

Abstract
Negative pressure pulmonary edema (NPPE) following the use of the laryngeal mask airway (LMA) is an uncommon and under-reported event. We present a case of a 58-year-old male, who developed NPPE following LMA use. After biting vigorously on his LMA, the patient developed stridor upon emergence, with concurrent appearance of blood-tinged, frothy sputum and pulmonary edema. He subsequently required three days of mechanical ventilation. After discontinuation of mechanical ventilation the patient continued to require additional pulmonary support using continuous positive airway pressure, with a full facemask, to correct the persistent hypoxemia. His roentgenographic findings demonstrated an accelerated improvement with judicious administration of intravenous furosemide.
Keywords: Airway, edema, education, hypoxia, ventilation
INTRODUCTION
Negative pressure pulmonary edema (NPPE) is an uncommon, but potentially life-threatening complication of general anesthesia. It is usually seen during emergence from anesthesia. In healthy adults undergoing general anesthesia, the incidence of NPPE is 0.05 to 0.1%.[1–3] It is even less common with the use of a laryngeal mask airway (LMA).[4–8] The sequelae of acute NPPE can be devastating with mortality as high as 11 – 40%.[9] NPPE can complicate the clinical course and outcome of otherwise healthy patients undergoing simple surgical procedures. Requirement for prolonged endotracheal intubation and intensive care is not uncommon. Early recognition and institution of appropriate positive pressure ventilation is important to ensuring successful outcomes. An unusual case of acute NPPE associated with a patient forcefully biting down on the LMA during emergence is presented. A review of pathophysiology, risk factors, and management strategies for NPPE then follows.
CASE REPORT
A 58-year-old male presented to the Emergency Department with a three-day history of fever, worsening perineal erythema, and pain. His medical history revealed only mild seasonal allergies; his functional status was very good. His surgical history was significant for an inguinal herniorrhaphy three years prior to his current presentation. His medications consisted of fexofenadine and pseudoephedrine for seasonal allergies. He was not in acute distress. On physical examination, he was noted to have significant perineal cellulitis and pain. His preoperative chest X-ray revealed no infiltrates, pulmonary edema or cardiomegaly. Computed tomography (CT) demonstrated extensive inflammation of the perineum and scrotum as well as subcutaneous air, consistent with the diagnosis of Fournier's gangrene. Preoperatively the patient was normotensive, but tachycardic (heart rate 110 – 120 beats / minute). No cardiac murmurs were identified. His electrocardiogram on admission to the Intensive Care Unit (ICU) did not show any ST, T-wave changes. He did not demonstrate a third heart sound, peripheral edema, or jugular venous distention. His lungs were clear on auscultation bilaterally. His baseline SpO2 was 98% on room air. His hemoglobin and hematocrit were 14.9 g / dL and 44%, white blood cell count was 12.7 × 103 / mL, and platelet count was 146,000 / mL. His electrolyte panel featured sodium of 135 mmol / L, potassium 4.3 mmol / L, chloride 99 mmol / L, HCO3 26 mmol / L, blood urea nitrogen 16 mg / dL, and creatinine 1.14 mg / dL. He was without nourishment for > 8 hours preoperatively.
