

Abstract
The development of pulmonary arterial hypertension (PAH) in pediatric patients has been linked to the production of the arachidonic acid metabolite, thromboxane A2 (TxA2). The present study evaluated the therapeutic effect of furegrelate sodium, a thromboxane synthase inhibitor, on the development of PAH in a neonatal piglet model. Three-day-old piglets were exposed to 21 days of normoxia (N; 21% FIO2) or chronic hypoxia (CH; 10% FIO2). A third group of piglets received the oral TxA2 synthase inhibitor, furegrelate (3 mg/kg, 2 or 3 times daily) at the induction of CH. In vivo hemodynamics confirmed a 2.55-fold increase of the pulmonary vascular resistance index (PVRI) in CH piglets (104±7 WU) compared to N piglets (40±2 WU). The CH piglets treated twice daily with furegrelate failed to show improved PVRI, but furegrelate three times daily lowered the elevated PVRI in CH piglets by 34% to 69±5 WU and ameliorated the development of right ventricular hypertrophy. Microfocal X-ray computed tomography (CT) scanning was used to estimate the diameter-independent distensibility term, α (% change in diameter per Torr). Pulmonary arterial distensibility in isolated lungs of CH piglets (α=1.0±0.1% per Torr) was lower than that of N piglets (α=1.5±0.1% per Torr) indicative of vascular remodeling. Arterial distensibility was partially restored in furegrelate-treated CH piglets (α =1.2±0.1% per Torr) and microscopic evidence showing muscularization of small pulmonary arteries also was less prominent in these animals. Finally, isolated lungs of furegrelate-treated piglets showed lower basal and vasodilator-induced transpulmonary pressures compared to CH animals. These findings suggest that pharmacological inhibition of TxA2 synthase activity by furegrelate blunts the development of hypoxia-induced PAH in an established neonatal piglet model primarily by preserving the structural integrity of the pulmonary vasculature.
Keywords: pulmonary arterial hypertension, neonatal pulmonary arterial hypertension, hypoxia, thromboxane A2, thromboxane synthase, furegrelate, vasoconstriction
Cardiorespiratory pathologies of infancy including congenital heart defects and bronchopulmonary dysplasia may lead to chronic hypoxia (CH) and the development of pulmonary arterial hypertension (PAH).[1–4] An imbalance between vasodilator and vasoconstrictor substances in the pulmonary circulation that favors vasoconstriction contributes to increased vascular tone.[5–8] Therefore, treatment options have focused on reversing this imbalance by administering vasodilator substances or attenuating vasoconstrictor pathways. These therapies include calcium channel blockers, inhaled nitric oxide, intravenous prostacyclin, phosphodiesterase type-5 inhibitors, and endothelin (ET) receptor blockers.[1–4,9,10] However, the high cost, short half-lives, lack of vasodilator response, and serious side effects of these molecules dictate their use primarily as diagnostic or short-term interventions. Those drugs administered chronically to patients only slow disease progression that eventually culminates in right heart failure.
One alternative therapeutic target is the thromboxane A2 (TxA2)-mediated signaling pathway.[11,12] TxA2 is a potent vasoconstrictor, platelet aggregator, and mitogenic factor primarily produced in platelets and endothelial cells. The enzymatic conversion of the arachidonic acid metabolite, endoperoxide PGH2, to TxA2 is accomplished by the enzyme TxA2 synthase.[13,14] The effects of TxA2 are mediated by its binding to the TxA2/PGH2 receptor (TP), a Gq-protein coupled receptor that activates phospholipase 2 to mobilize intracellular calcium.[15] Importantly, children with PAH from congenital heart defects show elevated plasma and urinary levels of TxB2,[5,7] a stable metabolite of TxA2. Similarly, newborn piglets exposed to hypoxia to induce PAH show increased pulmonary vascular TxA2 levels,[16,17] which correlate with an increased expression of thromboxane synthase in the affected arteries.[17] Fike et al.[18] reported that the TP receptor blocking drug, terbogrel (10 mg/kg, p.o., twice daily), ameliorated the development of PAH in neonatal piglets exposed to three days of CH. Although the long-term efficacy of TxA2 modulation is unknown, potently blocking TxA2 synthesis as an early event may potentially blunt its vasoconstrictor and mitogenic effects and slow the progression of PAH.
To test this hypothesis, we treated neonatal piglets exposed to CH for 21 days with furegrelate sodium. Furegrelate is a thromboxane synthase inhibitor with a half-effective inhibitory concentration (IC50) of 15 nmol/l in human plasma.[19] Furegrelate was developed as an antiplatelet agent, but Phase 1 clinical trials in normal male patients revealed that it inhibited TxA2 synthesis in vivo without significantly affecting platelet aggregation.[20,21] Furegrelate also is orally available, has a long half-life of 4.2–5.8 hours in adult humans (compared to several other therapies for PAH including nitric oxide and prostacyclin analogs), and reportedly is highly specific for its target enzyme.[19,22] Considering its encouraging drug profile and its apparent safety in Phase 1 clinical trials in adults, the concept of “repositioning” furegrelate as a potential therapeutic agent for neonatal PAH is appealing. Thus in the present study, we designed preclinical studies to evaluate furegrelate as a therapeutic option for the treatment of neonatal PAH using an established piglet model of the disease.
