

Abstract
Glycemic control is an important aspect of patient care in the surgical Infections of the nervous system are among the most difficult infections in terms of the morbidity and mortality posed to patients, and thereby require urgent and accurate diagnosis. Although viral meningitides are more common, it is the bacterial meningitides that have the potential to cause a rapidly deteriorating condition that the physician should be familiar with. Viral encephalitis frequently accompanies viral meningitis, and can produce focal neurologic findings and cognitive difficulties that can mimic other neurologic disorders. Brain abscesses also have the potential to mimic and present like other neurologic disorders, and cause more focal deficits. Finally, other infectious diseases of the central nervous system, such as prion disease and cavernous sinus thrombosis, are explored in this review.
Keywords: Brain, encephalitis, infectious, meningitis, nervous
INTRODUCTION
Infections of the central nervous system (CNS) pose a unique challenge to physicians, due to both the potential morbidity and mortality that they cause as well as the inherent difficulties involved in their treatment. These infections mainly involve meningitis, encephalitis, and brain abscesses, and tend to cause more morbidity and mortality on average than infections involving other organ systems. Due to their potential for adverse consequences, it is important for the physician to be well versed in the presentations and care of the more common CNS infections, as these represent a not infrequent etiology of complaints seen from the primary care clinic to the surgeon's bed to even the psychiatric ward.
MENINGITIS
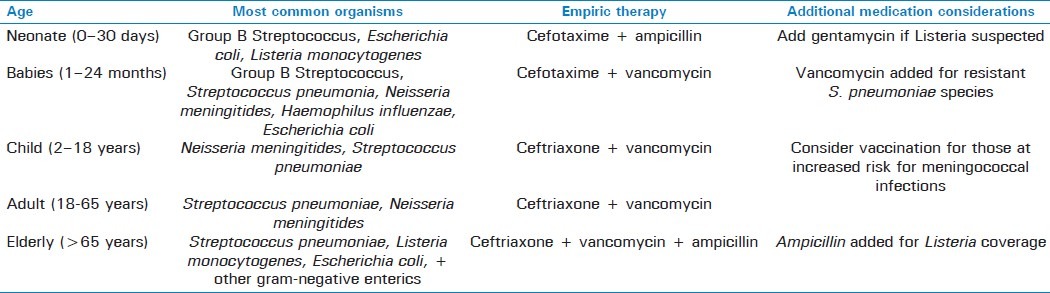
Among the bacterial meningitides, it is helpful to classify common agents according to population affected. In the first few months of life, the primary causative agents of neonatal meningitis include Group B Streptococcus, Escherichia coli, and Listeria monocytogenes. After this period, Neisseria meningitides and Streptococcus pneumoniae are the most frequent causes followed by Haemophilus influenzae, with Neisseria being more prevalent in children, whereas Streptococcus is more prevalent in adults and the elderly. Although each of these infections presents with similar features, and requires similar workup, including cerebrospinal fluid (CSF) analysis via lumbar puncture (LP), there are several important differences between the various types that are outlined in the following sections [Tables 1 and 2].
Table 1.

Table 2.
Empiric therapy for treatment of meningitis by age group[3]
Neisseria meningitides
Infection with N. meningitides, also known as meningococcal meningitis, can cause an acute fulminant meningitis, which is a medical emergency. There are 5 main serogroups that are responsible for the majority of meningococcal illness—A, B, C, Y, and W-135. It is the most common form of meningitis in children from 2–18 years of age. Risk factors include living in close quarters, such as dormitory settings. Typical symptoms and signs of the disease include a stiff neck, high fever, headache, and altered mental status (AMS), with at least 2 out of these 4 symptoms being found in more than 90% of all patients.[1] Other signs and symptoms include nausea and vomiting, radicular pain, signs of increased intracranial pressure, such as papilledema, and often focal neurologic deficits, such as cranial nerve VI palsy.[1] Complications of acute fulminant meningitis include Waterhouse–Friderichsen syndrome—bilateral adrenal insufficiency from hemorrhage into the adrenal glands, disseminated intravascular coagulation, coma, and rapid death.
Physical diagnostic tests include examining the patient for Kernig and Brudzinski signs. The Kernig test involves attempting to straighten the patient's leg out from a flexed knee and hip position, with pain upon passive stretch indicative of a positive sign. The Brudzinski test involves flexing the head of a supine patient while examining to see if the legs flex upward at the hip as well.
Unlike other noninfectious cases of increased intracranial pressure, if an infectious etiology is suspected, then diagnostic LP ought to be performed immediately without a preceding computed tomography (CT) scan to obtain cultures and to determine susceptibility; empiric antibiotic treatment should be begun shortly thereafter. If clinical examination of the patient reveals significant papilledema and clinical judgment predicts a high probability of cerebral herniation from LP, then LP ought to be delayed and empiric treatment started immediately.[2]
One absolute contraindication to LP is localized infection around the LP site.[3] LP performed around 4–8 h after antibiotic treatment has been administered has around a 50% sensitivity for revealing the causative organism. CSF, however, will still reveal features characteristic of a bacterial meningitis, including a neutrophilic pleocytosis, increased protein count, with a decreased glucose count. When performed in a timely manner, LP will reveal gram-negative diplococci in the CSF.
In a patient presenting with petechiae, an often neglected diagnostic procedure involved obtaining skin films or aspiration of petechiae, using Periappuram et al's method of finding specimens from skin scrapings, which revealed gram-negative diplococci in 80% of patients sampled, with positive skin films obtained from 85% CSF culture-negative cases.[4] Sensitivity of needle aspiration of petechiae ranged from 46% to 51%, whereas skin biopsy was significantly more sensitive, ranging from 72% to 80%.[4–6] These additional modalities are often positive even when CSF findings are negative, and hence represent an important method of being able to rapidly and effectively diagnose patients with meningitis and sepsis, even after the treatment of antibiotics.
Empiric antibiotic treatment for suspected bacterial meningitis centers around the treatment for meningococcal meningitis, owing to the severity of this infection, and consists of intravenous administration of a third-generation cephalosporin, such as ceftriaxone or cefotaxime with good CNS penetration. Duration of treatment for meningococcus tends to be shorter than that for other bacterial meningitis infections, averaging around 7 days.
Streptococcus pneumoniae
Streptococcal meningitis from the causative organism S. pneumoniae is the leading cause of meningitis in the elderly, and one of the leading causes of bacterial meningitis in all adults and children older than 2 months. The illness is usually not as rapid and fulminant as that caused by meningococcal meningitis, but nonetheless can present with similar symptoms. Workup is similar to those patients suspected of having meningococcal meningitis, with LP preceding empiric treatment with a third-generation cephalosporin. LP should return gram-positive diplococci. In cases where resistance to cephalosporin is suspected, vancomycin should be added for additional coverage.[7] Out of the several species of bacterial meningitis, clinical trials to date have provided the greatest benefits for adjuvant dexamethasone therapy for pneumococcal meningitis.[8–10] An estimated 1 in 12 cases of streptococcal meningitis is fatal, with 1 in 3–4 survivors suffering neurologic sequelae, including deafness, persistent seizures, and mental retardation in children.[11] Since the advent of the heptavalent pneumococcal vaccine in 2000, pneumococcal meningitis has decreased significantly in children, estimated by the Center for Disease Control (CDC) to be around 77% lower than immediately before vaccine introduction, while diminishing in adults as well due to herd immunity.[12–14]
Haemophilus influenzae
Haemophilus influenzae, particularly serotype B (HiB), is a frequent cause of meningitis in children under the age of 5 years.[15] This organism is a gram-negative rod and a frequent inhabitant of the sinuses, inner and middle ear, respiratory tract, and bloodstream; it commonly causes meningitis via a combination of both direct sinus and hematogenous spread. The frequency of HiB strain in the United States has decreased dramatically, however, in the past couple of years since the advent of the HiB vaccine, and is now a distant third behind streptococcus and meningococcus in childhood meningitis.[16] It continues, however, to be a major cause of childhood meningitis worldwide, with 386,000 deaths per year being attributed to the combination of HiB meningitis and pneumonia.[15] Furthermore, 15%–35% of all survivors of HiB meningitis are left with permanent neurologic sequelae, including deafness and mental retardation.[15] Timely administration of corticosteroids has been shown to help decrease the risk of subsequent neurologic deficits.[7,8] Expanding vaccine coverage into developing countries has the potential to reduce these numbers.
Diagnosis of bacterial meningitis in children is similar to that in adults, with neurologic examination of the patient determining the safety of performing an LP in children, and antibiotics should not be delayed for lengthy diagnostic procedures, such as imaging. CT, in fact, has generally not been shown to be of additional value in diagnosis of meningitis in children; a prospective study of 41 children diagnosed with bacterial meningitis showed that CT failed to reveal any clinically significant abnormalities, which were not already detected upon neurologic testing.[2] All children suspected of bacterial meningitis should receive empiric therapy with a third-generation cephalosporin due to the threat of meningococcus. Upon specific diagnosis of HiB meningitis, it is acceptable to continue treatment with a third-generation cephalosporin, but local antibiotic-resistance patterns ought to be taken into account.
Group B Streptococcus
Group B Streptococcus is one of the leading causes of meningitis in neonates. Its causative agent, Streptococcus agalactiae, is a gram-positive round bacterium with beta-hemolytic properties, also capable of causing sepsis and pneumonia in the newborns.[17–19] Neonates are most frequently infected with this organism during birth during passage through the vaginal canal, as Group B Streptococcus (GBS) is a frequent colonizer of the maternal vaginal canal. It is estimated that 10%–40% of all pregnant women in the United States are colonized with GBS in either the vagina or the rectum, and about half those infants exposed during birth will be colonized, while transmission to newborns with subsequent clinically significant sequelae occurs in about 1.8 out of every 1000 live births.[18,19]
Neonatal GBS infection is characterized as either early, occurring in the first 7 days of life or late, occurring anytime afterward but usually within the first 2 months of life.[20] Risk factors for early infection include prematurity, low birth weight, preterm labor, premature rupture of membranes, previous infant affected with Group B Streptococcus, intrapartum fever, prolonged rupture of membranes exceeding 18 h, or a history of maternal GBS infection.[17–20] A combination of these factors may increase the risk for neonatal infection up to 45.5 per 1000 live births.[19] Risk factors for late infection are lesser known, but could involve vertical transmission from mother to neonate or include community or nosocomial sources.[19] Pathogen-specific risk factors include maternal colonization with the serotype III variety of GBS.[17]
The occurrence of meningitis in a newborn is not as specific as in the adult, as the infant may present simply with vague, nonlocalizing signs of irritability, without the characteristic stiff neck or accompanying Kernig and Brudzinski signs, and unable to voice a headache.[5] Due to the potential severity of meningitis, all infants under the age of 1 year presenting with a fever above 100.4F should receive a LP to examine for possible meningitis.
