone. It has been demonstrated that osteoclastogenesis is inhibited primarily by reduction of IL-1 induced COX2 activities and PGE2 production and only marginally by suppression of RANKL.[16,17] Parathyroid regulates the productions of IL-6 and RANKL by osteoblasts, thus facilitating differentiation, multiplication and formation of osteoclasts. Thus PTH and PTHrP (PTH related proteins) perform bone resorption to increase the blood level of calcium ions.[18–20]
Activated vitamin D, Calcitriol (1,25 dihydroxyvitamin D3) is essential for mineralization of osteoid. But it has been seen that they alone in presence of hypocalcemia will not be so helpful. Experiments have shown that vitamin D deficient mice had high numbers with lower number of osteoblasts and with high levels of alkaline phosphatase, because of the low production of RANKL and increased production of OPG.[21]
Sex hormones (oestrogen / progesterone / testosterone) do affects bone remodeling but their effects are apposite to PTH.[22] Their deficiency causes increased number of osteoblast precursor cells and as well as osteoclasts precursors with increased production of RANKL and decreased production of OPG. Decreased sex hormones also enhances the production of proinflamatory and pro-resorptive cytokines in lymphocytes such as IL-1, IL-6 and tumor necrosis factor-α (TNF-α).[23] It has been proved that the bone protective properties of estrogen is mediated by transforming growth factor-β (TGF-β), inducing apoptosis in osteoclasts.[24–26]
ROLE OF RANKL / OPG / RANK AXIS ON BONE REMODELING
RANKL and its receptors RANK and Osteoprotegerin (OPG); all belongs to super TNF family. Their discovery has raised the possibility to inhibit the development of osteoclasts.[27,28] Osteoprotegerin (OPG) a protein is secreted by stromal cells has a variety of possible functions (bone protection, endothelial cell survival and vascular calcification) as it is expressed in lung, heart, kidney, liver, thyroid, spinal cord, brain, intestine and bone.[29–34] RANKL protein and its m RNA too expressed in a variety of tissues, including bone, heart, brain, kidney, liver, lung, intestine, skeletal muscles, placenta, spleen, thymus and testis.[35] RANKL is mainly expressed in stromal cells / preosteoblasts as well as on activated T cells.[31,32] The human RANKL gene is located on chromosome 13q14 and encodes for three isoforms: RANKL1, RANKL2 and RANKL3. RANK deficiency represents the same phenotype as those with RANKL deficiency. However, nuclear factor of activated T cells c1 (NFATc1) is considered as master regulator of osteoclastogenesis.[36–40] Most important work of RANKL is the induction of osteoclastogenesis (bone remodeling).
ROLE OF CYTOKINES IN BONE REMODELLING
It has been established that IL-1α, IL-1β, IL-6, IL-7 and TNF-α directly and indirectly promote osteoclastogenesis,[41–44] where as interferon-β (IFN-β), IFN-γ, IL-3, IL-4, IL-10, IL-13, IL-12 alone or in synergy with IL-18, inhibits osteoclasts formation.[45–51] Among the osteoclastogenesis-inhibiting cytokines, interferons have attracted significant attention and thus well studied. IFN-γ and IFN-β, produced by activated T-cells, strongly suppress osteoclastogenesis by inhibiting RANKL. INF-β is induced by RANLK, but acts as negative regulator of RANKL signaling. IL-6 is produced by stromal cells /osteoblasts.[52] TGF- β, PTH, PTH related peptide (PTHrP), low density lipoprotein receptor related protein-5 (LRP-5) and osteopontein are known to promote osteoblats proliferation and differentiation.[53] Bone morphogenic protein-6 (BMP-6) is most potent regulator of osteoblasts differentiation.[54]
INFLUENCE OF LYMPHOCYTES ON REMODELLING
Lymphocytes (T cells) influence bone remodeling through its regulation on osteoclatogenesis [Figure 1]. Though the type and amount of impact of these lymphocytes on osteoclastogenesis varied from in vitro to in vivo, the same is true about the type of lymphocyte too. The data concerning the effect of CD4 and CD8 lymphocytes on osteoclastogenesis is not consistent. In one study,[55] it was showed that vitamin D3 stimulated osteoclast like cell formation was increased in presence of lymphocyte depletion. It was postulated that it was due to more PGE2 production, leading to upregulated RANKL and downregulated OPG expression. Resting T cells exert no effect on osteoclastogenesis.[55] In other studies, these resting T cells were found to exert a negative influence on osteoclastogenesis via production of granulocyte/monocyte colony stimulating factor (GM-CSF) and IFN-γ by CD-4 but not CD8 T cells.[56] Some other researchers showed in an in vitro study that a downregulation effect of lymphocytes, which was due to the CD8 T cell subset, and independent of IL-4 and TGF-β. On the other side, activated T cell promotes osteoclastogenesis. It was concluded that the osteoclastogenic effect was CD4+- dependent.[57]
Figure 1.