The patient was taken to the Operating Room for emergency surgical debridement of the Fournier's gangrene. He was premedicated with 2 milligrams (mg) of midazolam intravenously (IV) and anesthesia was induced with IV propofol (200 mg) and fentanyl (100 micrograms [mcg]). There was no central venous pressure monitor placed. A laryngeal mask airway (LMA, size number 4) was placed without difficulty. Anesthesia was maintained with sevoflurane 2 – 3%, with 50% oxygen and air. The patient received fentanyl 500 mcg and 1 mg of dilaudid for intraoperative analgesia. He remained hemodynamically stable throughout, but was tachypneic, with a respiratory rate of 35 breaths / minute. Surgical blood loss was ~200 mL and the procedure was otherwise uneventful. At the conclusion of surgery sevoflurane was discontinued and the patient was able to follow commands. However, before LMA removal the patient forcefully bit down on the LMA. Immediately after LMA removal, a laryngospasm was noted, with concurrent tachycardia and hypertension. Application of positive pressure via face mask (FiO2 100%) was unsuccessful in providing effective ventilation. Intravenous propofol and succinylcholine were administered to facilitate orotracheal intubation with a 7.5 endotracheal tube (ETT). Auscultation revealed bilateral rales, and pink frothy secretions were suctioned from the ETT. He did develop a brief episode of tachycardia (120 – 130 beats per minute) and hypertension (170 – 180 / 90 – 100 mmHg) on extubation. His SpO2 remained at 85 – 88% despite an FiO2 of 100%. His arterial blood gas (ABG) in the operating room showed pH 7.27, PaCO2 59 mmHg, PaO2 46 mmHg, HCO3 of 16 mmol / L, base excess of –1.9, and oxygen saturation of 75%, with a lactate of 1.27 mmol / L. He was on synchronized intermittent mechanical ventilation (SIMV) at 12 cycles / minute, tidal volume 750 mL, pressure support 10 cm H2O, with positive end-expiratory pressure (PEEP) of 10 cm H2O. After PEEP was increased to 12 cm H2O, his SpO2 recovered to > 90%. Of note, his peak and plateau pressures were only minimally elevated and his ability to generate a negative pressure was adequate (> –25 cm H2O). He was transferred to the intensive care unit (ICU) where he was continued on SIMV, with a tidal volume of 580 mL, FiO2 100%, pressure support 15 cm H2O, and PEEP of 12 cm H2O. His initial ABG in the ICU showed pH 7.6, PaCO2 25 mmHg, PaO2 165 mmHg, HCO3 27 mmol / L, and SpO2 99% (FiO2 of 80%). Appropriate ventilatorory adjustments were made. An ICU admission chest radiograph demonstrated bilateral patchy infiltrates, no pneumothoraces or effusions, and a normal heart size; these changes were central and peripheral and not in the dependent areas of the lung [Figure 1]. Over the subsequent 12 hours his FiO2 was weaned to 40%, and his ABG showed pH 7.45, PCO2 41 mmHg, PaO2 78 mmHg, HCO3 28 mmol / L, base excess 3.8, and oxygen saturation 96%. Postoperatively, during the first few days he was in positive fluid balance, however, he was in a negative fluid balance thereafter. His arterial line did demonstrate pulse wave variability.
Figure 1.
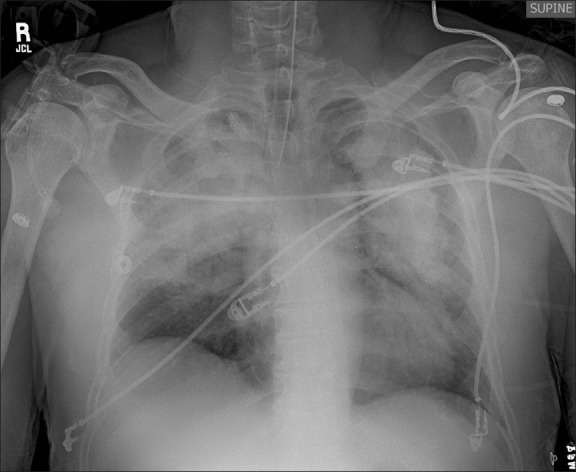
Chest X-ray one hour postoperatively
The patient was taken for a planned re-debridement on postoperative day #2 and was not extubated until postoperative day #3. Shortly after extubation, he complained of acute shortness of breath, with an ABG showing pH 7.49, PCO2 39 mmHg, PaO2 62 mmHg, HCO3 29 mmol / L, and SpO2 93% (FiO2 of 80%). He was placed on intermittent continuous positive airway pressure (CPAP, 10 cm H2O) via full face mask. Although the chest radiogram showed an improvement from the index postoperative film, the patchy infiltrates persisted [Figure 2]. After ensuring adequate volume status, furosemide (40 mg intravenous) was administered, with resultant improvement in oxygenation, shortness of breath, and subsequent roentgenographic examination. The patient received two additional doses of furosemide (20 mg intravenous) over the next 24 hours, as determined on the basis of clinical re-evaluation. He was weaned off oxygen on postoperative day six as his chest radiogram showed near complete resolution of the pulmonary edema and his oxygenation was appropriate [Figure 3]. He was discharged from the hospital on postoperative day seven. The patient was doing well on follow-up at three months, and underwent uneventful skin grafting of the residual perineal wound without any general anesthesia-related complications.