Empiric treatment of neonatal meningitis consists of ampicillin and cefotaxime or similar third-generation cephalosporin; steroid therapy is often indicated as well, given with or before the administration of antibiotics.[7] The case fatality rates of early-onset GBS infections have decreased dramatically over the past couple of decades, dropping from nearly over half a few decades ago to its present range of 3%–13%.[19,21–24]
Prevention of Group B streptococcal meningitis includes universal screening of all mothers late in pregnancy, around 35–37 weeks, with chemoprophylactic intrapartum administration of antibiotics indicated for those women either colonized or at a higher risk for perinatal infection, possessing the risk factors mentioned above.[18–20] First line antibiotic therapy involves intravenous administration of 5 million units of penicillin G, with 2.5 million units every 4 h afterward; backup to penicillin G is ampicillin, with cefazolin recommended for those with mild penicillin allergies and clindamycin, erythromycin, or vancomycin recommended for those with anaphylactic reactions to penicillin.[19] Adverse reactions to chemoprophylaxis include allergic or anaphylactic reactions to medication, as well as development of resistant GBS or other strains of bacteria leading to neonatal meningitis with antimicrobial-resistant pathogens.[18]
Escherichia coli
Along with Group B Streptococcus, Escherichia coli is one of the leading causes of meningitis in neonates up to 3 months of age, and should be suspected in all infants in this age range presenting with fever of unexplained origin. It is estimated that while 34.1% of neonatal meningitis is caused by Group B Streptococcus, E. coli is the responsible agent for 28.5% of cases.[25] Neonatal E. coli meningitis is transmitted vertically from mother to fetus, and is found more commonly in premature infants and those with low birth weight.[3,25] The K1 capsular antigen strain of E. coli is particularly associated with increased morbidity and mortality, and is the most common etiologic agent of gram-negative neonatal meningitis.[25,26]
Fever, failure to thrive, focal neurologic deficits, irritability, decreased feeding, jaundice, vomiting, seizures, apnea, and neurologic sequelae commonly accompany neonatal E. coli meningitis.[5,25] Diagnostic workup for suspected infant patients includes blood cultures, throat swab, obtaining a complete blood count, blood ethylenediaminetetra-acetic acid specimen for polymerase chain reaction (PCR) studies, erythrocyte sedimentation rate and C-reactive protein, examining the urine for bacterial antigens, and LP.[5] CSF is reflective of bacterial meningitis with high protein, low glucose, and neutrophil predominance, and should reveal gram-negative rods. The blood or CSF latex agglutination test is an alternative diagnostic modality that can be used to diagnose both E coli and GBS infection, but its lack of sensitivity limits its clinical usage.[5] A CT scan is of limited diagnostic value.[5] Empiric treatment consists of ampicillin and cefotaxime or ceftriaxone.[5,7,25]
Listeria monocytogenes
Listerial meningitis is most frequently seen in infants from birth to 3 months of age, and again in elderly patients above the age of 65 years; other populations at risk include immunocompromised patients, pregnant women, and alcoholics. Although meningitis is the most common cerebral manifestation of listeriosis, encephalitis and brain abscess are possible as well.[27] Common sources of Listeria infection include dairy products, such as unpasteurized milk and soft cheese. Neonatal infection is believed to occur either during passage through the birth canal or by way of transplacental infection.[3] Predisposing factors include prematurity and low birth weight.[3]
Symptoms are similar to other forms of meningitis, and include fever, nuchal rigidity in adults and elderly, headache in the elderly, irritability and poor feeding in infants, lethargy, and seizures, with seizures being more commonly found than in other forms of meningitis.[27] Findings on LP reveal gram-positive rods, although the characteristic polymorphonuclear pleocytosis may not be present.
Empiric treatment in the elderly includes covering against both Listeria and S. pneumoniae, and therefore ampicillin ought to be administered in addition to a third-generation cephalosporin. Specific treatment of listerial meningitis includes ampicillin with an aminoglycoside, such as gentamycin, and therapy should be administered for 2–3 weeks owing to the poor CSF penetration of ampicillin.[28]
Prognosis of GBS, E. coli, and Listeria neonatal meningitis is good, with mortality rates at experienced tertiary care centers ranging around 3%–13%, but morbidity from these early setbacks on the other hand may be significant.[21–23] In a cohort of 1717 children with meningitis during the first year of life vs 1485 controls, 1584 and 1391, respectively, that were successfully followed-up with at 5 years of age, Bedford et al. demonstrated that almost one-fifth of children with meningitis during the first year of life exhibited permanent, severe, or moderate disabilities, including learning difficulties, visual and hearing disorders, motor difficulties, seizure disorders, speech and language problems, and behavioral difficulties.[21] Mortality in elderly patients with listeriosis is significantly higher than that in the neonates, owing to the frequency of comorbid debilitating conditions found in this group.[29]
Mycobacterium tuberculosis
Mycobacterial meningitis is frequently present in immunocompromised hosts, such as HIV patients or the homeless population, and results from hematogenous spread of the organism through the blood-brain barrier. The causative agent is Mycobacterium tuberculosis, an acid-fast slow-growing aerobic bacterium that primarily affects humans.[30] The transmission of tuberculi is primarily via airborne droplets, and primary site of infection therefore tends to be the lungs.[30] From the lungs, hematogenous spread into the meninges is then possible, especially in patients with poor cell-mediated immunity. CNS tuberculous disease begins with the development of small tuberculous foci in either the brain, spinal cord, or the meninges.[30] Characteristic signs and symptoms are typical of those for meningitis, including headache, stiff neck, a slightly lower fever than in fulminant bacterial meningitis, coupled with the additional findings of AMS, possible focal neurologic deficits, and nausea and vomiting.
A quick and easy test to examine for lifetime exposure is the tuberculin PPD test, which takes advantage of cell-mediated immunity to detect if the individual has ever been exposed. This test has appreciably less utility for detecting active infection, however, and furthermore is of no use in those individuals who have already received the Bacillus–Calmette–Guerin vaccine. LP is of utility in testing for meningeal infection, and CSF usually reveals a lymphocytic pleocytosis and a modest increase in protein coupled with low glucose levels.[31] Antibody detection, antigen detection, and molecular methods are also utilized, and can help differentiate acute from chronic infections, with varying degrees of specificity for CNS infection vs systemic infection.[30] Contrast-enhanced magnetic resonance imaging (MRI) is the imaging study of choice, and is preferred over CT due to enhanced specificity and sensitivity, as well as better delineation of the lesion.
Standard international guidelines for treatment of tuberculosis advise 4-drug therapy with isoniazid, rifampin, pyrazinamide, and ethambutol for a period of 2 months, followed by an additional 6-month period of just isoniazid and rifampin. Isoniazid and pyrazinamide have the best CNS penetration out of the 4 drugs.[30] Although isoniazid is the mainstay of therapy, resistance to it is very common, and several alternative agents have been recommended in the case of isoniazid-resistant strains; these agents include the fluoroquinolones, ethionamide, streptomycin, and amikacin.[30]
FUNGAL MENINGITIS
Common fungal agents of meningitis include Cryptococcus neoformans, Candida albicans, Histoplasma capsulatum, Blastomycoses, and Coccidiodes immitis [Tables 3 and 4]. Susceptible patients are usually either immunocompromised or have undergone direct neurosurgical interventions, such as shunt placement. The primary method of spread in most cases involves respiratory infection with subsequent hematogenous dissemination.[32] Symptoms are typical of those of meningitis, including fever, headache, AMS, nausea, vomiting, and neck stiffness, in addition to complications of abscess, papilledema, seizures, and focal neurologic deficits.[32] LP for fungal meningitis usually reveals a lymphocytic prevalence with mild depressions in glucose and increased protein.[29]
Table 3.
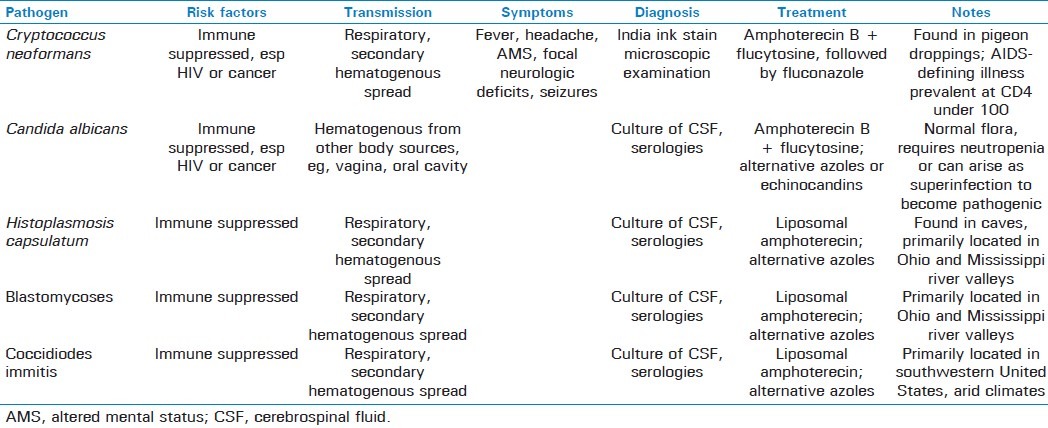
Table 4.