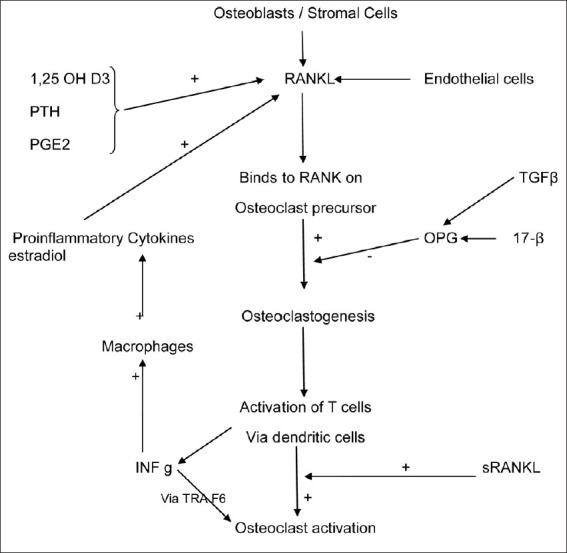
Cellular Regulation of Osteoclastogenesis
In mice, it was found that T cells are not only one which was required for osteoclastogesis. Fibroblasts, the main source of RANKL are stimulated by the proinflammatory cytokines. It has also been seen that RANKL is also produced by T cells.[57] As already described before, the RANKL regulates the differentiation of osteoclasts.
It has been established that the osteoblasts seem to have antigen-presenting properties, since these are shown to express MCHII molecules, besides the adhesions to CD54 and CD166, which could activate T cells.[56] Osteoblasts can produce IL-6 upon encountering T cells and stimulation by IL-17.[58] Osteoblasts play a crucial role in stem cell maintenance in bone marrow due to an intimate cell to cell contact via integrins.[59]
IMMUNOLOGICAL RESPONSE TO BONE TRAUMA
Fracture healing is a unique physiological process resulting into restoration of bone tissues. Growing evidence indicates that the immune cells and secreted factors are crucial for the physiological response following injury.[59–63] In a traumatic fracture, there is not only injury to bones, but the surrounding soft tissue damage also occurs, resulting into damage to local blood supply. Thus the fractured site and its surrounding develop hypoxia with nutrition deficiency.[64] So, the fracture haematoma is not only characterized by local hypoxia, but also by low pH with high concentration of lactate. Thus the fracture initiated a local inflammatory response.[65] Following this inflammatory response, the soluble factors interact with progenitor cells, located in the periosteum, endosteum, bone marrow and surrounding soft tissues.[64]
In one of the study,[66] it has been discussed that IL8 upregulation in fracture haematoma (FH) and surrounding haematoma (SH) indicates the same type of inflammatory reaction. The hypoxia-inducible factor (HIF) and its regulation play an important role for the functions of innate and adaptive immune cells. Investigations have proved that it is the initial inflammatory response of fracture, which is crucial for final clinical bone healing. HIF regulates this adaptation by switching the cellular energy metabolism from oxidative phosphorylation towards glycolysis. Hypoxia / HIF re-establishes the normal oxygen supply by promoting angiogenesis via VEGF and IL-8. Limited and reduced immune response with increasing age may explain the higher chances of delayed / nonunions in these patients.[66]
The exact interplay of immune function and bone regeneration in early (inflammatory) phase is currently unclear.[67–70] Due to 1) bleeding (peripheral blood leaks through vessel damage into the gap) and 2) broken bone itself (bone marrow flows into the gap) various cells reach into the fracture haematoma.[71] Innate immune cells and CD4+ T cells adapts well to the energy insufficiencies.[72] Progress has been made in understanding the link between adaptive and innate immune system in bone, with emphasis on osteoclasts.[73,74] The function of the adaptive immune system in fracture healing is less understood. Effector cells of the adaptive immune system are lymphocytes. A role of lymphocytes in fracture healing has been suggested but has not been characterised in depth yet. It has already been shown that CD8+ cytotaxic T cells have a counter regulatory role in wound healing because depletion of CD8+ T lymphocytes has a positive influence on wound healing.[75,76] It has also been established that lack of B lymphocytes can lead to higher bone formation in mice.[75–77]