Figure 2.
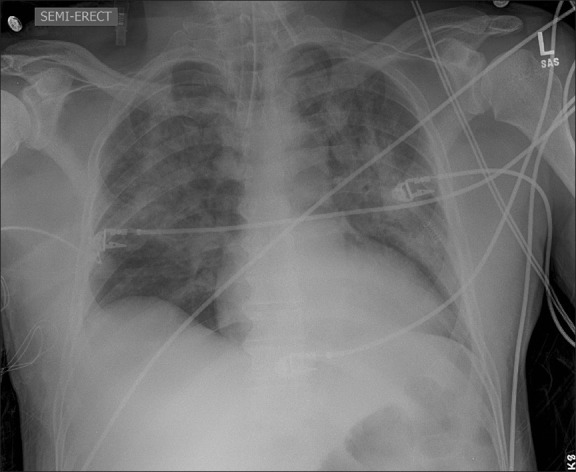
Chest X-ray six hours postoperatively
Figure 3.
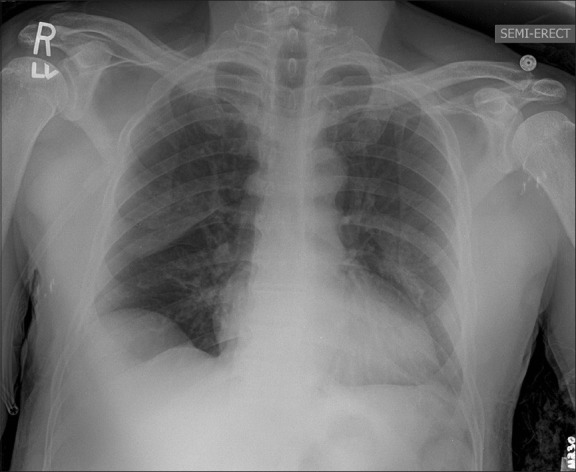
Chest X-ray 36 hours postoperatively
DISCUSSION
Ezri et al., first reported the possible association of LMA use and pulmonary edema.[4] Shankar et al. suggested that forceful inspiration against an LMA could lead to large subatmospheric pressure changes, resulting in a clinical appearance similar to that of pulmonary edema.[5] NPPE has been previously reported after biting on an LMA during emergence that resulted in dyspnea and hemoptysis.[6] The ProSeal™ LMA has also been associated with airway obstruction and the potential for NPPE.[7] Unilateral NPPE has also been reported with LMA use in a patient in the lateral position.[8] In this report, acute obstruction associated with forceful inspiration after biting of the LMA was the most likely ‘trigger’ event for NPPE.
Negative pressure pulmonary edema was first described in 1927, when researchers noted that prolonged inspiration against fixed airway obstruction resulted in pulmonary edema in the canine model.[10] NPPE was formally described by Oswalt et al., in three adult patients who experienced acute onset of pulmonary edema minutes to hours after severe acute upper airway obstruction.[11] Since then, only a few case reports / series of NPPE were published, and our understanding of the pathogenesis and treatment of NPPE remains limited.[12–21]
Negative pressure pulmonary edema has been subdivided by Guffin et al. into two distinct classes. Type I, associated with forceful inspiratory effort in the context of an acute upper airway obstruction, and Type II, which occurs after the relief of chronic partial airway obstruction.[20] Common etiologies for Type I NPPE include laryngospasm, epiglottitis, croup, choking / foreign body aspiration, strangulation, hanging, endotracheal tube obstruction, laryngeal tumor, goiter, postoperative vocal cord paralysis, and near drowning. Of these, the laryngospasm remains the most common cause, accounting for ~50% of the reported cases.[2,17,20,22,23] Type II NPPE can be seen after adenoidectomy / tonsillectomy, laryngeal mass resection, correction of choanal stenosis or reduction of a hypertrophic redundant uvula.