MATERIALS AND METHODS
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committees at the Zablocki Veterans Administration Medical Center and the Medical College of Wisconsin, or at the University of Arkansas for Medical Sciences. Briefly, three-day-old piglets were exposed in pairs to 21 days of normoxia (N; 21% FIO2) or chronic hypoxia (CH; 10% FIO2) in environmental chambers.[23] Experimental groups of CH piglets received furegrelate (3 mg/kg, p.o. by syringe) either twice daily (FBID) or three times daily (FTID) with the first dose started just prior to the introduction of CH. The dose of furegrelate was chosen based on the literature[19,21,23] and a pilot study (Fig. 1A) in which we observed that furegrelate at a dose of 3 mg/kg three times daily attenuated the development of CH-induced PAH, whereas twice daily administration had no therapeutic benefit.
Figure 1.
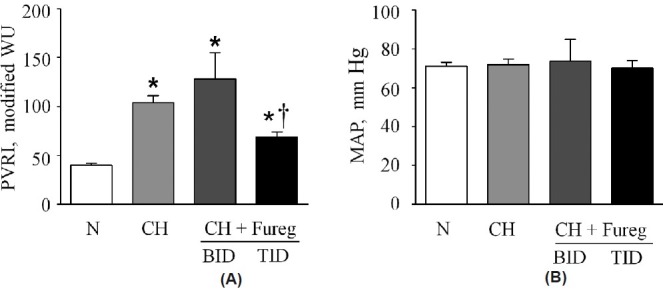
(A) Values of resting pulmonary vascular resistance index (PVRI) in neonatal piglets exposed for three weeks to normoxia (N), chronic hypoxia (CH) or chronic hypoxia + furegrelate twice daily (CH + FBID) or three times daily (CH + FTID). Sample sizes were 19, 26, 6 and 13, respectively. FNx01Significant difference (P<0.05) from PVRI of N piglets. †Significant difference (P<0.05) from PVRI of CH piglets. (B) Mean arterial pressure (MAP) in the same groups of animals showed no significant difference between groups. Values are mean±S.E.M.
Right ventricular hypertrophy
Right ventricular hypertrophy (RVH) was calculated in dissected hearts as the wet weight ratio of right ventricular free wall to left ventricular wall plus septum (RV/LV + S).[24]
Urinary 11-dehydro thromboxane B2
Urine was withdrawn at the end of hemodynamic studies from a subset of piglets and immediately stored at -80°C. Urinary 11–dehydro thromboxane B2, a stable metabolite of TxA2, was measured using a commercial enzyme immunoassay kit.[25]
In vivo hemodynamics
In order to evaluate the level of PAH, in vivo hemodynamics were measured in anesthetized piglets ventilated with room air (FIO2=0.21) at a rate of 15–18 breaths per minute with a peak airway pressure of 10–15 mmHg and a positive end-expiratory pressure (PEEP) of 3 mmHg.[24] Measurements included pulmonary arterial pressure (PAP), pulmonary capillary wedge pressure (PCWP), and cardiac output (CO) measured by thermodilution. CO was divided by body weight to obtain cardiac index (CI). Pulmonary vascular resistance indexed to body weight was then calculated as PVRI (Modified Wood Units) = (PAP – PCWP)/CI. After completing these measurements, piglets were given an additional dose of pentobarbital, heparinized (5000 units i.v.), and exsanguinated. Lungs or lung lobes were dissected for in vitro studies.
Isolated perfused lungs
Pulmonary vascular reactivity was evaluated in left lungs perfused with a mixture of autologous blood and artificially ventilated at a tidal volume of ~15 ml/kg and at a rate of 20 breaths per minute in normoxic conditions. Pulmonary artery pressure (PAP), left atrial pressure (PLA), and airway pressure (PAW) were measured on-line and the transpulmonary pressure gradient (ΔPtp=PAP – PLA) was calculated. In some lungs, baseline PAP was obtained before the addition of nifedipine (10 μmol/l in perfusate) and papaverine (15 mg bolus i.v.) to elicit a maximal vasodilator response.[24]
Morphometric analysis
Tissue cubes (1 cm) were dissected from a mid-sagittal slice of the left or right lower lung lobe. The tissues were embedded in paraffin, sectioned, and stained with hemotoxylin and eosin. The %MT (%MT=2× muscle thickness/external diameter) was measured for arteries ranging from 50 μm to 500 μm in outer diameter using a Nikon E600 microscope and MetaMorph software.