An experiment[66] was conducted to study the response to hypoxia in fracture haematomain immunological patients. It was demonstrated that a higher mRNA expression of several genes from FH and SF cells than those in peripheral blood (e.g. osteogenic SSP1, IL8, CXCR4). It was also seen in the same study that there was decreased expression of the osteoblast differentiation transcription factor RUNX2 in FH and SH from immunologically compromised patients, but there was no difference in SSP1 expression. It was postulated that this fact could indicate that bone regeneration is induced in FH in immunologically compromised patients (but so not in SF). It had been documented that a higher inflammatory response (higher IL8 and CXCR4 expression) occured bone regeneration in immunologically restricted patients, but near normal angiogenesis (VEGF) occurs in these patients. It had been postulated that ongoing angiogenesis could be the one of the pathways in bone regeneration. Researchers concluded that there occurs a disturbed (higher and prolonged) inflammatory response with inadequate response to hypoxia (lower expression level of LDHA and PGK1 in fracture haematoma) with normal angiogenesis in immunologically disturbed patients, resulting into prolonged bone healing (delayed union) or nonunion.[66]
In another experiment,[78] the absence of mature lymphocytes was modeled in recombination activation gene 1 knockout mice to look further into the role of lymphocytes to fracture healing. It was postulated that fracture healing would be delayed in the absence of lymphocytes as effector cells of adaptive immune system. The underlying defect in these was loss of recombination activation gene, which encodes for a recombinase that is crucial for the ‘somatic variable diverse joining’ (VDJ) recombination in the development of B- and T- cell receptors. It was found that lack of lymphocytes led to accelerated bone regeneration in RAG-/- mice. It has also been reported that γ/δ T cells negatively influence fracture healing. It was suggested that probably B cells and not the T cells are predominant during immunoregulation of bone repair.[78]
It has already been discussed that during bone formation, cytokines secreted by lymphocytes have paracrine effects on bone cell differentiation and function. T helper lymphocyte 1 (THs1) cells promote cellular immunity via secretion of cytokines such as TNF-α and LT-β in the callus.[79] It has been conceived that lower levels of TNF-α favours osteogenesis as it acts as a proapoptotic factor on osteoblasts. But it has also been shown that TNF-α signaling impairs fracture healing.[80,81] The positive and negative effects of TNF-α depend upon its concentration. Activated natural killer cells of innate immune system produce IFN-γ in response to bone fracture and maintained its expression for a prolonged period. It causes macrophage activation, leading to tissue destruction.[78]
Lymphopenic mice have displayed strinkingly higher expression of IL-10, which produced by either myeloid cells or regulatory T cells. IL-10 is a potent inhibitor of secretion of proinflammatory cytokines such as TNF-α, IL-1, IL-6 and T cell activation. Experiment had shown that during fracture healing, lymphocytes can negatively regulate the amount of IL-10 in callus. IL-10 knock out mice have osteopenic and fragile bones.[82–85]
The role of IL-2 has not yet been investigated in fracture healing. But in some experiments on mice, it has been seen that lack of IL-2 enhanced bone formation as IL-2 infusion leads to bone resorption.[78]
The Th2 cytokine IL-4 is considered to exert anti-inflammatory function. It was discovered as a 'B cell growth factor’[86] and inhibits monocyte production of IL-1, TNF-α and PGE2. IL-4 inhibits bone resorption and is a chemoattractant for osteoblasts, stimulating proliferation and inhibiting differentiation. During physiologic fracture healing, expression of IL-4 is unregulated.[87]
An experiment[88] demonstrated that cells in fracture haematoma adapts to hypoxic conditions, which leads to angiogenesis, chemotaxis and osteogenesis (upregulation of VEGF) and IL6.