The most common clinical setting for NPPE is during emergence from general anesthesia. During this critical period, upper airway obstruction is usually followed by the rapid onset of respiratory distress, with or without hemoptysis, and clinical / radiological features consistent with bilateral pulmonary edema.[18] However, in some patients, the onset may be considerably delayed, up to a few hours.[18] Therefore, close postoperative observation must be considered for an extended period of time in patients experiencing respiratory difficulty or laryngospasm.
Patients with a short neck, difficult intubation, endotracheal tube obstruction, history of obstructive sleep apnea, obesity, acromegaly, and upper aerodigestive tract surgery have increased susceptibility to this condition.[10,21] This syndrome is also noticed more frequently in young healthy, and muscular patients, because of their ability to generate high amounts of negative intrathoracic pressures during an obstructing event.[3,19]
The patient presented herein had Type I NPPE. The pathogenesis of type 1 NPPE is multifactorial.[10–12,21–28] The mechanism revolves around a large inspiratory force generated against an obstructed upper airway (modified Mueller maneuver) that leads to an acute and unusual elevation of negative intrathoracic pressure. Healthy human subjects can generate very high levels of negative inspiratory pressure with a reported maximum of – 140 cm H2O from a baseline of – 4 cm H2O.[23] The first of the two proposed pathophysiological mechanisms is that NPPE is a type of hydrostatic pulmonary edema caused by the significant fluid shifts from the microvasculature to the peri-microvascular interstitium. There is alteration in the Starling forces (i.e., intra- / extracapillary hydrostatic and oncotic pressures) that influence the movement of fluid across the pulmonary capillaries. Negative intrathoracic pressure will be transmitted to the interstitial space and alveoli, causing an increase in the hydrostatic pressure gradient favoring transudation of the fluid from the pulmonary capillary to the pulmonary interstitial space. It also causes an increase in the venous return to the heart resulting in an increase in the pulmonary blood volume, pulmonary venous pressures again causing an increase in the hydrostatic pressure gradient, and edema formation. The legitimate question of whether our patient had a history of underlying heart or lung disease may arise. He denied any history of heart or lung disease, and as mentioned previously, he had a very good functional status. Thus, our suspicion for edema of cardiac origin was very low. The acute onset of airway obstruction at the time of extubation, followed by desaturation, was more consistent with the clinical picture of NPPE.
The concurrent development of hypoxemia during NPPE triggers hypoxic pulmonary vasoconstriction and raises pulmonary vascular resistance, thus increasing transmural hydrostatic pressures. As the right ventricular volume increases, the interventricular septum may shift leftward, indicative of reduced left ventricular diastolic compliance. Simultaneously, this negative intra-pleural pressure also has a direct depressing effect on the cardiac output by increasing the cardiac afterload.[27] The cardiac afterload is also increased secondary to hypoxia-induced systemic and pulmonary vasoconstriction. There is an accompanying hyperadrenergic response. The increase in afterload, in combination with hypoxemia of the myocardial muscle, leads to a decrease in the left ventricular function, which in turn increases pulmonary venous pressure and further aggravates the pulmonary edema.
The second theoretical mechanism of pathogenesis involves the loss of capillary integrity, leading to increased capillary permeability and edema formation.[12,15,25,28] The extreme elevation in transmural pressure causes mechanical stress on the alveolar capillary membrane, leading to the transudation of erythrocytes into the alveoli, a process termed as ‘stress failure’.[28–30] Alveolar hemorrhage, the hallmark of capillary failure has been documented by bronchoscopy and bronchoalveolar lavage during the episodes of NPPE. The classic descriptions of serosanguinous or pink frothy secretion observed in our case as well as several other case reports, indicate that this mechanism may be important in the pathogenesis of NPPE.