Signs and symptoms of central nervous system infections

AIDS and cancer patients are especially prone to cryptococcal meningitis, with the incidence of cryptococcosis dramatically increasing since the rise of the AIDS epidemic.[33,34] In a multistate case–control study performed by Hajjeh et al, 86% of patients sampled with cryptococcosis had HIV.[35] These patients often do not present with the same symptoms as found in immunocompetent patients, often experiencing a relatively sudden onset of symptoms vs a more subacute course in immunocompetent patients.[32,36] Pigeon droppings are one of the important carriers of Cryptococcus; smoking is another significant risk factor.[35] Diagnosis can be made via direct microscopic examination using India Ink staining; cytology or histopathology, serologies, and culture after 48 h can also be used.[33]
Amphoterecin is the agent of choice in cryptococcal meningitis, and should be administered along with flucytosine for 2 weeks, followed by fluconazole for a minimum of 10 weeks.[37,38] Highly active antiretroviral therapy for HIV-infected patients is the most efficacious method for cryptococcal prevention.[32,39] Once infected, however, lifelong fluconazole (or itraconazole in patients that cannot tolerate fluconazole) therapy is required, especially in AIDS patients with CD4 cell counts below 100.[32,37,40]
Coccidioidal meningitis usually occurs in epidemics, but affects a few hundred people a year at baseline, primarily in the southwestern United States.[41,42] Diagnosis is usually via serology, as meningitis is usually the result of disseminated pulmonary infection. Although previously treated with amphoterecin, these patients often require chronic therapy, prompting a switch to azoles, including fluconazole and itraconazole as the agent of choice for both induction and maintenance; addition of amphoterecin to treatment regimen is still indicated for azole failure.[42,43] Amphoterecin can be administered either intravenously or intrathecally. Histoplasmoses and blastomycoses produce a meningitis similar to coccidiomycoses, and are transmitted similarly, that is, respiratory infection followed by blood-borne spread into the meninges and occasionally brain parenchyma, due to the genetic similarities between these species.[44]
However, unlike coccidiomycosis, histoplasmosis and blastomycosis species tend to be found in central United States around the Mississippi and Ohio river valleys. Compared with histoplasmosis, blastomycosis is less prevalent yet poses more serious infection in those that manage to acquire it. Diagnosis for isolated CNS histoplasmosis infections may require multiple diagnostic modalities, including CSF testing, serologies, or culture.[44,45] Culture is the definitive method in the diagnosis of most fungal meningitis, as serologies may reveal false positives and PCR remains unproven, yet may necessitate obtaining several tissue or fluid samples before one returns positive for a culture.[44–46] There are no reliable skin or serologic tests to confirm previous blastomycosis infection, and half of the patients infected suffer symptoms for almost a month before diagnosis is made.[44] Due to its less common incidence combined with potentially severe sequelae, clinical suspicion must be high for blastomycosis. Treatment for both consists of liposomal amphoterecin B, which has been found to achieve better brain tissue concentrations with less nephrotoxicity than standard deoxycholate amphoterecin formation.[45] Itraconazole and fluconazole are recommended in patients unable to tolerate amphoterecin, while there have been several case studies reporting successful treatment of blastomycosis with vorizonazole.[47–49] AIDS patients may frequently require an azole for lifelong suppressive therapy, or until CD4 counts rise above 200.[46]
Out of the 150 species of Candida known, Candida albicans is the major human pathogen.[50] Although frequently present as a vaginal infection in immunocompetent patients, often seen after antibiotic therapy, Candida has the potential to disseminate and cause meningitis primarily in immunosuppressed, especially neutropenic, individuals.[50] In addition to meningitis, candidiasis can cause a vertebral osteomyelitis manifested by chronic progressive lower back pain that can eventually lead to nerve root compression syndromes and loss of function, as well as an endophthalmitis manifest by retinal lesions, which can eventually lead to blindness.[50]
As blood cultures can be fairly unreliable with poor sensitivity, more reliable techniques, such as the 1,3-β-glucan assay, are used to diagnose invasive candidiasis.[50] Definitive diagnosis of candidal meningitis, however, is made similar to other fungal meningitis via culture and isolation of Candida species from the CSF.[50]
Treatment for Candida meningitis involves amphoterecin B and usually flucytosine due to its ability to penetrate the blood–brain barrier. In patients for whom amphoterecin-induced nephrotoxicity is a significant problem, fluconazole or the echinocandins may be used.[51]
LYME DISEASE
Lyme disease, classically obtained from bites from the Ixodes scapularis tick carrying the causative agent Borrelia burgdorferi, is primarily seen in the eastern United States, particularly in the northeastern regions. Neurologic symptoms of Lyme disease are preceded by an annular skin rash termed erythema migrans and nonspecific symptoms of a low-grade fever, malaise, and fatigue. The neurologic symptoms of Lyme disease begin to occur roughly about a month after initial tick bite, and frequently include focal neurologic findings as well as signs and symptoms of meningismus.[52] The pathogenesis of CNS disease includes direct invasion by the bacterium itself in addition to vascular invasion.[52] Focal neurologic findings seen typically include a sixth nerve palsy, but the organism can also affect cranial nerves III, IV, V, VII, and VIII.[53] Sensory deficits, especially in the face, are frequently seen. Papillitis and posterior uveitis have been causally linked to Lyme disease as well; optic neuritis, however, has not.[54]
Workup involves performing an LP, which reveals a lymphocytic pleocytosis with moderately elevated protein and decreased glucose.[53] Diagnosis is performed with serologies, first obtaining an enzyme-linked immunosorbent assay (ELISA) followed by confirmation with a Western blot.[55]
Treatment of neurologic Lyme disease involves a 14- to 28-day course of intravenous or intramuscular ceftriaxone or similar third-generation cephalosporin, although many recent studies have found the use of doxycycline to be equally efficacious in adults.[56–58] Prevention of Lyme disease involves wearing long, light colored clothing so as to better visualize ticks, while repellants containing DEET (diethyl-3-methylbenzamide) are highly effective at preventing tick bites.[59] A vaccine for Lyme disease, approved by the FDA in 1998, has since been taken off the market by its manufacturer due to lack of use.[59]
ENCEPHALITIS
Encephalites are inflammations due to infection of the brain parenchyma, and can be caused by many agents, such as bacterial, viral, fungal, and protozoan. The most common of these agents are viral, and include herpes simplex virus (HSV), cytomegalovirus (CMV), enteroviruses, mumps virus, varicella zoster virus, togaviruses, and flaviviruses.[60] Bacterial causes include syphilis, tuberculosis (discussed above), and many others. Additionally, fungal and protozoan agents are also capable of invading the brain parenchyma, producing rare yet serious illness.
Making the diagnosis of precise etiologic agent involves obtaining a careful history, including travel history, exposure to tick or mosquito bite, immunocompromised status, recent organ transplant, and other unusual exposures. Neuroimaging, especially via MRI, can often reveal lesions specific to particular etiologic agents, including temporal lobe lesions seen in HSV neuroinfection or lesions in the basal ganglia or thalamus seen in West Nile virus and eastern equine encephalitis (EEE).[61,62]
NEUROSYPHILIS
One of the world's oldest infectious diseases, syphilis is caused by the spirochete Treponema pallidum. Although initially spread often as a sexually transmitted infection, disseminated, chronic untreated syphilis can affect all aspects of the nervous system, from peripheral nerves to the brain and spinal cord. Although most chronic forms of syphilis tend to be asymptomatic, a variety of focal neurologic findings, including cranial nerve abnormalities as well as meningismal signs of fever, headache, and stiff neck, are often seen. Less common than these classical symptoms, neurosyphilis also has the capability of presenting with psychiatric disturbances, movement disorders, hearing loss, dementia, stroke-like syndrome, seizures, or even mimicking amyotrophic lateral sclerosis.[63–65]
Diagnosis of neurosyphilis can be quite challenging, as no single test is held to be diagnostic of neurosyphilis.[66] Screening in the population is commonly performed with rapid plasma regain or venereal disease research laboratory (VDRL) testing, usually detecting patients after a primary infection. Because neurologic symptoms typically succeed initial infection by many months or years, in a patient with a past history of syphilis, any abnormal neurologic findings should greatly raise clinical suspicion. CSF-VDRL levels is among the most specific tests for neurosyphilis and can be diagnostic in the proper clinical context, although this test lacks sensitivity.[66] Fluorescent treponemal antibody-absorption (FTA-Abs) testing of CSF has good sensitivity, on the other hand, but is plagued by many false positives.[64,66,67] HIV remains a significant risk factor for neurosyphilis, and clinical suspicion should be raised accordingly.
The treatment consists of aqueous crystalline penicillin, due to its superior penetration across the blood–brain barrier.[67] Patients with penicillin allergies and neurosyphilis should be desensitized to penicillin, as alternative antibiotics are incapable of penetrating the CNS in doses enough to be efficacious.[68]
Herpes simplex encephalitis
Viral encephalitis can often be distinguished from bacterial meningitis by the presence of focal neurologic findings in addition to AMS, especially if new-onset personality or behavioral changes are present [Table 5]. Herpes simplex encephalitis typically affects the temporal lobe. Focal neurologic findings are often found, including seizures originating from the temporal lobe. Symptoms typically begin with a prodrome of fever and headache, and later include memory loss, AMS, and confusion.[60] Psychiatric manifestations may be present, and patients generally present with a delirium, yet can have a variety of other psychiatric symptoms, including agitation, withdrawal, hallucinations—often olfactory, psychosis, or manic symptoms.[60,69] Over 90% of all cases of herpes encephalitis are due to herpes simplex virus type I, whereas the others are due to HSV II.[7,69] Most cases are believed to be due to reactivation of the herpes virus from the trigeminal ganglion or latent virus from the brain parenchyma.[60]
Table 5.
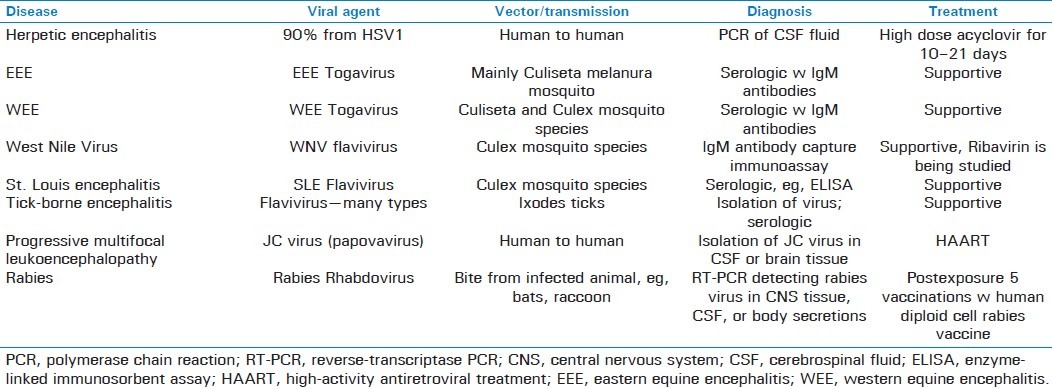
An LP is indicated to look for signs of viral infection in the CSF, such as a normal or reduced glucose level with a pleocytosis and increased number of red cells. Imaging of the brain, such as an MRI, will reveal medial temporal lobe involvement fairly early in the course of illness.[69] PCR assay of the CSF revealing herpes simplex viral DNA is the gold standard for specific diagnosis.[69]
Treatment involves high-dose acyclovir for 10–21 days. Steroids can be used adjunctively, especially if there is concern about herniation.[69] The course is often fatal, especially if treatment is delayed, and can lead to progressive neurologic dysfunction followed by death occurring within 1 month.[69,70] Mortality estimates of acute cases range from 20% to 50% with appropriate therapy.[8] Neonatal herpes encephalitis has a similar clinical picture, but can lead to more chronic neurologic deficits in survivors, with 56%–69% of all survivors suffering long-term neurologic complications after treatment.[70] One significant long-term complication of neonatal herpes encephalitis is the subsequent development of cerebral palsy.[70] Other complications include recurrent epilepsy, postinfectious encephalomyelitis, and permanent neuropsychiatric disorders.[60]
Equine encephalitis
The equine encephalites, including EEE, western equine encephalitis (WEE), and Venezuelan equine encephalitis (VEE) are caused by alphaviruses, single-stranded positive-sense RNA viruses belonging to the Togaviridae family. Although relatively uncommon, these infections, in particular EEE, have the potential to cause much morbidity and possess a high rate of mortality, demanding their urgent and accurate diagnosis by the emergency physician.
EEE has its major incidence along the southeastern United States found in hotter swampy locations inhabited by the mosquito Culiseta melanura, its primary vector; birds are the major reservoir of disease.[71] Clinical presentation is nonspecific, and can involve a typical viral prodrome of flu-like symptoms, including fever, chills, malaise, and myalgias lasting for up to a couple of weeks followed by encephalitis involving severe headache, nausea, vomiting, AMS, and focal neurologic deficits with complications of seizures, nerve palsies, coma, and death.[71,72]
WEE has traditionally been more prevalent than its eastern counterpart, as Sejvar reports that during 1964–2002, 640 human cases of WEE were reported to CDC compared with 200 human cases for EEE during that time span, although Davis et al. revealed that the incidence has dramatically slowed down with only five reported cases in the United States in the past 20 years and none since 1999.[71,73] WEE is typically found in the western and midwestern United States. Symptoms found in WEE are similar to those of EEE, although they tend to be milder, with fewer than 10% developing coma and most recovering fully after several weeks.