Besides numerous other factors, fibroblast growth factor receptor (FGFR) signaling is involved in fracture healing and bone remodelling.[88,89] FGF23 is a phosphatonin produced by osteoblastic cells, which signals via FGFR1, thereby exerting effects in bone and kidney. A study analyzed if serum FGF23 levels might be an indicator to predict fracture healing and union. It was concluded that FGF23 was involved in bone healing, could be measured by a sensitive assay in peripheral blood, and was a promising candidate as an indicator for healing processes prone to reunion versus nonunion.[90]
CONCLUSION
In view of the intricate interrelationships between the immune and skeletal system it may be concluded that immunological effects on bone remodeling, including fracture healing can be divided into noxious or beneficial. In case of bone injury, host immune mechanism produces impact on physiological defense mechanism and reparative potential at the same time. Any immunomodulatory intervention strategy to improve bone remodeling including fracture healing could specifically target lymphocyte subsets or cytokines to eliminate any detrimental action and to preserve necessary physiological leverage. Though presently, the clinical implications of above observations are unclear, but we speculate facilitating these effects therapeutically may improve remodeling including fracture healing. It would be worth taking a close look at molecular mechanisms regulating osteoclastogenesis and their correlation with environmental factors affecting the remodeling process.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Parfitt AM. Osteonal and hemiosteonal remodeling: The spatial and temporal framework for signal traffic in adult human bone. J Cell Biochem. 1994;55:273–86. doi: 10.1002/jcb.240550303. [DOI] [PubMed] [Google Scholar]
- 2.Praffit AM. Pharmacological manipulations of bone remodeling in calcium homeostasis. In: Kanis, editor. Progress in basic and clinical pharmacology. Basel: Karger; 1990. pp. 1–27. [Google Scholar]
- 3.Power J, Loveridge N, Rushton N, Parker M, Reeve J. Osteocyte density in aging subjects is enhanced in bone adjacent to remodeling haversian systems. Bone. 2002;30:859–65. doi: 10.1016/s8756-3282(02)00731-7. [DOI] [PubMed] [Google Scholar]
- 4.Cowin SC. Mechanosensation and fluid transport in living bone. J Musculoskelet Neuronal Interact. 2002;2:256–66. [PubMed] [Google Scholar]
- 5.Marom R, Shur I, Solomon R, Benayahu D. Characterisation of adhesion and differentiation markers of osteogenic marrow stromal cells. J Cell Physiol. 2005;202:41–8. doi: 10.1002/jcp.20109. [DOI] [PubMed] [Google Scholar]
- 6.Noble BS, Peet N, Steevens HY, Brabbs A, Mosley JR, Reilly GC, et al. Mechanical loading: Biphasic osteocyte survival and targeting osteoclasts for bone destruction in bone cortical bones. Am J Physiol. 2003;284:C934–44. doi: 10.1152/ajpcell.00234.2002. [DOI] [PubMed] [Google Scholar]
- 7.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow derived stromal cells. Proc Natl Acad Sci U S A. 1990;87:7260–4. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurihara N, Chenu C, Miller M, Civin C, Roodman GD. Identification of committed mononuclear precursors for osteoclasts-like cells formed in long term human marrow cultures. Endocrinology. 1990;126:2733–41. doi: 10.1210/endo-126-5-2733. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida S, Domon T, Wakita M. Studies of the clear zone of osteoclasts: Immunohistological aspects of its form and distribution. Arch Histol Cytol. 1989;52:513–20. doi: 10.1679/aohc.52.513. [DOI] [PubMed] [Google Scholar]
- 10.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The noval zinc finger containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 11.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa 1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 12.Gu G, Mulari M, Peng Z, Hentunen TA, Väänänen HK. Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Cell. 2005;335:1095–101. doi: 10.1016/j.bbrc.2005.06.211. [DOI] [PubMed] [Google Scholar]
- 13.Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res. 2006;21:67–77. doi: 10.1359/JBMR.051007. [DOI] [PubMed] [Google Scholar]