Microfocal X-ray computed tomography imaging
We adapted the rat lung microfocal X-ray CT imaging technique to piglet lungs.[26] After flushing the pulmonary artery with a papaverine-saline mixture to remove all blood and minimize active vascular tone, the lobe was suspended in the imaging field and inflated with a 15% O2, 6% CO2, balance N2 gas mixture at a constant airway distending pressure of 5 mmHg. The papaverine-saline solution was replaced with the radiopaque contrast agent perfluorooctyl bromide (PFOB). Over the range of pressure studied, PFOB does not traverse the capillaries to the veins due to the surface tension at the PFOB-aqueous interface. Thus, only the arterial tree was filled. The lung lobe was then imaged at four intraarterial pressures (6, 12, 21, and 30 mmHg) referenced to the middle of the lobe. At each pressure, planar images were collected as a seven frame average in 1° intervals over 360° of rotation. The set of images was preprocessed, compensating for spatial distortions introduced by the imaging system, and then reconstructed using a Feildkamp cone-beam algorithm.[26] The reconstructed volume was 4973 with a isotropic resolution of a typical pixel size of 160 μm. Arterial vessel diameters were identified and measured from the reconstructed volumes. Diameter measurements ranging from ~150 μm to ~3000 μm were made on 90 arterial vessel segments per lung, along the main pulmonary trunk and branches at four pressures (6, 12, 21, and 30 mmHg) referenced to the top of the lung. The actual intravascular pressure relative to atmosphere within each artery was obtained from the measured vessels’ vertical distance from the reference pressure level and the PFOB density. The pressure–diameter (P–D) relationship of each of the 90 arteries was then calculated and the slope was estimated by linear regression. Subsequently, the slope of each P–D curve (β) was plotted against its respective undistended vessel diameter intercept at 0 pressure (Do). The trend relating β to Do was analyzed by linear regression through the origin and the diameter-independent distensibility term, α, (percent change in diameter per Torr) was calculated for each lobe.
Statistical analysis
Data were displayed as mean±S.E.M. Comparison of a single variable between groups was subjected to one-way ANOVA with post hoc multiple comparison test (Student–Newman–Keuls method). Differences were judged to be significant at the level of P<0.05.
RESULTS
Furegrelate blunts the development of neonatal PAH
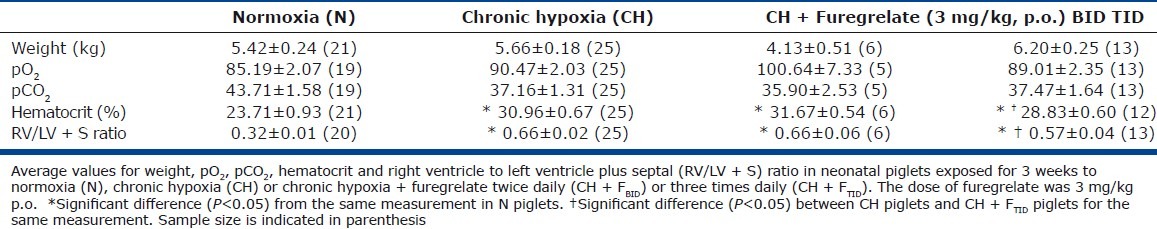
Table 1 compares data between N piglets and untreated and furegrelate-treated CH piglets after three weeks in environmental chambers. Furegrelate was administered orally by syringe to take advantage of its oral bioavailability. Weight, arterial pO2, and arterial pCO2 were not significantly different between the three groups of animals. However, the CH piglets showed a higher hematocrit, RV/LV + S ratio (Table 1) and pulmonary vascular resistance index (PVRI; Fig. 1A) compared to N piglets, indicating the development of PAH. In initial therapeutic studies, the oral administration of 3 mg/kg furegrelate orally twice daily (CH + Fureg, BID) failed to lower the elevated hematocrit and RV/LV + S ratio (Table 1) observed in untreated CH piglets. Similarly, furegrelate BID also failed to blunt the elevated PVRI induced by hypoxia that averaged 128±27 WU in treated piglets and 104±7 WU in untreated CH piglets (Fig. 1A; CH + Fureg). However, CH piglets treated with furegrelate three times daily (TID) showed a markedly reduced PVRI of 69±5 WU compared to untreated CH animals. In addition, the RV/LV + S ratio was significantly reduced in CH + FTID piglets (0.57±.04) compared to untreated CH animals (0.66±.02) and hematocrit was partially restored to normal values (Table 1). Importantly, there was no change in the systemic mean arterial pressure between N and CH+FTID piglets, suggesting the absence of a pronounced systemic dilator effect of furegrelate (Fig. 1B). Collectively, these findings suggest that oral administration of furegrelate three times daily reduces the clinical signs of PAH in CH piglets without inducing systemic hypotension. Thus, the remainder of our studies used the dosing regimen of furegrelate, 3 mg/kg orally three times daily.