Laboratory studies in both diseases reveal a hyponatremia, believed to be secondary to a syndrome of inappropriate antidiuretic hormone (SIADH), in addition to leukocytosis.[71] CSF studies typically show a neutrophil-predominant pleocytosis with elevated protein levels and mildly decreased to normal glucose levels.[71] Additionally, Deresiewicz et al. have demonstrated that MRI can be a valuable modality in the diagnosis of EEE, as patients with EEE demonstrate focal radiologic findings of early involvement of the basal ganglia and thalami visible on T2-weighted imaging.[61] Definitive diagnosis is usually through serology detecting IgM antibodies to viral antigen in the CSF.[16,72]
No specific antiviral therapies currently exist, and management is geared toward treating accompanying complications, such as seizures and increased intracranial pressure.[71] Mortality for EEE estimates a minimum of one out of every three affected, with another third ending up significantly disabled, whereas prognosis is much better in WEE with approximately one in every 20 patients affected succumbing to disease.[16,61,71,72] Although a vaccine has been developed by the United States Army against the VEE virus, no vaccines currently exist for the viruses causing EEE or WEE.[71]
FLAVIVIRUS ENCEPHALITIS
The flaviviruses encompass a large group of viruses causing a widespread range of diseases with high global morbidity and mortality, including the causative agents of West Nile encephalitis, St. Louis encephalitis, Tick-borne encephalitis, Japanese encephalitis, and others.[74]
West Nile
Since its introduction into New York in 1999, the West Nile virus has spread rapidly throughout the United States, with several thousand reported cases since then and the number only expected to rise.[75] Initial presentation of the West Nile virus usually includes nonspecific flu-like symptoms, such as low-grade fever, malaise, headache, nausea, vomiting, and anorexia, in addition to less common symptoms of eye pain, rash, lymphadenopathy; only around 20% of those infected develop symptomatic disease, however.[75,76] Diagnosis is usually serologic, made by the detection of West Nile IgM antibodies in the serum or CSF.[71] False negatives may result, especially early in illness, and therefore serologies ought to be repeated 3–7 days later in clinically suspected patients attaining negative serologies. False positives may result as well, especially in patients with previous exposure to other flavivirus infections, which may cause West Nile ELISA testing to detect cross-reacting antibodies. PCR assay of CSF, and especially blood, is less sensitive than serologies and therefore not relied upon.[71]
Treatment consists of initially placing the affected patient in an isolated room away from mosquitoes, with supportive care measures of fluid, oxygen, and pressors to prevent against hypotension and lactic acidosis.[74] Specific antiviral therapies have not been developed. A live attenuated 17-D vaccine has been developed conferring immunity in 95% of those vaccinated.[74]
St. Louis encephalitis
St. Louis encephalitis is transmitted by a flavivirus primarily carried by mosquito vectors of the Culex genus that often feed on birds. Although prevalent throughout the western hemisphere, St. Louis encephalitis predominantly sees outbreaks occur in the central United States around the Mississippi and Ohio river valleys, as well as parts of Texas, Florida, and the Caribbean.[77] Clinical features include either an encephalitis and/or a viral meningitis picture, often preceded by 1–3 weeks of nonspecific flu-like symptoms, including fever, headache, malaise, myalgia, nausea, vomiting, drowsiness, sore throat, and photophobia. Neurologic deficits are seen in about a quarter of those infected, while seizures present in a tenth of infected patients and indicate a poorer prognosis. Occasionally, a Guillain-Barre syndrome may occur, requiring respiratory support.[77] Laboratory abnormalities include an elevated CSF pressure with a mild increase in protein yet normal glucose levels. Peripherally, an increase in neutrophils with a left shift may be seen. The kidneys are also affected in a quarter of those infected, revealing hematuria, proteinuria, or pyuria with viral antigen frequently being detected in the urinary sediment. Finally, an SIADH picture with decreased sodium in the blood may be seen.
MRI scans of the brain in patients affected by St. Louis encephalitis typically show nonspecific edema, whereas CT scans are normal. Electroencephalograms (EEGs) of the brain often show polymorphic delta activity.[77] Diagnosis can be made via ELISA demonstrating rising antibody titers, and more specifically by RT-PCR.[77] Treatment of this disease is largely supportive, with no specific antivirals devised against the causative agent. The case fatality rate ranges from 7% to 9%, with many experiencing a prolonged convalescence marked by irritability, tremulousness, insomnia, and depression.[77,78]
Tick-borne encephalitis
The tick-borne encephalites comprise a group of several closely related flaviviruses that are carried on forest ticks primarily of the various Ixodes species. Cases mainly occur in the warmer spring and summer months, and the majority of the burden of illness is located in Europe and Russia. Clinical symptoms include a biphasic illness, marked by flu-like symptoms, an encephalitic or meningitic picture, nausea, vomiting, fever, headache, and in more severe cases hemorrhagic fever, focal deficits, stiff neck, paralysis, visual disturbances, convulsions, and coma may occur. European strains are typically less virulent than eastern Russian strains, with case fatality rates around 10% for the European strains while ranging from 20% to 60% in the eastern Russian strains.[77,78] MRI of the brain shows diffuse edema of deeper structures, whereas spinal MRI might show anterior horn lesions.[77] Diagnosis may be made via ELISA or direct isolation of virus from blood. Similar to other flavivirus infections, treatment is largely supportive. Vaccines to many species, however, have been devised and utilized in Europe for higher risk groups.
Progressive multifocal leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease of the brain seen primarily in immunocompromised patients, in particular those with HIV. The causative agent is the JC virus, a papovavirus with a double-stranded circular DNA genome that has a penchant to invade oligodendrocytes. The most common presenting symptom is limb weakness, followed by cognitive disorders.[79] Other symptoms include disorders of speech, gait abnormalities, visual deficits, limb incoordination, seizures, headache, and sensory deficits. On MRI, lesions do not take up gadolinium and are not space-occupying. A prominent diagnosis in the differential is multiple sclerosis (MS), and must be ruled out. Seizures, speech disorders, and aphasia are far more prevalent in progressive multifocal encephalopathy than in MS, whereas sensory deficits and limb incoordination are significantly more prevalent in MS than PML.[79] Interestingly, the treatment of MS can often lead to the acquisition of the JC virus and subsequent PML, due to immune suppression caused by agents, such as natalizumab and other CD4 suppressants.[80,81]
Definitive diagnosis of PML requires the isolation of the JC virus in CSF or brain tissue.[82] There is no effective treatment for progressive multifocal leukoencephalopathy. Although few agents, such as mefloquine and cytosine arabinoside have generated excitement through in vitro experiments, there have been no clinical trials as of yet establishing any agent's efficacy in human cases of PML.[77,83] The most effective treatment strategy in those patients co-infected with HIV is to initiate effective highly active antiretroviral therapy with the goal of improving CD4 counts to levels capable of battling the JC virus.[77]
RABIES
The rabies virus is part of the Rhabdoviridae family of viruses, which comprises a linear, single-stranded negative sense RNA virus found within a bullet-shaped envelop. It is a zoonotic infection, spreading from animals to humans mainly by way of bite, with the primary culprits in the United States being bats, raccoons, and skunks, whereas the primary culprits worldwide are dogs. Rodents and hares on the other hand have not been known to carry or transmit the virus to humans.[84] Although animal bite is the primary mode of transmission, infected aerosolized bat urine from entering heavily infested caves is a significant source of disease. Incubation of the virus typically takes place over 1–3 months, and CNS infection takes place through the process of retrograde spread of the virus from peripheral nerves unto the central nerves and finally into the brain parenchyma.[84,85] Infection with the rabies virus is always fatal once encephalitis occurs, and therefore once a suspected exposure occurs, immediate treatment is of utmost importance.
Diagnosis may be made clinically, often including quarantine or killing of the infected animal while observing for signs of infection in the animal. Definitive diagnosis in humans may be made through reverse-transcriptase PCR detecting rabies virus in CNS tissue, CSF, or body secretions.[77] Management consists of thorough wound cleaning, urgent administration of human rabies immune globulin at both the site of infection and one shot intramuscularly, followed by a set of five postexposure vaccinations within the first month. Vaccination is performed via administration of human diploid cell rabies vaccine, 1.0 mL given intramuscularly.[84] Pre-exposure vaccination involves similar dosing and administration, but employs four shots rather than five, and is recommended for those at a high risk of occupational or recreational exposure.
Primary amoebic meningoencephalitis
Naegleria fowleri, the causative agent of primary amoebic meningoencephalitis, is a common fresh-water amoebic protozoan found throughout shallow bodies of water in the United States. The typical means of obtaining this infection is by diving in fresh water wherein the organism goes up the patient's nasal cavity and penetrates the cribiform plate, making its way to the brain.
LP often reveals amoebas in the CSF. Although infections with this protozoan are very rare, with a little over 300 cases being reported worldwide, they are extremely deadly, with no effective or specific therapies devised against this organism.[86,87]
There have been less than a dozen case reports in the medical literature of cured cases, although high-dose amphoterecin combined with flucytosine or fluconazole and possibly rifampicin confer the best chance of survival.[86,88] Death usually occurs in 1 week.
Cerebral abscess
Cerebral abscesses can frequently present with a new-onset headache that can evolve over several hours to several weeks, accompanied by focal neurologic deficits. Lethargy, nuchal rigidity, nausea, vomiting, and new-onset seizures often accompany the development of abscesses.[89,90] The two major causes of brain abscesses include hematogenous spread of pathogens across the blood–brain barrier and via direct contiguous spread from the sinuses after a sinus infection, with the latter accounting for over 70% of all brain abscesses.[89] A minor etiology involves direct trauma to the skull implanting the pathogenic organism. The differential diagnosis for brain abscesses always involves neoplasm, although abscesses tend to be more acute in their onset and are often accompanied by other systemic symptoms, such as fever and leukocytosis, although these are usually present in only half of all affected patients, and positive blood cultures found in far fewer. History will also be of paramount importance in detecting the likelihood of abscess vs tumor, and it is important to ask about preceding sinus infections, systemic infections, and possible head trauma.