- 14.Luchin A, Purdom G, Murphy K, Clark MY, Angel N, Cassady AI, et al. The microphthalmia transcription factor regulates the expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J Bone Miner Res. 2000;15:451–60. doi: 10.1359/jbmr.2000.15.3.451. [DOI] [PubMed] [Google Scholar]
- 15.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulates bone loss and joint destruction in aduvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 16.Ha H, Lee JH, Kim HN, Kim HM, Kwak HB, Lee S, et al. Alpha-Lipoic acid inhibits inflammatory bone resorption by suppressing prostaglandin E2 synthesis. J Immunol. 2006;176:111–7. doi: 10.4049/jimmunol.176.1.111. [DOI] [PubMed] [Google Scholar]
- 17.Shoji M, Tanabe N, Mitsui N, Tanaka H, Suzuki N, Takeichi O, et al. Lipopolysaccharide stimulates the productions of prostaglandin E2 and the receptor Ep4 in osteoblasts. Life Sci. 2006;78:2012–8. doi: 10.1016/j.lfs.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Dai J, He P, Chen X, Greenfield EM. TNFa and PTH utize distinct mechanisms to induce IL-6 and RANKL expression with markedly different kinetics. Bone. 2006;38:509–20. doi: 10.1016/j.bone.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Greenfield EM, Horowitz MC, Lavis SA. Stimulation by parathyroid hormone of interleukin-6 and leukemia inhibitory factor expression in osteoblasts is an immediate-early gene response induced by cAMP signal transduction. J Biol Chem. 1996;271:10984–9. doi: 10.1074/jbc.271.18.10984. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Harrison J, Lorenzo J, Kream BE. Parathyroid hormone induces interleukin-6 heterogeneous nuclear and messanger RNA expression in murine calvarial organ cultures. Bone. 1998;23:327–32. doi: 10.1016/s8756-3282(98)00115-x. [DOI] [PubMed] [Google Scholar]
- 21.Kitazawa S, Kajimoto K, Kondo T, Kitazawa R. Vitamin D3 supports osteoclastogenesis via functional vitamin D response element of human RANKL gene promoter. J Cell Biochem. 2003;89:771–7. doi: 10.1002/jcb.10567. [DOI] [PubMed] [Google Scholar]
- 22.Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC. Osteoblast programmed cell death (apoptosis): Modulation by growth factors and cytokines. J Bone Miner Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- 23.Eghbali FG, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early post menopausal women. J Clin Invest. 2003;111:1221–30. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jilk RL, Passeri G, Girasole G, Cooper S, Abrams J, Broxmeyer H, et al. Estrogen loss upregulates hematopoiesis in the mouse: A mediating role of IL-6. Exp Hematol. 1995;23:500–6. [PubMed] [Google Scholar]
- 25.Jilka RL, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, et al. Increased osteoclast development for estrogen loss: Mediation by interleukin-6. Science. 1992;257:88–91. doi: 10.1126/science.1621100. [DOI] [PubMed] [Google Scholar]
- 26.Oursler MJ, Cortese C, Keeting PE, Anderson MA, Bonde SK, Riggs BL, et al. Modulation of transforming growth factor-beta production in normal human osteoblast-like cells by 17β-estrodiol and parathyroid hormone. Endocrinology. 1991;129:3313–20. doi: 10.1210/endo-129-6-3313. [DOI] [PubMed] [Google Scholar]
- 27.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collin OP, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. 2001;276:20659–72. doi: 10.1074/jbc.M010153200. [DOI] [PubMed] [Google Scholar]
- 29.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–37. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 30.Yun TJ, Chaudhary PM, Shu GL, Frazer JK, Ewings MK, Schwartz SM, et al. OPG/FDCR-1, a TFN receptor family member, is expressed lymphoid cells and is up-regulated by ligating CD-40. J Immunol. 1998;161:6113–21. [PubMed] [Google Scholar]
- 31.Hofbauer LC, Shui C, Riggs BL, Dunstan CR, Spelsberg TC, O’Brien T, et al. Effects of immunosuppressants on receptor of NF-kappaB ligand and osteoprotegerin production by human osteoblastic and coronary artery smooth muscle cells. Biochem Biophys Res Commun. 2001;280:334–9. doi: 10.1006/bbrc.2000.4130. [DOI] [PubMed] [Google Scholar]
- 32.Mayankar UM, Scatena M, Suchland KL, Yun TJ, Clark EA, Giachelli CM. Osteoprotegerin is a alpha vs beta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J Biol Chem. 2000;275:20959–62. doi: 10.1074/jbc.C000290200. [DOI] [PubMed] [Google Scholar]