Table 1.
Profiles of normoxic (N), chronic hypoxic (CH), and CH piglets treated with furegrelate
The efficacy of furegrelate (3 mg/kg, p.o., TID) to reduce the synthesis of TxA2 was initially evaluated by enzyme immunoassay (EIA) of TxB2, a stable TxA2 metabolite in plasma of N, CH and CH + FTID piglets. However, due to a very high intra-assay coefficient of variation (>20%) these samples were not used. Subsequently, urine was obtained from the final animals studied and the level of 11-dehyro TxB2, a stable urinary TxA2 metabolite, was evaluated by EIA. The 11-dehydro TxB2 EIA showed a low intra-assay coefficient of variation (5%) after normalizing to creatinine to account for urine volume. Average 11-dehydro TxB2 levels were elevated in CH piglets (2.40±0.36 ng/mg creatinine, n = 8) compared to N piglets (1.83±0.21 ng/mg creatinine, n=6; Fig. 2A-B). The urinary 11-dehydro TxB2 level in CH + FTID piglets was 1.40±0.49 ng/mg creatinine (n=4), showing the lowest average value of the three animal groups (Fig. 2A and B). Thus we obtained initial evidence in this subset of animals that the dosing regimen of furegrelate we used (3 mg/kg, TID) inhibited the synthesis of TxA2 in CH piglets, although high animal-to-animal variability precluded statistical significance.
Figure 2.
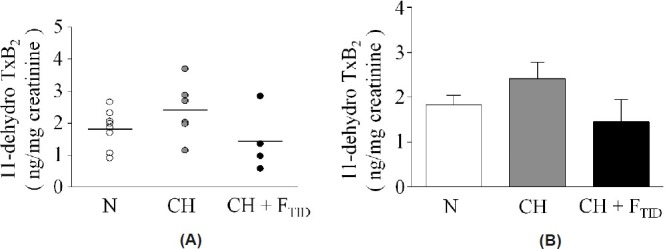
(A) Individual values of 11-dehydro TxB2, a stable metabolite of TxA2, in urine samples from neonatal piglets exposed for 3 weeks to normoxia (N), chronic hypoxia (CH) or chronic hypoxia + furegrelate three times daily (CH + FTID). Each symbol represents a single animal. (B) Average urinary 11-dehydro TxB2 values for the animals in A. Sample sizes were 8, 6 and 4, respectively. Values are mean±S.E.M.
Furegrelate attenuates pulmonary vascular remodeling
Hypoxia-induced vascular remodeling is an important feature of PAH that may limit responsiveness to vasodilator therapies. Thus, we compared pulmonary vascular distensibility and the percent muscular thickness (%MT) of pulmonary arteries between N, CH, and CH + FTID piglets. Microfocal X-ray CT imaging revealed overt vascular remodeling in lungs of CH piglets that was evident as a loss of PFOB-filled pulmonary arteries compared to normoxic lungs (Fig. 3A). Accordingly, the distensibility coefficient (α), an indicator of elasticity of the vasculature, was significantly reduced in the pulmonary arteries of CH piglets (1.0±0.1% per Torr) compared to N piglets (1.5±0.1% per Torr; Fig. 3B). Furegrelate therapy TID (FTID) blunted hypoxia-induced vascular remodeling in CH piglets (Fig. 3A) resulting in an improved α value in CH piglets of 1.2±0.1% per Torr, which was not significantly different than the α value of normoxic lungs (Fig. 3B). These findings suggested that 3 mg/kg furegrelate TID prevented the hypoxia-induced loss of distensibility in the pulmonary circulation, which retained vascular elasticity.
Figure 3.