CT or MRI are indicated whenever abscess is suspected, with MRI being more sensitive and specific for detecting various aspects of the abscess, including mass effects, surrounding edema, and response to therapy.[90] Abscesses tend to be located most commonly in the frontal and parietal lobes.[89] Fungal abscesses, though, are more poorly localized upon CT or MRI, and LP plays little value in the workup of brain abscesses as the abscess tends to be more focally confined to one part of the brain and organisms are rarely found in the CSF, leading to false negatives. Furthermore, the risk of possibly fatal transtentorial brain herniation far outweighs the meager sensitivity of detecting causative organism via LP.[89]
Treatment for bacterial abscesses includes either administration of antibiotics alone, or surgical removal of abscess via excision or aspiration. If surgery or aspiration is employed, antibiotics should concomitantly be administered for 3–6 weeks, whereas if antibiotics are given alone a longer course should be employed extending from 6 to 8 weeks.[90,91] Indications for surgical removal include when an abscess is in direct proximity to the ventricular system due to risk of spillage of organisms into the ventricles, when the abscess is in the posterior fossa due to potential mass effects, including compression of the brainstem, or when the abscess is sufficiently large enough, for example >3 cm in diameter, to warrant removal.[90] Serial CT or MRI scans are the diagnostic modality of choice to monitor treatment of abscesses, and the resolution of abscesses correlates proportionally to size seen on imaging, although in cases where antibiotics are given alone there is often a lag with imaging remaining positive weeks longer.[89] Recovery rate correlates with the size of abscess and degree of neurologic dysfunction primarily, with younger patients tending to fare best while immunosuppressed patients often fare worst.[90,92]
Before aspiration, a good history is helpful in identifying the likely causative organism of an abscess. Patients should be asked about recent infections, especially those affecting the sinus cavities, as well as about any potential intravenous drug usage, immunocompromised states, steroid usage, and congenital cardiac conditions, all of which can predispose to the formation of particular abscesses. Streptococcal infections are among the most common causes of brain abscesses, and frequently result from parameningeal spread into the cranial cavity. Anaerobic organisms, such as Bacteroides, Peptococcus, Peptostreptococcus, and Prevotella also frequently enter the brain by way of contiguous spread from either nasal or oropharyngeal origins.[89,90] Staphylococcus aureus, however, while possibly residing the patient's nares, is frequently induced via intravenous drug usage and resulting hematogenous metastases and is far more common in the heroin and steroid-injecting population.
Less frequent causes including Candida are also found in increased amounts in IV drug users, as well as chronically immunosuppressed patients. Although fungal abscesses are relatively rare in the general population, in a study of 58 patients with histology- or culture-proven brain abscess diagnosed between January 1984 and March 1992 at the Fred Hutchinson Cancer Research Center in Seattle, a fungus was isolated in 92% of the cases, with Aspergillus being the most frequent culprit at 58% followed by Candida, underscoring the need to suspect alternative causes of brain abscess in specific patient populations.[92]
The treatment of choice for streptococcal abscesses includes a third-generation cephalosporin, such as ceftriaxone. Anaerobic abscesses respond best to metronidazole, whereas empiric therapy for the treatment of brain abscesses usually involves a combination of both anaerobic and streptococcal coverage. In patients suspected of being infected with S. aureus, the prospect of methicillin resistance must be taken into account, and empiric treatment with vancomycin is indicated. Suspected Candida infections should employ an antifungal agent, such as amphoterecin B and flucytosine, often followed by an oral agent, such as fluconazole, which has good penetration into the CSF.
TOXOPLASMOSIS
Toxoplasmosis, from the causative agent Toxoplasma gondii, frequently occurs in immunocompromised individuals, such as end-stage HIV patients, those on systemic chemotherapy, in patients with hereditary immunologic disorders, or in the infants of pregnant women exposed to cat litter. Congenital toxoplasmosis presents with mental retardation, seizures due to calcification of basal ganglia, blindness, and death in infants.[93] It is also a frequent cause of cerebral palsy, hypothesized to be caused by inflammatory cytokines secondary to the infection; periventricular leukomalacia appears to be the primary identifiable risk factor portending the development of cerebral palsy.[93–98] The classic triad seen involves chorioretinitis, hydrocephalus, and intracellular calcification appearing as ring-enhancing lesions seen on CT scan with contrast.[99] In patients with known history of HIV, toxoplasmosis is considered as an AIDS-defining illness, and is usually seen after the CD4 cell count diminishes to 100 or lower.
Prevention against toxoplasmosis in susceptible patients requires the combination of trimethoprim and sulfamethoxazole. Once toxoplasmosis has already been diagnosed, specific therapy with pyrimethamine and sulfadiazine should be initiated. In neonatal infections, the prospect of “brain cooling” or induction of cerebral hypothermia has recently gained much attention as a potential method of tempering the effects of cytokine-mediated brain injury, thereby diminishing the likelihood of development of cerebral palsy.[95,100] Although studies have failed to reveal a clinically significant effect of treating mothers to prevent perinatal spread to fetus, antibiotic treatment of infected newborns in the first year of life has shown to significantly improve clinical outcome.[99,101–103]
Spinal abscesses
In addition to cerebral abscesses, epidural abscesses may also be a cause of fever accompanied by focal neurologic deficits, with the distinguishing feature of back pain commonly present. Although rare with a presumed occurrence of less than 1 in 5000 hospital admissions, these infections require urgent diagnosis and treatment as they have the potential to lead to permanent neurologic complications.[104,105] Similar to cerebral abscesses, surgical instrumentation is also a frequent etiology, especially in patients undergoing prolonged spinal surgery procedures or those receiving spinal anesthesia. Intravenous drug abuse is a significant risk factor as well, as is diabetes, older age, any immunocompromised state, or spinal penetrating trauma.[105–107] Men tend to be more frequently affected than women.[107] The causative organism most frequently implicated is S. aureus.[104–106] Staphylococcus epidermidis is also frequently prevalent in instrumentation procedures, whereas Pseudomonas aeruginosa plays more of a role in IV drug users and diabetics.[105] In patients with tuberculous spondylitis, M. tuberculosis is the primary organism identified.[105]
Common symptoms of spinal abscesses, in addition to fever and back pain, include sensory and motor deficits, pain in a radicular distribution, paresthesias, meningeal symptoms, and often loss of bowel or bladder function. Back pain is the most common symptom present in nearly 85% of the patients.[105] Sepsis may often result, and initial symptoms may be nonspecific.[106] Physical signs may reveal focal tenderness and warmth, erythema, or edema over the involved area. Clinical suspicion must remain high in appropriate patients, especially those having undergone recent spinal instrumentation.
Whenever spinal abscess is suspected, an urgent workup, including spinal imaging along with neurosurgical consultation ought to be obtained. The imaging technique of choice is MRI, which has greater specificity and sensitivity than contrast-enhanced CT; plain radiographs are typically not helpful.[105] After confirming the diagnosis, empiric antibiotics with staphylococcal coverage ought to be administered intravenously, followed by early neurosurgical decompression.[104,106] Blood cultures should be obtained before the onset of antibiotic therapy, but therapy should not be delayed awaiting results.
Antibiotic therapy ought to be continued for an average of 6 weeks, tailored to the offending organism.[104,105] Mortality estimates range from 6% to 32%, and frequently depend on the onset of initiation of therapy.[105] Recurrence is uncommon in the absence of ongoing risk factors.
Arachnoiditis
Another location for spinal inflammation includes the area surrounding the arachnoid space, which also has the potential for infection known as arachnoiditis. Similar to epidural abscesses, spinal instrumentation is a frequently implicated pathology, with immunocompromise, tuberculosis, and trauma comprising the additional common etiologies of infectious arachnoiditis. Symptoms include burning back pain with paresthesias, fever, neurologic defects, and back spasms.[108] Pain may frequently be worse with movement, and often does not follow radicular distribution.[108] Upon physical examination, both upper and lower motor neuron findings may be present.[108] Although symptoms may frequently be nonspecific, clinical suspicion following spinal instrumentation should be high and radiographic studies with MRI and blood cultures should be obtained immediately.
Treatment with broad-spectrum antibiotics, especially possessing antistaphylococcal coverage, should be initiated. For patients with tuberculous arachnoiditis, treatment with four-drug therapy should be initiated, following de la Blanchardiere et al's demonstration of the value of steroids in preventing neurologic complications in these patients.[109,110] In patients outside the United States, neurocysticercosis represents a significant cause of arachnoiditis, and requires antiparasitic treatment, such as praziquantel, along with steroids if significant edema exists.[111] In addition to antibiotics, treatment with tricyclic antidepressants, anticonvulsants, such as gabapentin, and nonsteroidals may be of benefit in alleviating symptoms.[108,112] Although usually not necessary, surgery may be required in cases of rapidly progressive neurologic deterioration.[108] Physical therapy can be of benefit to patients with more chronic conditions.
Prions
Prions are infective, malfolding protein particles representing a rare cause of encephalopathic disease that can arise as either sporadic mutations in human beings, can be inherited, derived from contaminated meat, or iatrogenically introduced. Diseases such as kuru have resulted from the consumption of human brain, historically affecting the Fore tribe of Papua New Guinea up until the 1950s, whereas other diseases including bovine spongiform encephalopathy (BSE) and scrapie have resulted from the consumption of infected beef. Creutzfeldt–Jakob disease (CJD) is the most common form of prion disease with an incidence of around one per million, and is primarily acquired in 80%–85% by spontaneous malfolding in the PrPc protein causing it to become the abnormal prion protein PrPsc.[113] About 10%–15% of CJD is inherited in an autosomal dominant format, and in several well-documented albeit rare cases, CJD has been spread iatrogenically via mainly corneal transplants, dural graft transplants, and intranasal human growth hormone infusions.[113] Sporadic CJD occurs on average in the sixth to seventh decade of life, whereas inherited conditions tend to become symptomatic in the fourth decade of life.[113] Additionally, there have been documented cases of a new variant CJD, related to BSE, arising over the past 15 years that is under greater risk for spread through blood products; traditional CJD is not believed to be readily transmissible through blood.[114,115] The average age of onset for variant CJD, including BSE-related cases, is significantly lower than spontaneous CJD at 29 years of age. In each case, regardless of the transmission, the pathophysiology of disease revolves around a misfolded prion protein that possesses the capability of enzymatically converting other identical proteins nearby into its own protein configuration, conferring upon it additional properties not seen in native properly folded proteins. These properties include resistance to most common techniques of heat destruction and sterilization, resistance to proteases, enhanced stability, propensity to accumulate in lysosomes, and polymerization into amyloid-like structures in the brain, giving the brain its characteristic spongiform appearance.[113]
Earliest symptoms of infection with prion proteins and resulting encephalopathy present are nonspecific, and include sleep disturbances, lack of coordination, fatigue, cognitive changes, and weight loss. As the disease progresses, the characteristic symptoms of myoclonal jerks and subacute yet rapidly progressive dementia are seen, as well as the typical periodic slow wave complexes seen upon EEG. Diagnosis is made clinically. CSF analysis is typically normal, but may be useful for containing the sensitive but less specific 14-3-3-protein.[113] Furthermore, beta-amyloid peptide 1-42, characteristically decreased in patients with Alzheimer's disease, is also found decreased in CJD.[116,117] Neuroimaging usually reveals no changes in the early stages, but later MRI may show basal ganglia hypersensitivity best seen with diffusion-weighted imaging, and finally followed by diffuse atrophy. Variant CJD due to BSE gives a more characteristic appearance on MRI, with 80% of infected patients revealing a positive Pulvinar sign, indicating prominent enhanced signal in the posterior thalamus.[113]
There are no effective treatments for prion-based diseases, and the clinical course is characterized by a rapidly progressive encephalopathy with dementia and cerebral wasting, with death usually occurring within 1 year.[118] A single randomized double-blinded placebo-controlled trial performed by Otto and colleagues in 2004, however, revealed that the compound flupiritine appeared to be effective in slowing down cognitive deterioration.[119] Vaccines currently being created against prion diseases, in particular against variant CJD, hold promise yet face challenges in their development.[120–122]
Cavernous sinus thrombosis
Although there are many noninfectious causes for cerebral venous sinus thrombosis, infections remain a major and still devastating cause of this condition, occurring in about 10%–15% of cases and previously being associated with a 100% mortality before the advent of antibiotics.[123,124] Septic cavernous sinus thrombosis usually occurs secondarily to a suppurative process of the head or face, including orbital, sphenoid, ethmoid, or other facial sinus involvement that subsequently leads to septic thrombosis of the cavernous sinus.[124] The most frequent causative organism is S. aureus, implicated in 60%–70% of cases.[123] Patients usually present with fever, headache, cranial nerve deficits (mainly involving the third or sixth cranial nerves), and a multitude of other eye symptoms, including ptosis, proptosis, chemosis, orbital pain, double vision, edema, exophthalmos, ophthalmoplegia, or diminished pupillary responses.[123,125] If infection is allowed to spread, mental status changes, including drowsiness, confusion, or coma may result, in addition to meningitis, abscesses, and sepsis.[124]
Cavernous sinus thrombosis may be diagnosed by way of high-resolution CT scan or MRI, in addition to magnetic resonance venography.[123,126] A complete blood count is likely to reveal a leukocytosis, whereas blood cultures are found to be positive with the causative agent in about 70% of the patients.[123] CSF findings resemble those of aseptic meningitis, and may show increased white blood cells with normal protein, glucose, and opening pressure.