- 33.Srivastava S, Matsuda M, Hou Z, Bailey JP, Kitazawa R, Herbst MP, et al. Receptor activator of NF-kappaB ligand induction via Jak2 and Stat5a in mammary epithelial cells. J Biol Chem. 2003;278:46171–8. doi: 10.1074/jbc.M308545200. [DOI] [PubMed] [Google Scholar]
- 34.Cross SS, Yang Z, Brown NJ, Balasubramanian SP, Evans CA, Woodward JK, et al. Osteoprotegerin (OPG)-a potential new role in the regulation of endothelial cell phenotype and tumor angiogenesis? Int J Cancer. 2006;118:1901–8. doi: 10.1002/ijc.21606. [DOI] [PubMed] [Google Scholar]
- 35.Kartsogiannis V, Zhou H, Horwood NJ, Thomas RJ, Hards DK, Quinn JM, et al. Localisation of RANKL mRNA and protein in skeletal and extraskeletal tissue. Bone. 1999;25:525–34. doi: 10.1016/s8756-3282(99)00214-8. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki J, Ikeda T, Kuroyama H, Seki S, Kasai M, Utsuyama M, et al. Regulation of osteoclastogenesis by three human RANKL isoforms expressed in NIH3T3 cells. J Biol Chem. 2001;276:20659–72. doi: 10.1016/j.bbrc.2003.12.191. [DOI] [PubMed] [Google Scholar]
- 37.Ikeda T, Kasai M, Utsuyama M, Hirokawa K. Determination of three isoforms of the receptor activator of nuclear factor-kappaB ligand and their differential expression in bone and thymus. Endocrinology. 2001;142:1419–26. doi: 10.1210/endo.142.4.8070. [DOI] [PubMed] [Google Scholar]
- 38.Lum L, Wong BR, Joseign R, Becherer JD, Erdjument-Bromage H, Schlöndorff J. Evidence for a role of a tumor necrosis factor-alpha-converting enzyme-likeprotease in the shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dentritic cell survival. J Biol Chem. 1999;274:13613–8. doi: 10.1074/jbc.274.19.13613. [DOI] [PubMed] [Google Scholar]
- 39.Kanamaru F, Iwai H, Ikeda T, Nakajima A, Ishikawa I, Azuma M. Expression of membrane bound and soluble receptor activator of NF-kB ligand in human T cells. Immunol Lett. 2004;94:239–46. doi: 10.1016/j.imlet.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 41.Takayanagi T, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 integrate RANKL signaling in the terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 42.Kudo O, Fuzikawa Y, Itonaga I, Sabokbar A, Torisu T, Athanasou NA. Proinflammatory cytokine (TNF/IL-1) induction of human osteoclast formation. J Pathol. 2002;198:220–7. doi: 10.1002/path.1190. [DOI] [PubMed] [Google Scholar]
- 43.Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of RANKL, osteoprotegerin, RANK in mouse calvariae. J Immunol. 2002;169:3353–62. doi: 10.4049/jimmunol.169.6.3353. [DOI] [PubMed] [Google Scholar]
- 44.Sabokbar A, Kudo O, Athanasou NA. Two distinct cellular mechanisms of osteoclast formation and bone resorption in periprosthetic osteolysis. J Orthop Res. 2003;21:73–80. doi: 10.1016/S0736-0266(02)00106-7. [DOI] [PubMed] [Google Scholar]
- 45.Toraldo G, Roggia C, Qian WP, Pacifici R, Weitzmann MN. IL-7 induces bone loss in vivo by induction of receptor activator of RANKL and TNFα from T cells. Proc Natl Acad Sci U S A. 2003;100:125–30. doi: 10.1073/pnas.0136772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roodman GD. Cell biology of osteoclasts. Exp Hematol. 1999;27:1229–41. doi: 10.1016/s0301-472x(99)00061-2. [DOI] [PubMed] [Google Scholar]
- 47.Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD, Pike JW. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator –activated receptor γ 1. Proc Natl Acad Sci U S A. 2001;98:2443–8. doi: 10.1073/pnas.041493198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fox SW, Haque SJ, Lovibond AC, Chambers TJ. The possible role of TGF-β-induced suppressors of cytokine signaling expression in osteoclast/ macrophage lineage commitment in vitro. J Immunol. 2003;170:3679–87. doi: 10.4049/jimmunol.170.7.3679. [DOI] [PubMed] [Google Scholar]
- 49.Horwood NJ, Elliott J, Martin TJ, Gillespie MT. IL-12 alone and in synergy with IL-18 inhibits osteoclast formation in vitro. J Immunol. 2001;166:4915–21. doi: 10.4049/jimmunol.166.8.4915. [DOI] [PubMed] [Google Scholar]
- 50.Huang W, O’Keefe RJ, Schwarz EM. Exposure to RANKL renders pre-osteoclasts resistant to IFN-γ by inducing terminal differentiation. Arthritis Res Ther. 2003;5:49–59. doi: 10.1186/ar612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khapli SM, Mangashetti LS, Yogesha SD, Wani MR. IL-3 acts directlyon osteoclast precursors and irreversibly inhibits receptor activators of NFkB ligands induced osteoclast differentiation by diverting the cells to macrophage lineage. J Immunol. 2003;171:142–51. doi: 10.4049/jimmunol.171.1.142. [DOI] [PubMed] [Google Scholar]