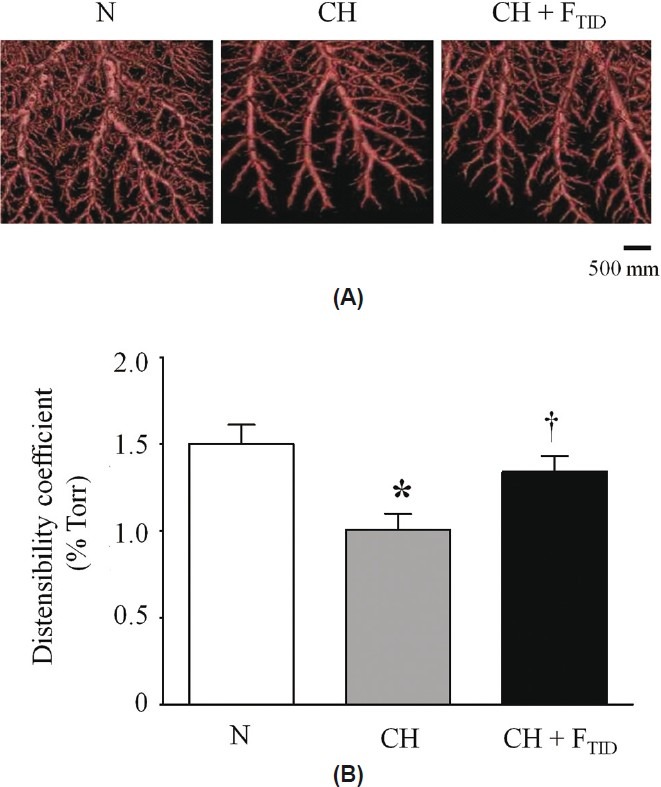
(A) Computer-generated three-dimensional images of pulmonary arterial trees from neonatal piglets exposed for 3 weeks to normoxia (N), chronic hypoxia (CH) or chronic hypoxia + furegrelate three times daily (CH + FTID). (B) Comparison of distensibility coefficients between the pulmonary circulations of N, CH and CH + FTID piglets. Sample sizes were 15, 7 and 6, respectively. *Significant difference (P<0.05) between N and CH. †Significant difference (P<0.05) between CH and CH + FTID. Values are mean±S.E.M.
To further corroborate our distensibility findings, we compared the percent muscular thickness (%MT) of pulmonary arteries (o.d., 50–500 μm) between lungs of N, CH and CH + FTID piglets. Lung sections stained with hemotoxylin and eosin revealed thickened arterial walls in untreated CH piglets compared to N piglets. This abnormality was less evident in arteries of CH + FTID piglets (Fig.4A). The calculated %MT was 2.3-fold higher in arteries of CH piglets (20.5±0.6%) compared to N piglets (8.82±0.2%) suggestive of extensive pulmonary vascular remodeling in the CH piglets with PAH. The %MT value for small pulmonary arteries was significantly lower (14.2±0.6) in CH + FTID piglets compared to untreated CH animals. Thus, we obtained evidence that oral furegrelate therapy mitigated the development of hypoxia-induced pulmonary vascular remodeling in CH piglets.
Figure 4.
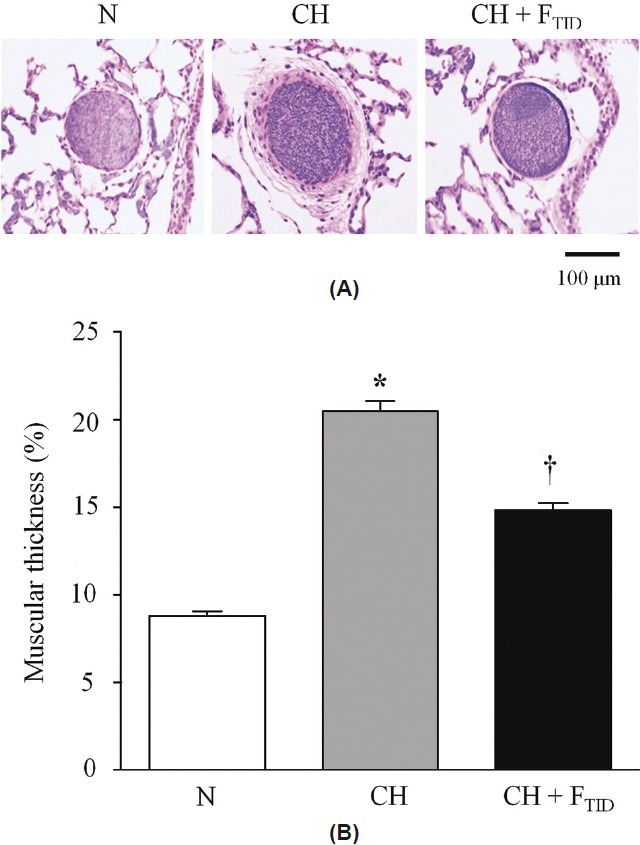
(A) Histological sections comparing percent medial thickness (%MT) of small pulmonary arteries in lungs from neonatal piglets exposed for 3 weeks to normoxia (N), chronic hypoxia (CH) or chronic hypoxia + furegrelate three times daily (CH + FTID). (B) Average %MT values for arteries of N, CH and CH + FTID piglets. *Significant difference (P<0.05) between N and CH. † = Significant difference (P<0.05) between CH and CH + FTID. Sample sizes were 103 arteries from 4 piglets (N), 192 arteries from 4 piglets (CH), and 184 arteries from 4 piglets (CH + FTID). Values are mean±S.E.M.