Empiric antibiotic therapy should be administered while attempting to diagnose the causative agent, and ought to consist of staphylococcal coverage in addition to a broad-spectrum penicillinase-resistant penicillin or third- or fourth-generation cephalosporin. Intravenous antibiotics should be administered for at least 3–4 weeks; surgical debridement should be performed as needed for primary infections or abscess complications.[123,124] Additionally, anticoagulants, such as heparin—either subcutaneous low–molecular-weight heparins or unfractionated—may be considered.[127,128] Despite our current collection of antibiotics, prognosis for this once universally deadly disease still remains poor, with estimated mortality measuring up to 30% of those infected.[125,129]
CONCLUSION
Infections of the CNS represent a significant source of morbidity and mortality throughout the world, and demand that physicians in a variety of specialties are familiar enough with their presenting signs and symptoms and can formulate a diagnosis in time before further damage arises. Despite advances in vaccinations, meningitis arising from N. meningitides and S. pneumoniae species in adults and children and from Group B Streptococcus, E. coli, and Listeria species in neonates remain common reasons for presentation to the emergency room. Bacterial meningitis must be differentiated from viral meningitis and encephalitis, commonly caused by herpes simplex viruses, varicella, cytomegalovirus, togaviruses, and flaviviruses. Tuberculosis and various fungal meningitis may also present with similar symptoms, although tend to affect debilitated populations. The LP is a valuable method of differentiating empirically between common bacterial vs other causes of meningitis and encephalitis. Abscesses tend to present with focal neurologic deficits, and contain unique precipitating events leading to their formation. Prions and cavernous sinus thrombosis represent rare yet devastating sources of infection. MRI and CT are important imaging techniques typically capable of detecting many infections, although a careful history and physical exam remain invaluable.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Gaston I, Muruzabal J, Quesada P, Maravi E. Infections of the central nervous system in emergency department. An Sist Sanit Navar. 2008;31:99–113. [PubMed] [Google Scholar]
- 2.Cabral DA, Flodmark O, Farrell K, Speert DP. Prospective study of computed tomography in acute bacterial meningitis. J Pediatr. 1987;111:201–5. doi: 10.1016/s0022-3476(87)80067-7. [DOI] [PubMed] [Google Scholar]
- 3.Mace SE. Acute bacterial meningitis. (viii).Emerg Med Clin North Am. 2008;26:281–317. doi: 10.1016/j.emc.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Periappuram M, Taylor MR, Keane CT. Rapid detection of meningococci from petechiae in acute meningococcal infection. J Infect. 1995;31:201–3. doi: 10.1016/s0163-4453(95)80027-1. [DOI] [PubMed] [Google Scholar]
- 5.El Bashir H, Laundy M, Booy R. Diagnosis and treatment of bacterial meningitis. Arch Dis Child. 2003;88:615–20. doi: 10.1136/adc.88.7.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Deuren M, Van Dijke BJ, Koopman RJ, Horrevorts AM, Meis JF, Santman FW, et al. Rapid diagnosis of acute meningococcal infections by needle aspiration or biopsy of skin lesions. BMJ. 1993;306:1229–32. doi: 10.1136/bmj.306.6887.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitch MT, Abrahamian FM, Moran GJ, Talan DA. Emergency department management of meningitis and encephalitis. (v-vi).Infect Dis Clin North Am. 2008;22:33–52. doi: 10.1016/j.idc.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 8.De Gans J, Van de Beek D. Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347:1549–56. doi: 10.1056/NEJMoa021334. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen TH, Tran TH, Thwaites G, Ly VC, Dinh XS, Ho Dang TN, et al. Dexamethasone in Vietnamese adolescents and adults with bacterial meningitis. N Engl J Med. 2007;357:2431–40. doi: 10.1056/NEJMoa070852. [DOI] [PubMed] [Google Scholar]
- 10.Tyler KL. Neurological infections: Advances in therapy, outcome, and prediction. Lancet Neurol. 2009;8:19–21. doi: 10.1016/S1474-4422(08)70279-5. [DOI] [PubMed] [Google Scholar]
- 11.Weisberg S. Pneumococcus. Disease-a-Month. 2007;53:495–502. [Google Scholar]
- 12.Centers for Disease Control and Prevention (CDC). Invasive pneumococcal disease in children 5 years after conjugate vaccine introduction--eight states, 1998-2005. MMWR Morb Mortal Wkly Rep. 2008;57:144–8. [PubMed] [Google Scholar]
- 13.Black S, France EK, Isaacman D, Bracken L, Lewis E, Hansen J, et al. Surveillance for invasive pneumococcal disease during 2000-2005 in a population of children who received 7-valent pneumococcal conjugate vaccine. Pediatr Infect Dis J. 2007;26:771–7. doi: 10.1097/INF.0b013e318124a494. [DOI] [PubMed] [Google Scholar]
- 14.Lynch JP, 3rd, Zhanel GG. Streptococcus pneumoniae: Epidemiology, risk factors, and strategies for prevention. Semin Respir Crit Care Med. 2009;30:189–209. doi: 10.1055/s-0029-1202938. [DOI] [PubMed] [Google Scholar]
- 15.Henry M. Haemophilus influenza type B. World Health Organization fact sheet. 2005. [Accessed June 4, 2010]. Retrieved from http://www.who.int/mediacentre/factsheets/fs294/en/
- 16.Hans D, Kelly E, Wilhelmson K, Katz ED. Rapidly fatal infections. (vii).Emerg Med Clin North Am. 2008;26:259–79. doi: 10.1016/j.emc.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koenig JM, Keenan WJ. Group B streptococcus and early-onset sepsis in the era of maternal prophylaxis. Pediatr Clin North Am. 2009;56:689–708. doi: 10.1016/j.pcl.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrag S, Gorwitz R, Fultz-Butts K, Schuchat A. Prevention of perinatal group B streptococcal disease. MMWR Recomm Rep. 2002;51:1–22. [PubMed] [Google Scholar]
- 19.Winn HN. Group B streptococcus infection in pregnancy. Clin Perinatol. 2007;34:387–92. doi: 10.1016/j.clp.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 20.Beckmann C, Ling F, Smith R, Barzansky B, Herbert W, Laube D. Obstet Gynecol. 5th edition. 2006. Medical and surgical conditions of pregnancy; pp. 179–81. [Google Scholar]
- 21.Bedford H, De Louvois J, Halket S, Peckham C, Hurley R, Harvey D. Meningitis in infancy in England and Wales: Follow up at age 5 years. BMJ. 2001;323:533–6. doi: 10.1136/bmj.323.7312.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bizzarro MJ, Raskind C, Baltimore RS, Gallagher PG. Seventy-five years of neonatal sepsis at Yale: 1928-2003. Pediatrics. 2005;116:595–602. doi: 10.1542/peds.2005-0552. [DOI] [PubMed] [Google Scholar]
- 23.Galiza EP, Heath PT. Improving the outcome of neonatal meningitis. Curr Opin Infect Dis. 2009;22:229–34. doi: 10.1097/QCO.0b013e32832ad49e. [DOI] [PubMed] [Google Scholar]
- 24.Heath PT, Balfour G, Weisner AM, Efstratiou A, Lamagni TL, Tighe H, et al. Group B streptococcal disease in UK and Irish infants younger than 90 days. Lancet. 2004;363:292–4. doi: 10.1016/S0140-6736(03)15389-5. [DOI] [PubMed] [Google Scholar]
- 25.Madappa T. Escherichia Coli Infections. (eMedicine) February 19, 2009. [Accessed June 3, 2009]. Retrieved from < http://emedicine.medscape.com/article/217485-overview. > .