- 52.Takayanagi H, Sato K, Takoaka A, Taniguchi T. Interplay between interferon and other cytokine systems in bone metabolism. Immunol Rev. 2005;208:181–93. doi: 10.1111/j.0105-2896.2005.00337.x. [DOI] [PubMed] [Google Scholar]
- 53.Thomson BM, Bennett J, Dean V, Triffitt J, Meikle MC, Loveridge N. Preliminary characterization of porcain bone marrow stromal cells: Skeletogenic potential, colony forming activity and response to dexamethasone, transforming growth factor β, and basic fibroblast growth factor. J Bone Miner Res. 1993;8:1173–88. doi: 10.1002/jbmr.5650081004. [DOI] [PubMed] [Google Scholar]
- 54.Mackie EJ. Cells in focus-osteoblasts: Novel roles in orchestration of skeletal architecture. Int J Biochem Cell Biol. 2003;35:1301–5. doi: 10.1016/s1357-2725(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 55.Grcevic D, Sun KL, Marusic A, Lorenzo JA. Depletion of CD4 and CD8 T lymphocytesin mice in vivo enhances 1,25-dihydroxyvitamin D3-stimulated osteoclast-like cell formation in vitro by a mechanism that is dependent on prostaglandin synthesis. J Immunol. 2000;165:4231–8. doi: 10.4049/jimmunol.165.8.4231. [DOI] [PubMed] [Google Scholar]
- 56.Shinoda K, Sugiyama E, Taki H, Harada S, Mino T, Maruyama M, et al. Resting T cells negatively regulate osteoclast generation from peripheral blood monocytes. Bone. 2003;33:711–20. doi: 10.1016/s8756-3282(03)00230-8. [DOI] [PubMed] [Google Scholar]
- 57.Martina R, Wolfgang S, Peter P. Osteoimmunology. Int Arch Allergy Immunol. 2007;143:31–48. doi: 10.1159/000098223. [DOI] [PubMed] [Google Scholar]
- 58.Stanley KT, Van DC, Motyl C, Endres J, Fox DA. Immunocompetent properties of human osteoblasts: Interactions with interaction with T lymphocytes. J Bone Miner Res. 2006;1:29–36. doi: 10.1359/JBMR.051004. [DOI] [PubMed] [Google Scholar]
- 59.Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop Relat Res. 1998;355:7–21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 60.McKibbin B. The biology of fracture healing in long bone. J Bone Joint Surg Br. 1978;60B:150–62. doi: 10.1302/0301-620X.60B2.350882. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt BK, Schell H, Kolar P, Kolar P, Pfaff M, Perka C, et al. Cellular composition of the initial fracture hematoma compared to a muscle hematoma: A study in sheep. J Orthop Res. 2009;27:1147–51. doi: 10.1002/jor.20901. [DOI] [PubMed] [Google Scholar]
- 62.Timlin M, Toomey D, Condron C, Power C, Street J, Murray P, et al. Fracture hematoma is a potent proinflammatory mediator of neutriphil function. J Trauma. 2005;58:1223–29. doi: 10.1097/01.ta.0000169866.88781.f1. [DOI] [PubMed] [Google Scholar]
- 63.Matin P, Leibovich SJ. Inflammatory cells during wound repair: The good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Schindeler A, Liu R, Little DG. The contribution of different cell lineage to bone repairexploring a role for muscle stem cells. Differentiation. 2009;77:12–8. doi: 10.1016/j.diff.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 65.Taguchi K, Ogawa R, Migita M, Hanawa H, Ito H, Orimo H. The role of bone marrow derived cells in bone fracture repair in a green fluorescent protein chimeric mouse model. Biochem Biophys Res Commun. 2005;331:31–6. doi: 10.1016/j.bbrc.2005.03.119. [DOI] [PubMed] [Google Scholar]
- 66.Hoff P, Gaber T, Bleek KS, Sentürk U, Tran CL, Blankenstein K, et al. Immunologically restricted patients exhibit a pronounced inflammation and inadequate response to hypoxia in fracture hematoma. Immunol Res. 2011;51:116–22. doi: 10.1007/s12026-011-8235-9. [DOI] [PubMed] [Google Scholar]
- 67.Braun W, Rutter A. Fracture healing: Morphologic and physiologic aspects. Unfallchirurg. 1996;99:59–67. [PubMed] [Google Scholar]
- 68.Phillips AM. Overview of the fracture healing cascade. Injury. 2005;36S:5–7. doi: 10.1016/j.injury.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 69.Remedios H. Bone and Bone healing. Vet Clin North Am Small Anim Pract. 1999;29:1029–44. doi: 10.1016/s0195-5616(99)50101-0. [DOI] [PubMed] [Google Scholar]