Furegrelate improves pulmonary pressure profiles in isolated lungs
We also evaluated the effect of furegrelate (3 mg/ kg, p.o., TID) on the resting and vasodilator-induced transpulmonary pressure gradient (ΔPtp) in isolated perfused lungs. The isolated lung avoids the systemic hypotensive effect of vasodilators that can confound the interpretation of hemodynamic measurements in vivo; it thereby allows pulmonary vascular tone to be assessed independently. Similar to our previous finding,[24] control ΔPtp was profoundly elevated in the isolated lungs of CH piglets (23.46±3.05 mmHg) compared to N piglets (11.80 ± 1.09 mmHg; Fig. 5). In contrast, ΔPtp in isolated lungs of CH + FTID piglets was significantly lower (14.81±0.63 mmHg) than ΔPtp in untreated CH piglets, corresponding to a 74% improvement (Fig. 5). Maximal pulmonary dilator responses were obtained in isolated lungs by adding nifedipine (N, 10 mmol/l) and papaverine (P, 15 mg bolus) to the perfusate for additive block of voltage-gated L-type Ca2+ channels and vascular contractile mechanisms, respectively.[24] The difference between the control ΔPtp value and the ΔPtp value after vasodilator challenge was regarded as the active pulmonary vascular tone; the residual tone may relate to structural limitations that confer vascular resistance. Calculated accordingly, 43% of active tone was sensitive to vasodilator challenge in isolated lungs of N piglets, resulting in a residual ΔPtp of 6.42±0.34 mmHg (Fig. 5). Isolated lungs of CH piglets showed a similar vasodilator-induced fall of 39% resulting in a residual ΔPtp of 13.71±1.20 mmHg after loss of active tone. This value was significantly (2.1–fold) higher than the residual ΔPtp of N piglets, suggesting that structural changes in the pulmonary circulation of CH piglets limited a further reduction of vascular resistance. Isolated lungs of CH + FTID piglets responded to the vasodilator challenge with a 33% fall in ΔPtp resulting in a residual ΔPtp value of 9.25±0.65 mmHg, a value significantly lower than the residual ΔPtp of 13.71±1.20 mmHg in lungs of untreated CH piglets. Collectively, these findings show that furegrelate therapy blunts the development of elevated ΔPtp in response to 3 weeks of hypoxia. After a strong vasodilator challenge, the elevated ΔPtp in CH piglets still persisted as evidence of structural remodeling and this vasodilator–resistant component was ameliorated by furegrelate treatment.
Figure 5.
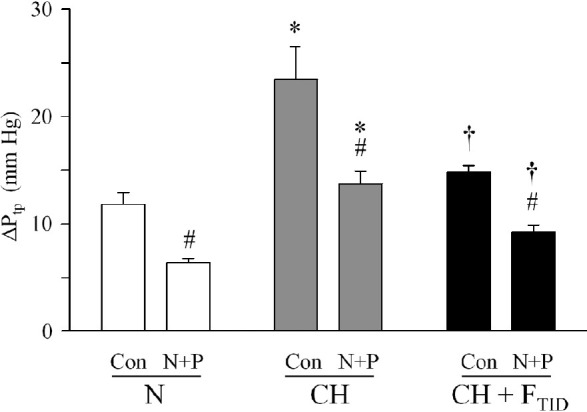
Resting ∆Ptp in isolated lungs of piglets exposed to normoxia (N), chronic hypoxia (CH) or CH + furegrelate three times daily (CH + FTID). Lungs were perfused with control (Con) solution before nifedipine (10 μmol/L) and papaverine (15 mg/kg bolus) were added to induce dilation (N+P). *Significant difference (P<0.05) between N and CH piglets for the same measurement. †Significant difference (P<0.05) between CH and CH + FTID piglets for the same measurement. #Significant difference (P<0.05) between control (Con) and vasodilator challenge (N+P) in the same animal group.
DISCUSSION
The treatment of pediatric PAH has focused on correcting the imbalance between vasodilator and vasoconstrictor pathways in the pulmonary circulation that favors the development of anomalous pulmonary vascular tone.[1–4,9,10] Therapeutic options have attempted to reverse this imbalance by administering vasodilator substances or attenuating vasoconstrictor pathways. Unfortunately, existing therapies are often short acting, have potentially severe side effects, or are ineffective. For example, the use of inhalational nitric oxide to ameliorate the abnormal vasoconstrictor tone of PAH is limited by its extremely short half-life and the potential for rebound hypertension upon discontinuation.[3] Prostacyclin analogs including epoprostenol also are very short-acting therapeutics that additionally cause the off-target effect of systemic hypotension.[2,3] Finally, the endothelin receptor blockers including bosentan, sitaxsentan, and ambrisentan are used on a limited basis in adults, but results from a randomized controlled trial in children with PAH are not yet available.[4,9,10] Since treatment options for pediatric patients with PAH are limited, the potent pulmonary smooth muscle constrictor and mitogen, thromboxane A2, has drawn attention as a potential drug target to develop primary or adjunct therapeutic agents for PAH. Several lines of evidence implicate the thromboxane signaling pathway as a contributor to pediatric PAH. Infants with PAH secondary to meconium aspiration show elevated levels of plasma TxB2, a stable metabolite of TxA2 that positively correlates with pulmonary artery pressure.[27] Newborns with congenital heart disease also show elevated plasma TxB2 and elevated urinary TxB2 levels.[5,7] Additionally, TxB2 is increased in broncho-alveolar lavage samples and in the plasma of infants with persistent PAH. In these patients, TxB2 levels positively correlate with negative outcome after extracorporeal membrane oxygenation.[28] However, despite clinical studies implicating TxA2 in pediatric PAH, preclinical studies to evaluate inhibitors of the TxA2 signaling pathway have been limited, due in large part to the rigorous care of infant piglets that is required to carefully evaluate drug effects in this standard model of neonatal PAH. Only Fike and colleagues[18] recently reported that thromboxane synthase was upregulated in pulmonary arteries of CH piglets after 10 days of hypoxia. Earlier, the same authors showed that oral terbogrel, a TxA2 synthase inhibitor and thromboxane (TP) receptor antagonist, blunted the development of early-stage PAH in piglets exposed to hypoxia for three days.[17] However, terbogrel did not pass Phase 1 clinical trials for use in adults with PAH because it caused the off-target effect of leg pain.[12] Thus, the search continues to identify an inhibitor of the TxA2 signaling pathway that has a positive drug profile and retains the ability to ameliorate the development of neonatal PAH.