Negative pressure pulmonary edema is characterized by a rapid onset (within minutes) and a relatively quick resolution, with significant clinical and radiographic improvement in 12 to 24 hours.[17,18] NPPE, if recognized and treated early, is usually a self-limited condition and is reversible.[2,24] Treatment is mainly supportive. Removal of the obstructive event and maintaining patent airway and oxygenation should be the initial steps. Not all patients will require reintubation.[18] It is important to apply positive pressure to the airway early. Nasal bi-level positive airway pressure, mask continuous positive airway pressure (CPAP), and intubation and ventilation with positive end-expiratory pressure (PEEP) have been used successfully.[2,26] In this case we chose CPAP because the patient did not fail to ventilate, but was hypoxemic. However, it has been demonstrated that both Biphasic Intermittent Positive Airway Pressure (BIPAP) and CPAP are effective modalities for treating patients with acute pulmonary edema associated with hypercapnea.[31]
Diuretics were often administered, but their benefit in patients with NPPE was controversial. This was because the primary problem was not fluid overload, but interstitial fluid shifts, induced by the negative intrathoracic pressure.[18,28,32] If the patient was euvolemic, diuretic treatment was usually not required and most patients recovered quickly after the airway obstruction was resolved.[33,34] Diuretic therapy was an essential component in treating hydrostatic pulmonary edema, and it was also used in some patients with acute lung injury. Hence, we decided not to administer diuretics initially. Another complicating factor in our patient was that he had a septic focus, and therefore, had an increased risk for septic shock and had just undergone surgery. As his chest radiogram failed to improve as expected, and the patient was still requiring CPAP support on postoperative day three, furosemide was cautiously given. It did improve his oxygenation, and was therefore continued for an additional 24 hours. Bronchodilators were often used and were shown to be beneficial (our patient was treated with albuterol nebulization). Patients with NPPE could have wheezing secondary to interstitial edema-induced narrowing of the bronchial lumina. In vitro and in vivo studies in human and animal models had shown that beta agonists could increase the rate of alveolar fluid clearance via increased active cation transport, which accelerated the regression of symptoms of pulmonary edema.[35] The theory of vascular leak and the use of diuresis in patients with acute lung injury was tied to this case. Zumsteg et al. demonstrated that when a critical volume of fluid collected interstitially, alveolar flooding commenced.[36] Here, we administered a diuretic on the third day to improve oxygenation, with good result, after three days of cautious observation.
In a patient developing acute onset of perioperative pulmonary edema, other causes of pulmonary edema should be considered. The differential diagnosis should include cardiogenic edema, neurogenic edema, aspiration pneumonitis, acute respiratory distress syndrome (ARDS), pulmonary embolism, anaphylaxis, drug-induced non-cardiogenic pulmonary edema, and postoperative residual curarization. These other causes were all considered in our patient. In NPPE, an increase in extravascular lung water decreased the compliance and increased shunting. Our patient's original arterial blood gas demonstrated respiratory acidosis. At that time our patient was on a spontaneous mode of ventilation, and prior to that had received high doses of narcotics intraoperatively. This could have resulted in the retention of CO2. He had no history of cardiac pathology in the past and had a good functional status, and his electrocardiogram was normal. He had no evidence of neurogenic pathology. There were no other features of anaphylaxis. As he had a rapid onset of bilateral diffuse infiltrates on chest X-ray, aspiration pneumonitis was unlikely. Postoperative residual curarization was also identified as one of the risk factors for upper airway obstruction, as it typically impaired the upper airway dilator muscle strength, while preserving inspiratory muscle function.[37,38]
This patient did have an increased risk for acute lung injury (ALI) or ARDS, secondary to his underlying infectious process. NPPE has been described as one of the imitators of ALI. Patients with NPPE can develop diffuse alveolar injury through damage to the pulmonary capillaries by mechanical disruption of the alveolar-capillary membrane. Many of these patients may fulfill the clinical, physiological, and radiographic criteria for ALI / ARDS. Our patient did have a septic focus and did fulfill the SIRS criteria. His PaO2/ FiO2 was indeed < 200 and his chest X-ray did show bilateral infiltrates. However, he did not develop bacteremia, he did not develop septic shock, and he was oxygenating adequately throughout the case. His onset of hypoxemia and respiratory distress was precipitous and coincided with the extubation. He showed immediate improvement with CPAP. This clinical picture was more consistent with NPPE, than ALI / ARDS.