- 70.Schindeler A, Mc Donald MM, Bokko P, Little DG. Bone remodeling during fracture repair: The cellular picture. Semin Cell Dev Biol. 2008;19:459–66. doi: 10.1016/j.semcdb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 71.Cramer T, Yamanishi Y, Clausen BE, Johnson RS. HIF-1 alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tripmacher R, Gaber T, Dziurla R, Häupl T, Erekul K, Grützkau A, et al. Human CD4(+) T cell maintain specific functions even under conditions of extremely restricted ATP production. Eur J Immunol. 2008;38:1631–42. doi: 10.1002/eji.200738047. [DOI] [PubMed] [Google Scholar]
- 73.Horowitz M, Vignery A, Gershon RK, Baron R. Thymus derived lymphocytes and their interaction with macrophages are required for the production of osteoclasts activating factor in the mouse. Proc Natl Acad Sci U S A. 1984;81:2181–5. doi: 10.1073/pnas.81.7.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97:1566–71. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg. 2004;187:11S–6S. doi: 10.1016/S0002-9610(03)00296-4. [DOI] [PubMed] [Google Scholar]
- 76.Schaffer M, Barbul A. Lymphocyte function in wound healing and following injury. Br J Surg. 1998;85:444–60. doi: 10.1046/j.1365-2168.1998.00734.x. [DOI] [PubMed] [Google Scholar]
- 77.Schaffer M, Bongartz M, Hoffmann W, Viebahn R. MCH-class-11-deficiency impairs wound healing. J Surg Res. 2007;138:100–5. doi: 10.1016/j.jss.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 78.Toben D, Schroeder I, Khassawna TE, Mehta M, Hoffmann JE, Frisch JT, et al. Fracture healing is accelerated in the absence of the adaptive immune system. J Bone Miner Res. 2011;26:113–24. doi: 10.1002/jbmr.185. [DOI] [PubMed] [Google Scholar]
- 79.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th 17 function as an osteoclastogenic helper T cell subset that links T cells activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, et al. Impaired fracture healing in the absence of TNF-alpha signaling-the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. 2003;18:1584–92. doi: 10.1359/jbmr.2003.18.9.1584. [DOI] [PubMed] [Google Scholar]
- 81.Lehmann W, Edgar CM, Wang K, Cho TJ, Barnes GL, Kakar S, et al. TNF-alpha coordinately regulates the expression of specific matrix metalloproteinases and angeogenic factors during fracture healing. Bone. 2005;36:300–10. doi: 10.1016/j.bone.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 82.Grutz G. New insights into the molecular mechanism of IL-10 mediated immunosuppression. J Leukoc Biol. 2005;77:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- 83.Liu D, Yao S, Wise GE. Effect of IL-10 on gene expression of osteoclastogenic regulatory molecules in the rat dental follicle. Eur J Oral Sci. 2006;114:42–9. doi: 10.1111/j.1600-0722.2006.00283.x. [DOI] [PubMed] [Google Scholar]
- 84.Sasaki H, Hou L, Belani A, Wang CY, Uchiyama T, Müller R, et al. IL-10, but not IL-4, suppresses infection stimulated bone resorption in vivo. J Immunol. 2000;165:3626–30. doi: 10.4049/jimmunol.165.7.3626. [DOI] [PubMed] [Google Scholar]
- 85.Dresner PR, Gelb N, Rachmilewitz D, Karmeli F, Weinreb M. IL-10 deficient mice develop Osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology. 2004;127:792–801. doi: 10.1053/j.gastro.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 86.Brown MA, Hural J. Functions of IL-4 and control of its expression. Crit Rev Immunol. 1997;17:1–32. doi: 10.1615/critrevimmunol.v17.i1.10. [DOI] [PubMed] [Google Scholar]
- 87.Watanabe K, Tanaka Y, Morimoto I, Yahata K, Zeki K, Fujihira T, et al. IL-4 as a potent inhibitor of bone resorption. Biochem Biophys Res Commun. 1990;172:1035–41. doi: 10.1016/0006-291x(90)91550-c. [DOI] [PubMed] [Google Scholar]
- 88.Kolar P, Gaber T, Perka C, Duda GN, Buttgereit F. Human early fracture haematoma is characterized by inflammation and hypoxia. Clin Orthop Relat Res. 2011;469:3118–26. doi: 10.1007/s11999-011-1865-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kwon BS, Wang S, Udagawa N, Haridas V, Lee ZH, Kim KK, et al. TRI, a new member of the tumor necrosis factor receptor superfamily, induces fibroblast proliferation and inhibits osteoclastogenesis and bone resorption. FASEB J. 1998;12:845–54. doi: 10.1096/fasebj.12.10.845. [DOI] [PubMed] [Google Scholar]
- 90.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, et al. Osteoprotegerin: A noval secreted protein involved in regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]