With this goal in mind, our study evaluated the effect of furegrelate sodium, a potent thromboxane synthase inhibitor,[19] on the development of hypoxia-induced PAH in the piglet model. Furegrelate was initially developed as an antiplatelet agent, but it failed to improve coagulation parameters in Phase 1 clinical trials in adult volunteers.[20,21] However, furegrelate has an appealing drug profile that includes oral bioavailability, a relatively long half-life of 4.2–5.8 hours compared to nitric oxide and prostacyclin analogs and high specificity for its target enzyme. Studies failed to detect major off-target effects in preclinical testing in dogs or in Phase 1 clinical trials using healthy adult human subjects.[20–23] These encouraging drug properties suggest that furegrelate may represent a valuable therapeutic agent if “repositioned” to treat PAH. Indeed, our findings provide initial evidence that oral administration of furegrelate blunts the development of hypoxia-induced PAH in neonatal piglets and attenuates the right ventricular hypertrophy and pulmonary vascular remodeling that are key detrimental components of the disease. Thus, we provide experimental evidence that furegrelate may represent a promising early intervention to mitigate pediatric PAH. Importantly, the dosing regimen of furegrelate (3 mg/kg, p.o., TID) that exerted beneficial pulmonary vascular effects failed to significantly lower systemic blood pressure. This feature may be an additional asset of the drug since other vasodilator drugs used to treat pediatric PAH including the prostacyclin analogs cause systemic hypotension in a subset of young patients as a limiting off-target effect.[3,4]
From a mechanistic standpoint, the ability of furegrelate to partially normalize PVRI in CH piglets may relate to at least two different mechanisms. First, furegrelate-induced block of thromboxane synthase would be expected to reduce circulating and local concentrations of TxA2 to attenuate active arterial tone. Second, TxA2 is a known mitogenic factor that promotes the proliferation of pulmonary VSMCs and structural remodeling. The TxA2 ligand binds to the thromboxane A2/prostaglandin H2 (TP) receptor to stimulate DNA synthesis, and to promote proto-oncogene expression and actin polymerization.[29–32] Of these two mechanisms, our data suggest that the major impact of furegrelate on the pulmonary circulation of CH piglets may relate more closely to the prevention of structural remodeling for several reasons: (1) the medial thickness (%MT) of small pulmonary arteries in lung sections from CH + FTID piglets was significantly less compared to untreated CH piglets; (2) the pulmonary circulation of CH + FTID piglets revealed an increased distensibility on X-ray CT scans compared to untreated CH animals, reflecting improved pulmonary vascular elasticity; and (3) isolated perfused lungs from furegrelate-treated CH piglets showed a lower transpulmonary pressure (Ptp) compared to similar lungs from untreated CH piglets, and this difference persisted after the pulmonary circulation of both animal groups was subjected to the potent vasodilators, nifedipine and papaverine, to minimize active arterial tone. Thus, in our experimental model, it appears that furegrelate ameliorates the earlier stages of hypoxia-induced structural remodeling that precede the final stages of PAH in which vasodilator responsiveness often converts to a fixed vascular lesion resistant to vasodilator therapies.[1] Future evaluations should include studies in which furegrelate is administered to experimental models of PAH in which the disease has progressed to a more recalcitrant stage.