CONCLUSIONS
The current case illustrates the development of NPPE as a complication of intermittent obstruction of LMA upon emergence from general anesthesia. The favorable outcome was likely due to early recognition and appropriate NPPE-directed therapy. The majority of the cases of NPPE in the literature are in association with endotracheal tube use, and the incidence in association with LMAs seems low. However, some patients emerging from anesthesia (especially in the second stage) have a tendency to bite on the airway, and if this obstruction is not relieved promptly, NPPE may develop. The increasing use of LMAs in the administration of anesthetics will provide more scenarios where NPPE can manifest. We encourage our colleagues to be vigilant in the recognition of NPPE, while using an LMA, and to be aware of the treatment modalities available to them, as well as the differential diagnoses involved when such a scenario is encountered.
ACKNOWLEDGMENTS
The authors would like to acknowledge the clinical expertise provided by Paul R. Beery II, M.D., Department of Surgery, Division of Critical Care, Trauma, and Burn, The Ohio State University Medical Center, Columbus, Ohio.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Olsson GL, Hallen B. Laryngospasm during anaesthesia. A computer-aided incidence study in 136,929 patients. Acta Anaesthesiol Scand. 1984;28:567–75. doi: 10.1111/j.1399-6576.1984.tb02121.x. [DOI] [PubMed] [Google Scholar]
- 2.McConkey PP. Postobstructive pulmonary oedema--a case series and review. Anaesth Intensive Care. 2000;28:72–6. doi: 10.1177/0310057X0002800114. [DOI] [PubMed] [Google Scholar]
- 3.Patton WC, Baker CL., Jr Prevalence of negative-pressure pulmonary edema at an orthopaedic hospital. J South Orthop Assoc. 2000;9:248–53. [PubMed] [Google Scholar]
- 4.Ezri T, Priscu V, Szmuk P, Soroker D. Laryngeal mask and pulmonary edema. Anesthesiology. 1993;78:219. doi: 10.1097/00000542-199301000-00045. [DOI] [PubMed] [Google Scholar]
- 5.Bhavani-Shankar K, Hart NS, Mushlin PS. Negative pressure induced airway and pulmonary injury. Can J Anaesth. 1997;44:78–81. doi: 10.1007/BF03014328. [DOI] [PubMed] [Google Scholar]
- 6.Devys JM, Balleau C, Jayr C, Bourgain JL. Biting the laryngeal mask: An unusual cause of negative pressure pulmonary edema. Can J Anaesth. 2000;47:176–8. doi: 10.1007/BF03018856. [DOI] [PubMed] [Google Scholar]
- 7.Stix MS, Rodriguez-Sallaberry FE, Cameron EM, Teague PD, O’Connor CJ., Jr Esophageal aspiration of air through the drain tube of the ProSeal laryngeal mask. Anesth Analg. 2001;93:1354–7. doi: 10.1097/00000539-200111000-00065. [DOI] [PubMed] [Google Scholar]
- 8.Sullivan M. Unilateral negative pressure pulmonary edema during anesthesia with a laryngeal mask airway. Can J Anaesth. 1999;46:1053–6. doi: 10.1007/BF03013201. [DOI] [PubMed] [Google Scholar]
- 9.Goldenberg JD, Portugal LG, Wenig BL, Weingarten RT. Negative-pressure pulmonary edema in the otolaryngology patient. Otolaryngol Head Neck Surg. 1997;117:62–6. doi: 10.1016/S0194-59989770208-0. [DOI] [PubMed] [Google Scholar]