- 26.Kim BY, Kang J, Kim KS. Invasion processes of pathogenic Escherichia coli. Int J Med Microbiol. 2005;295:463–70. doi: 10.1016/j.ijmm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Doganay M. Listeriosis: Clinical presentation. FEMS Immunol Med Microbiol. 2003;35:173–5. doi: 10.1016/S0928-8244(02)00467-4. [DOI] [PubMed] [Google Scholar]
- 28.Fanos V, Dall’Agnola A. Antibiotics in neonatal infections: A review. Drugs. 1999;58:405–27. doi: 10.2165/00003495-199958030-00003. [DOI] [PubMed] [Google Scholar]
- 29.Yildiz O, Aygen B, Esel D, Kayabas U, Alp E, Sumerkan B, et al. Sepsis and meningitis due to Listeria monocytogenes. Yonsei Med J. 2007;48:433–9. doi: 10.3349/ymj.2007.48.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rock RB, Olin M, Baker CA, Molitor TW, Peterson PK. Central nervous system tuberculosis: Pathogenesis and clinical aspects. Clin Microbiol Rev. 2008;21:243–61. doi: 10.1128/CMR.00042-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myers JN. Miliary, central nervous system, and genitourinary tuberculosis. Dis Mon. 2007;53:22–31. doi: 10.1016/j.disamonth.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Marx J. Rosen's emergency medicine: Concepts and clinical practice. Philadelphia: Mosby/Elsevier; 2006. May 8- Rosen P, Emergency M. [Google Scholar]
- 33.Chayakulkeeree M, Perfect JR. Cryptococcosis. (v-vi).Infect Dis Clin North Am. 2006;20:507–44. doi: 10.1016/j.idc.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell TG, Perfect JR. Cryptococcosis in the era of AIDS--100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev. 1995;8:515–48. doi: 10.1128/cmr.8.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hajjeh RA, Conn LA, Stephens DS, Baughman W, Hamill R, Graviss E, et al. Cryptococcosis: Population-based multistate active surveillance and risk factors in human immunodeficiency virus-infected persons. Cryptococcal Active Surveillance Group. J Infect Dis. 1999;179:449–54. doi: 10.1086/314606. [DOI] [PubMed] [Google Scholar]
- 36.Safdieh JE, Mead PA, Sepkowitz KA, Kiehn TE, Abrey LE. Bacterial and fungal meningitis in patients with cancer. Neurology. 2008;70:943–7. doi: 10.1212/01.wnl.0000305960.85546.5f. [DOI] [PubMed] [Google Scholar]
- 37.Saag MS, Graybill RJ, Larsen RA, Pappas PG, Perfect JR, Powderly WG, et al. Practice guidelines for the management of cryptococcal disease. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:710–8. doi: 10.1086/313757. [DOI] [PubMed] [Google Scholar]
- 38.Thompson GR, 3rd, Cadena J, Patterson TF. Overview of antifungal agents. (v).Clin Chest Med. 2009;30:203–15. doi: 10.1016/j.ccm.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Friedman GD, Jeffrey Fessel W, Udaltsova NV, Hurley LB. Cryptococcosis: The 1981-2000 epidemic. Mycoses. 2005;48:122–5. doi: 10.1111/j.1439-0507.2004.01082.x. [DOI] [PubMed] [Google Scholar]
- 40.Van der Horst CM, Saag MS, Cloud GA, Hamill RJ, Graybill JR, Sobel JD, et al. Treatment of cryptococcal meningitis associated with the acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Mycoses Study Group and AIDS Clinical Trials Group. N Engl J Med. 1997;337:15–21. doi: 10.1056/NEJM199707033370103. [DOI] [PubMed] [Google Scholar]
- 41.Anstead GM, Graybill JR. Coccidioidomycosis. Infect Dis Clin North Am. 2006;20:621–43. doi: 10.1016/j.idc.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Williams PL. Coccidioidal meningitis. Ann N Y Acad Sci. 2007;1111:377–84. doi: 10.1196/annals.1406.037. [DOI] [PubMed] [Google Scholar]
- 43.Ampel NM. Coccidioidomycosis: A review of recent advances. (v).Clin Chest Med. 2009;30:241–51. doi: 10.1016/j.ccm.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 44.McKinnell JA, Pappas PG. Blastomycosis: New insights into diagnosis, prevention, and treatment. (v).Clin Chest Med. 2009;30:227–39. doi: 10.1016/j.ccm.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 45.Wheat LJ, Musial CE, Jenny-Avital E. Diagnosis and management of central nervous system histoplasmosis. Clin Infect Dis. 2005;40:844–52. doi: 10.1086/427880. [DOI] [PubMed] [Google Scholar]
- 46.Kauffman CA. Endemic mycoses: Blastomycosis, histoplasmosis, and sporotrichosis. (vii).Infect Dis Clin North Am. 2006;20:645–62. doi: 10.1016/j.idc.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 47.Bakleh M, Aksamit AJ, Tleyjeh IM, Marshall WF. Successful treatment of cerebral blastomycosis with voriconazole. Clin Infect Dis. 2005;40:e69–71. doi: 10.1086/429319. [DOI] [PubMed] [Google Scholar]
- 48.Borgia SM, Fuller JD, Sarabia A, El-Helou P. Cerebral blastomycosis: A case series incorporating voriconazole in the treatment regimen. Med Mycol. 2006;44:659–64. doi: 10.1080/13693780600803870. [DOI] [PubMed] [Google Scholar]
- 49.Panicker J, Walsh T, Kamani N. Recurrent central nervous system blastomycosis in an immunocompetent child treated successfully with sequential liposomal amphotericin B and voriconazole. Pediatr Infect Dis J. 2006;25:377–9. doi: 10.1097/01.inf.0000207475.89745.51. [DOI] [PubMed] [Google Scholar]
- 50.Pappas PG. Invasive candidiasis. Infect Dis Clin North Am. 2006;20:485–506. doi: 10.1016/j.idc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Patel NP, Malangoni MA. Antimicrobial agents for surgical infections. (vii).Surg Clin North Am. 2009;89:327–47. doi: 10.1016/j.suc.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 52.Feder HM., Jr Lyme disease in children. (vii).Infect Dis Clin North Am. 2008;22:315–26. doi: 10.1016/j.idc.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 53.Halperin JJ. (2008) Nervous system Lyme disease. (vi).Infect Dis Clin North Am. 2008;22:261–74. doi: 10.1016/j.idc.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol. 2005;25:71–82. doi: 10.1097/01.wno.0000166060.35366.70. [DOI] [PubMed] [Google Scholar]
- 55.DePietropaolo DL, Powers JH, Gill JM, Foy AJ. Diagnosis of lyme disease. Am Fam Physician. 2005;72:297–304. [PubMed] [Google Scholar]
- 56.Borg R, Dotevall L, Hagberg L, Maraspin V, Lotric-Furlan S, Cimperman J, et al. Intravenous ceftriaxone compared with oral doxycycline for the treatment of Lyme neuroborreliosis. Scand J Infect Dis. 2005;37:449–54. doi: 10.1080/00365540510027228. [DOI] [PubMed] [Google Scholar]
- 57.Halperin JJ, Shapiro ED, Logigian E, Belman AL, Dotevall L, Wormser GP, et al. Practice parameter: Treatment of nervous system Lyme disease (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2007;69:91–102. doi: 10.1212/01.wnl.0000265517.66976.28. [DOI] [PubMed] [Google Scholar]
- 58.Ljostad U, Skogvoll E, Eikeland R, Midgard R, Skarpaas T, Berg A, et al. Oral doxycycline versus intravenous ceftriaxone for European Lyme neuroborreliosis: A multicentre, non-inferiority, double-blind, randomised trial. Lancet Neurol. 2008;7:690–5. doi: 10.1016/S1474-4422(08)70119-4. [DOI] [PubMed] [Google Scholar]
- 59.Clark RP, Hu LT. Prevention of lyme disease and other tick-borne infections. (vii).Infect Dis Clin North Am. 2008;22:381–96. doi: 10.1016/j.idc.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziai WC, Lewin JJ., 3rd Update in the diagnosis and management of central nervous system infections. (viii).Neurol Clin. 2008;26:427–68. doi: 10.1016/j.ncl.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 61.Deresiewicz RL, Thaler SJ, Hsu L, Zamani AA. Clinical and neuroradiographic manifestations of eastern equine encephalitis. N Engl J Med. 1997;336:1867–74. doi: 10.1056/NEJM199706263362604. [DOI] [PubMed] [Google Scholar]
- 62.Wright EJ, Brew BJ, Wesselingh SL. Pathogenesis and diagnosis of viral infections of the nervous system. (vii).Neurol Clin. 2008;26:617–33. doi: 10.1016/j.ncl.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 63.Friedrich F, Geusau A, Greisenegger S, Ossege M, Aigner M. Manifest psychosis in neurosyphilis. Gen Hosp Psychiatry. 2009;31:379–81. doi: 10.1016/j.genhosppsych.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 64.Madhusudhan M. Neurosyphilis. Neurol India. 2009;57:233–4. doi: 10.4103/0028-3886.53259. [DOI] [PubMed] [Google Scholar]
- 65.Tso MK, Koo K, Tso GY. Neurosyphilis in a Non-HIV Patient: More than a Psychiatric Concern. Mcgill J Med. 2008;11:160–3. [PMC free article] [PubMed] [Google Scholar]
- 66.Domantay-Apostol GP, Handog EB, Gabriel MT. Syphilis: The international challenge of the great imitator. (v).Dermatol Clin. 2008;26:191–202. doi: 10.1016/j.det.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Kent ME, Romanelli F. Reexamining syphilis: An update on epidemiology, clinical manifestations, and management. Ann Pharmacother. 2008;42:226–36. doi: 10.1345/aph.1K086. [DOI] [PubMed] [Google Scholar]
- 68.Birnbaum NR, Goldschmidt RH, Buffett WO. Resolving the common clinical dilemmas of syphilis. (2245-6).Am Fam Physician. 1999;59:2233–40. [PubMed] [Google Scholar]
- 69.Moore DP, Jefferson JW, Boland R. Handbook of medical psychiatry. 2nd Ed. New York: Elsevier Mosby; 2004. Herpes simplex encephalitis. [Google Scholar]
- 70.Engman ML, Adolfsson I, Lewensohn-Fuchs I, Forsgren M, Mosskin M, Malm G. Neuropsychologic outcomes in children with neonatal herpes encephalitis. Pediatr Neurol. 2008;38:398–405. doi: 10.1016/j.pediatrneurol.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 71.Davis LE, Beckham JD, Tyler KL. North American encephalitic arboviruses. (ix).Neurol Clin. 2008;26:727–57. doi: 10.1016/j.ncl.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lury KM, Castillo M. Eastern equine encephalitis: CT and MRI findings in one case. Emerg Radiol. 2004;11:46–8. doi: 10.1007/s10140-004-0350-7. [DOI] [PubMed] [Google Scholar]
- 73.Sejvar JJ. The evolving epidemiology of viral encephalitis. Curr Opin Neurol. 2006;19:350–7. doi: 10.1097/01.wco.0000236613.74462.4c. [DOI] [PubMed] [Google Scholar]
- 74.Tsai T, Vaughn D, Solomon T, Mandell G, Bennett J, Dolin R. Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2005. Flaviviruses (yellow fever, dengue hemorrhagic fever, Japanese encephalitis, St Louis encephalitis, tick-borne encephalitis) pp. 1926–50. [Google Scholar]