- 10.Udeshi A, Cantie SM, Pierre E. Postobstructive pulmonary edema. J Crit Care. 2010;25(508):e501–5. doi: 10.1016/j.jcrc.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Oswalt CE, Gates GA, Holmstrom MG. Pulmonary edema as a complication of acute airway obstruction. JAMA. 1977;238:1833–5. [PubMed] [Google Scholar]
- 12.Mussi RK, Toro IF. Negative-pressure pulmonary edema and hemorrhage associated with upper airway obstruction. J Bras Pneumol. 2008;34:420–4. doi: 10.1590/s1806-37132008000600013. [DOI] [PubMed] [Google Scholar]
- 13.Albergaria VF, Soares CM, Araujo Rde M, de Mendonca WL. Negative-pressure pulmonary edema after transsphenoidal hypophysectomy. Case report. Rev Bras Anestesiol. 2008;58:391–6. doi: 10.1590/s0034-70942008000400009. [DOI] [PubMed] [Google Scholar]
- 14.Mamiya H, Ichinohe T, Kaneko Y. Negative pressure pulmonary edema after oral and maxillofacial surgery. Anesth Prog. 2009;56:49–52. doi: 10.2344/0003-3006-56.2.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel AR, Bersten AD. Pulmonary haemorrhage associated with negative-pressure pulmonary oedema: A case report. Crit Care Resusc. 2006;8:115–6. [PubMed] [Google Scholar]
- 16.Massad I, Halawa SA, Badran I, Al-Barzangi B. Negative pressure pulmonary edema-five case reports. Middle East J Anesthesiol. 2006;18:977–84. [PubMed] [Google Scholar]
- 17.Westreich R, Sampson I, Shaari CM, Lawson W. Negative-pressure pulmonary edema after routine septorhinoplasty: Discussion of pathophysiology, treatment, and prevention. Arch Facial Plast Surg. 2006;8:8–15. doi: 10.1001/archfaci.8.1.8. [DOI] [PubMed] [Google Scholar]
- 18.Krodel DJ, Bittner EA, Abdulnour R, Brown R, Eikermann M. Case scenario: Acute postoperative negative pressure pulmonary edema. Anesthesiology. 2010;113:200–7. doi: 10.1097/ALN.0b013e3181e32e68. [DOI] [PubMed] [Google Scholar]
- 19.Holmes JR, Hensinger RN, Wojtys EW. Postoperative pulmonary edema in young, athletic adults. Am J Sports Med. 1991;19:365–71. doi: 10.1177/036354659101900407. [DOI] [PubMed] [Google Scholar]
- 20.Guffin TN, Har-el G, Sanders A, Lucente FE, Nash M. Acute postobstructive pulmonary edema. Otolaryngol Head Neck Surg. 1995;112:235–7. doi: 10.1016/S0194-59989570242-3. [DOI] [PubMed] [Google Scholar]
- 21.Lorch DG, Sahn SA. Post-extubation pulmonary edema following anesthesia induced by upper airway obstruction. Are certain patients at increased risk? Chest. 1986;90:802–5. doi: 10.1378/chest.90.6.802. [DOI] [PubMed] [Google Scholar]
- 22.Broccard AF, Liaudet L, Aubert JD, Schnyder P, Schaller MD. Negative pressure post-tracheal extubation alveolar hemorrhage. Anesth Analg. 2001;92:273–5. doi: 10.1097/00000539-200101000-00055. [DOI] [PubMed] [Google Scholar]
- 23.Fremont RD, Kallet RH, Matthay MA, Ware LB. Postobstructive pulmonary edema: A case for hydrostatic mechanisms. Chest. 2007;131:1742–6. doi: 10.1378/chest.06-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deepika K, Kenaan CA, Barrocas AM, Fonseca JJ, Bikazi GB. Negative pressure pulmonary edema after acute upper airway obstruction. J Clin Anesth. 1997;9:403–8. doi: 10.1016/s0952-8180(97)00070-6. [DOI] [PubMed] [Google Scholar]
- 25.Dolinski SY, MacGregor DA, Scuderi PE. Pulmonary hemorrhage associated with negative-pressure pulmonary edema. Anesthesiology. 2000;93:888–90. doi: 10.1097/00000542-200009000-00042. [DOI] [PubMed] [Google Scholar]
- 26.Lang SA, Duncan PG, Shephard DA, Ha HC. Pulmonary oedema associated with airway obstruction. Can J Anaesth. 1990;37:210–8. doi: 10.1007/BF03005472. [DOI] [PubMed] [Google Scholar]
- 27.Wise RA. Effect of alterations of pleural pressure on cardiac output. South Med J. 1985;78:423–8. doi: 10.1097/00007611-198504000-00017. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz DR, Maroo A, Malhotra A, Kesselman H. Negative pressure pulmonary hemorrhage. Chest. 1999;115:1194–7. doi: 10.1378/chest.115.4.1194. [DOI] [PubMed] [Google Scholar]
- 29.West JB, Tsukimoto K, Mathieu-Costello O, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol. 1991;70:1731–42. doi: 10.1152/jappl.1991.70.4.1731. [DOI] [PubMed] [Google Scholar]
- 30.Tsukimoto K, Mathieu-Costello O, Prediletto R, Elliott AR, West JB. Ultrastructural appearances of pulmonary capillaries at high transmural pressures. J Appl Physiol. 1991;71:573–82. doi: 10.1152/jappl.1991.71.2.573. [DOI] [PubMed] [Google Scholar]
- 31.Bellone A, Vettorello M, Monari A, Cortellaro F, Coen D. Noninvasive pressure support ventilation versus continuous positive pressure in acute hypercapnic pulmonary edema. Intensive Care Med. 2005;31:807–11. doi: 10.1007/s00134-005-2649-6. [DOI] [PubMed] [Google Scholar]
- 32.Stawicki SP, Sarani B, Braslow BM. Reexpansion pulmonary edema. OPUS 12 Sientist. 2008;2:29–31. [Google Scholar]
- 33.Koh MS, Hsu AA, Eng P. Negative pressure pulmonary oedema in the medical intensive care unit. Intensive Care Med. 2003;29:1601–4. doi: 10.1007/s00134-003-1896-7. [DOI] [PubMed] [Google Scholar]
- 34.Aggarwal D, Stawicki SP, Sarani B, Braslow BM. Point-counterpoint: The role of diuresis in management of reexpansion pulmonary edema. OPUS 12 Scientist. 2008;2:10. [Google Scholar]
- 35.Matthay MA, Fukuda N, Frank J, Kallet R, Daniel B, Sakuma T. Alveolar epithelial barrier. Role in lung fluid balance in clinical lung injury. Clin Chest Med. 2000;21:477–90. doi: 10.1016/s0272-5231(05)70160-x. [DOI] [PubMed] [Google Scholar]
- 36.Zumsteg TA, Havill AM, Gee MH. Relationships among lung extravascular fluid compartments with alveolar flooding. J Appl Physiol. 1982;53:267–71. doi: 10.1152/jappl.1982.53.1.267. [DOI] [PubMed] [Google Scholar]
- 37.Eikermann M, Vogt FM, Herbstreit F, Vahid-Dastgerdi M, Zenge MO, Ochterbeck C, et al. The predisposition to inspiratory upper airway collapse during partial neuromuscular blockade. Am J Respir Crit Care Med. 2007;175:9–15. doi: 10.1164/rccm.200512-1862OC. [DOI] [PubMed] [Google Scholar]
- 38.Herbstreit F, Peters J, Eikermann M. Impaired upper airway integrity by residual neuromuscular blockade: Increased airway collapsibility and blunted genioglossus muscle activity in response to negative pharyngeal pressure. Anesthesiology. 2009;110:1253–60. doi: 10.1097/ALN.0b013e31819faa71. [DOI] [PubMed] [Google Scholar]