- 75.Mazurek JM, Winpisinger K, Mattson BJ, Duffy R, Moolenaar RL. The epidemiology and early clinical features of West Nile virus infection. Am J Emerg Med. 2005;23:536–43. doi: 10.1016/j.ajem.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 76.Ferri F. Ferri's clinical advisor 2009. Philadelphia, Pa: Elsevier Mosby; 2009. West Nile Virus infection; pp. 1188–89. [Google Scholar]
- 77.Bleck TP. Cecil Medicine DOI. 23rd ed. Philadelphia, Pa: Elsevier; 2007. Arthropod-borne viruses affecting the central nervous system; pp. 2541–49. [Google Scholar]
- 78.Gould EA, Solomon T. Pathogenic flaviviruses. Lancet. 2008;371:500–9. doi: 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- 79.Weber T. Progressive multifocal leukoencephalopathy. (x-xi).Neurol Clin. 2008;26:833–54. doi: 10.1016/j.ncl.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 80.Stuve O, Gold R, Chan A, Mix E, Zettl U, Kieseier BC. Alpha4-Integrin antagonism with natalizumab: Effects and adverse effects. J Neurol. 2008;255:58–65. doi: 10.1007/s00415-008-6011-0. [DOI] [PubMed] [Google Scholar]
- 81.Letendre SL, Ellis RJ, Everall I, Ances B, Bharti A, McCutchan JA. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2009;17:46–56. [PMC free article] [PubMed] [Google Scholar]
- 82.Cinque P, Koralnik IJ, Gerevini S, Miro JM, Price RW. Progressive multifocal leukoencephalopathy in HIV-1 infection. Lancet Infect Dis. 2009;9:625–36. doi: 10.1016/S1473-3099(09)70226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berger JR, Houff SA. Neurological infections: The year of PML and influenza. Lancet Neurol. 2010;9:14–7. doi: 10.1016/S1474-4422(09)70337-0. [DOI] [PubMed] [Google Scholar]
- 84.Fulton S, Salata R. 10Infections of the nervous system. Andreoli and Carpenter's Cecil Essentials of Medicine. 2004:933–44. [Google Scholar]
- 85.Somand D, Meurer W. Central nervous system infections. (ix).Emerg Med Clin North Am. 2009;27:89–100. doi: 10.1016/j.emc.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 86.Jain R, Prabhakar S, Modi M, Bhatia R, Sehgal R. Naegleria meningitis: A rare survival. Neurol India. 2002;50:470–2. [PubMed] [Google Scholar]
- 87.Wiwanitkit V. Review of clinical presentations in Thai patients with primary amoebic meningoencephalitis. Med Gen Med. 2004;6:2. [PMC free article] [PubMed] [Google Scholar]
- 88.Vargas-Zepeda J, Gomez-Alcala AV, Vasquez-Morales JA, Licea-Amaya L, De Jonckheere JF, Lares-Villa F. Successful treatment of Naegleria fowleri meningoencephalitis by using intravenous amphotericin B, fluconazole and rifampicin. Arch Med Res. 2005;36:83–6. doi: 10.1016/j.arcmed.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 89.Chun CH, Johnson JD, Hofstetter M, Raff MJ. Brain abscess. A study of 45 consecutive cases. Medicine (Baltimore) 1986;65:415–31. [PubMed] [Google Scholar]
- 90.Nath A. (2010) Infectious diseases of the nervous system. Andreoli and Carpenter's Cecil Essentials of Medicine. 2010:1159–1164. [Google Scholar]
- 91.Buck DW, West MA. Contemporary Guide to Surgical Infections. 1st Ed. Newtown, Pa: 2008. Brain and central nervous system; pp. 69–75. [Google Scholar]
- 92.Hagensee ME, Bauwens JE, Kjos B, Bowden RA. Brain abscess following marrow transplantation: Experience at the Fred Hutchinson Cancer Research Center, 1984-1992. Clin Infect Dis. 1994;19:402–8. doi: 10.1093/clinids/19.3.402. [DOI] [PubMed] [Google Scholar]
- 93.Hermansen MC, Hermansen MG. Perinatal infections and cerebral palsy. Clin Perinatol. 2006;33:315–33. doi: 10.1016/j.clp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 94.Girard S, Kadhim H, Roy M, Lavoie K, Brochu ME, Larouche A, et al. Role of perinatal inflammation in cerebral palsy. Pediatr Neurol. 2009;40:168–74. doi: 10.1016/j.pediatrneurol.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 95.Gunn AJ, Bennet L. Brain cooling for preterm infants. Clin Perinatol. 2008;35:735–48. doi: 10.1016/j.clp.2008.07.012. vi-vii. [DOI] [PubMed] [Google Scholar]
- 96.Rezaie P, Dean A. Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology. 2002;22:106–32. doi: 10.1046/j.1440-1789.2002.00438.x. [DOI] [PubMed] [Google Scholar]
- 97.Schendel DE. Infection in pregnancy and cerebral palsy. J Am Med Womens Assoc. 2001;56:105–8. [PubMed] [Google Scholar]
- 98.Yoon BH, Park CW, Chaiworapongsa T. Intrauterine infection and the development of cerebral palsy. BJOG. 2003;20:124–7. doi: 10.1016/s1470-0328(03)00063-6. [DOI] [PubMed] [Google Scholar]
- 99.Gras L, Gilbert RE, Ades AE, Dunn DT. Effect of prenatal treatment on the risk of intracranial and ocular lesions in children with congenital toxoplasmosis. Int J Epidemiol. 2001;30:1309–13. doi: 10.1093/ije/30.6.1309. [DOI] [PubMed] [Google Scholar]
- 100.Berger R, Garnier Y, Jensen A. Perinatal brain damage: Underlying mechanisms and neuroprotective strategies. J Soc Gynecol Investig. 2002;9:319–28. [PubMed] [Google Scholar]
- 101.Gilbert RE, Gras L, Wallon M, Peyron F, Ades AE, Dunn DT. Effect of prenatal treatment on mother to child transmission of Toxoplasma gondii: Retrospective cohort study of 554 mother-child pairs in Lyon, France. Int J Epidemiol. 2001;30:1303–8. doi: 10.1093/ije/30.6.1303. [DOI] [PubMed] [Google Scholar]
- 102.Montoya JG, Remington JS. Management of Toxoplasma gondii infection during pregnancy. Clin Infect Dis. 2008;47:554–66. doi: 10.1086/590149. [DOI] [PubMed] [Google Scholar]
- 103.Thiebaut R, Leproust S, Chene G, Gilbert R. Effectiveness of prenatal treatment for congenital toxoplasmosis: A meta-analysis of individual patients’ data. Lancet. 2007;369:115–22. doi: 10.1016/S0140-6736(07)60072-5. [DOI] [PubMed] [Google Scholar]
- 104.Chao D, Nanda A. Spinal epidural abscess: A diagnostic challenge. Am Fam Physician. 2002;65:1341–6. [PubMed] [Google Scholar]
- 105.Pradilla G, Ardila GP, Hsu W, Rigamonti D. Epidural abscesses of the CNS. Lancet Neurol. 2009;8:292–300. doi: 10.1016/S1474-4422(09)70044-4. [DOI] [PubMed] [Google Scholar]
- 106.Chan YC, Dasey N. Iatrogenic spinal epidural abscess. Acta Chir Belg. 2007;107:109–18. [PubMed] [Google Scholar]
- 107.Reihsaus E, Waldbaur H, Seeling W. Spinal epidural abscess: A meta-analysis of 915 patients. Neurosurg Rev. 2000;23:175–204. doi: 10.1007/pl00011954. [DOI] [PubMed] [Google Scholar]
- 108.Osborne MD. Essentials of Physical Medicine and Rehabilitation. Philadelphia, Pa: Saunders; 2008. Arachnoiditis; pp. 565–68. [Google Scholar]
- 109.De La Blanchardiere A, Stern JB, Molina JM, Lesprit P, Gasnault J, De Kerviler E, et al. Spinal tuberculous arachnoiditis] Presse Med. 1996;25:1333–5. [PubMed] [Google Scholar]
- 110.Srivastava T, Kochar DK. Asymptomatic spinal arachnoiditis in patients with tuberculous meningitis. Neuroradiology. 2003;45:727–9. doi: 10.1007/s00234-003-1077-y. [DOI] [PubMed] [Google Scholar]
- 111.Mansur M. Cysticercosis. eMedicine. 2008. [Accessed July 19, 2009]. Retrieved from http://emedicine.medscape.com/article/215589-overview .
- 112.Covington E. Chronic pain management in spine disorders. Neurol Clin. 2007;25:539–66. doi: 10.1016/j.ncl.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 113.Eggenberger E. Prion disease. (viii).Neurol Clin. 2007;25:833–42. doi: 10.1016/j.ncl.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 114.Turner ML, Ironside JW. New-variant Creutzfeldt-Jakob disease: The risk of transmission by blood transfusion. Blood Rev. 1998;12:255–68. doi: 10.1016/s0268-960x(98)90007-8. [DOI] [PubMed] [Google Scholar]
- 115.Will RG, Kimberlin RH. Creutzfeldt-Jakob disease and the risk from blood or blood products. Vox Sang. 1998;75:178–80. [PubMed] [Google Scholar]
- 116.Otto M, Esselmann H, Schulz-Shaeffer W, Neumann M, Schroter A, Ratzka P, et al. Decreased beta-amyloid1-42 in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurology. 2000;54:1099–102. doi: 10.1212/wnl.54.5.1099. [DOI] [PubMed] [Google Scholar]
- 117.Wiltfang J, Esselmann H, Smirnov A, Bibl M, Cepek L, Steinacker P, et al. Beta-amyloid peptides in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Ann Neurol. 2003;54:263–7. doi: 10.1002/ana.10661. [DOI] [PubMed] [Google Scholar]
- 118.Stewart LA, Rydzewska LH, Keogh GF, Knight RS. Systematic review of therapeutic interventions in human prion disease. Neurology. 2008;70:1272–81. doi: 10.1212/01.wnl.0000308955.25760.c2. [DOI] [PubMed] [Google Scholar]
- 119.Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology. 2004;62:714–8. doi: 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- 120.Muller-Schiffmann A, Korth C. Vaccine approaches to prevent and treat prion infection: Progress and challenges. BioDrugs. 2008;22:45–52. doi: 10.2165/00063030-200822010-00005. [DOI] [PubMed] [Google Scholar]
- 121.Sakaguchi S. Prospects for preventative vaccines against prion diseases. Protein Pept Lett. 2009;16:260–70. doi: 10.2174/092986609787601804. [DOI] [PubMed] [Google Scholar]
- 122.Sakaguchi S, Arakawa T. Recent developments in mucosal vaccines against prion diseases. Expert Rev Vaccines. 2007;6:75–85. doi: 10.1586/14760584.6.1.75. [DOI] [PubMed] [Google Scholar]
- 123.Ebright JR, Pace MT, Niazi AF. Septic thrombosis of the cavernous sinuses. Arch Intern Med. 2001;161:2671–6. doi: 10.1001/archinte.161.22.2671. [DOI] [PubMed] [Google Scholar]
- 124.Laupland KB. Vascular and parameningeal infections of the head and neck. (viii).Infect Dis Clin North Am. 2007;21:577–90. doi: 10.1016/j.idc.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 125.Southwick FS, Richardson EP, Jr, Swartz MN. Septic thrombosis of the dural venous sinuses. Medicine (Baltimore) 1986;65:82–106. doi: 10.1097/00005792-198603000-00002. [DOI] [PubMed] [Google Scholar]
- 126.Hurley MC, Heran MK. Imaging studies for head and neck infections. (v-vi).Infect Dis Clin North Am. 2007;21:305–53. doi: 10.1016/j.idc.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 127.Bhatia K, Jones NS. Septic cavernous sinus thrombosis secondary to sinusitis: Are anticoagulants indicated? A review of the literature. J Laryngol Otol. 2002;116:667–76. doi: 10.1258/002221502760237920. [DOI] [PubMed] [Google Scholar]
- 128.Bousser MG, Ferro JM. Cerebral venous thrombosis: An update. Lancet Neurol. 2007;6:162–70. doi: 10.1016/S1474-4422(07)70029-7. [DOI] [PubMed] [Google Scholar]
- 129.DiNubile MJ. Septic thrombosis of the cavernous sinuses. Arch Neurol. 1988;45:567–72. doi: 10.1001/archneur.1988.00520290103022. [DOI] [PubMed] [Google Scholar]