The findings of our study point to the importance of defining the pharmacokinetic profile of furegrelate in neonates. We based our furegrelate dosing regimen on earlier preclinical studies in which the daily administration of furegrelate (3 mg/kg body weight, p.o.) produced >80% inhibition of TxA2 synthase in platelet-rich plasma of adult rhesus monkeys.[19] Additionally, a single oral dose of furegrelate in adult human subjects (200–1600 mg p.o.) dose-dependently inhibited thromboxane synthesis resulting in a 90% decline in the generation of TxB2 in platelet-rich plasma challenged with arachidonic acid.[20] Although these studies administered furegrelate once daily, we initially administered furegrelate twice daily (BID) to the CH piglets of our study since the reported drug half-life was only 4.2 to 5.8 hours in human adults. However, the twice daily dosing regimen did not blunt the development of PAH in 21-day CH piglets (CH + FBID). In contrast, the CH piglets that received furegrelate three times daily (CH + FTID) exhibited a 34% and 37% reduction in elevations of PVRI and Ptp, respectively, compared to values in untreated CH animals. Since we did not try to evaluate the beneficial effects of higher or more frequent doses of furegrelate in CH piglets, we may have underestimated its full therapeutic potential, and follow-up studies designed to define the pharmacokinetic profile of furegrelate will be necessary to optimize outcome. These efforts should also include assays to more accurately evaluate thromboxane synthase activity. Although we ultimately observed that average urinary levels of the stable TxA2 metabolite, 11-dehydro TxB2, were decreased in CH + FTID piglets compared to untreated CH animals, follow-up studies will be necessary to verify the statistical significance of this observation and ensure its association with beneficial outcome in larger sample sizes of experimental animals.
In conclusion, our study shows that the pharmacological inhibition of TxA2 synthase by oral administration of furegrelate blunts the development of CH-induced PAH in neonatal piglets. Furegrelate also attenuated other abnormalities of PAH including right ventricular hypertrophy, medial hypertrophy of small pulmonary arteries and loss of pulmonary vascular distensibility. Considering these findings, we propose that furegrelate should be explored further as a potentially effective therapeutic strategy to prevent the development of hypoxia-induced PAH in neonates.
ACKNOWLEDGMENTS
The authors would like to thank the staff of the Veterinary Medical Unit at the Zablocki Veterans Administration Medical Center in Milwaukee and Mr. Terry Fletcher at the University of Arkansas for Medical Sciences (UAMS) in Little Rock for their capable assistance.
Footnotes
Source of Support: Funding was provided by R01 HL-083013 (N.J.R. and J.B.G.) and R01 HL-19298 (J.B.G.) from the NIH. Additional support was provided from the UAMS Graduate Student Research Fund to D.K.H.-S., and stipend support was provided to N.D.D. from the UAMS Translational Research Institute supported by UL1 RR029884 from the NIH National Center for Research Resources.
Conflict of Interest: None declared.
REFERENCES
- 1.Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology. 2007;91:283–290. doi: 10.1159/000101343. [DOI] [PubMed] [Google Scholar]
- 2.Widlitz A, Barst RJ. Pulmonary arterial hypertension in children. Eur Respir J. 2003;21:155–76. doi: 10.1183/09031936.03.00088302. [DOI] [PubMed] [Google Scholar]
- 3.Porta NF, Steinhorn RH. Pulmonary vasodilator therapy in the NICU: Inhaled nitric oxide, sildenafil, and other pulmonary vasodilating agents. Clin Perinatol. 2012;39:149–64. doi: 10.1016/j.clp.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beghetti M. Current treatment options in children with pulmonary arterial hypertension and experiences with oral bosentan. Eur J Clin Invest. 2006;36(Suppl 3):16–24. doi: 10.1111/j.1365-2362.2006.01681.x. [DOI] [PubMed] [Google Scholar]
- 5.Adatia I, Barrow SE, Stratton P, Ritter JM, Haworth SG. Abnormalities in the biosynthesis of thromboxane A 2 and prostacyclin in children with cyanotic congenital heart disease. Br Heart J. 1993;69:179–82. doi: 10.1136/hrt.69.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–5. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- 7.Fuse S, Kamiya T. Plasma thromboxane B 2 concentration in pulmonary hypertension associated with congenital heart disease. Circulation. 1994;90:2952–5. doi: 10.1161/01.cir.90.6.2952. [DOI] [PubMed] [Google Scholar]
- 8.Adatia I, Barrow SE, Stratton PD, Miall-Allen VM, Ritter JM, Haworth SG. Thromboxane A2 and prostacyclin biosynthesis in children and adolescents with pulmonary vascular disease. Circulation. 1993;88:2117–22. doi: 10.1161/01.cir.88.5.2117. [DOI] [PubMed] [Google Scholar]
- 9.Rosenzweig EB, Ivy DD, Widlitz A, Doran A, Claussen LR, Yung D, et al. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46:697–704. doi: 10.1016/j.jacc.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 10.Barst RJ, Ivy D, Dingemanse J, Widlitz A, Schmitt K, Doran A, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;73:372–82. doi: 10.1016/s0009-9236(03)00005-5. [DOI] [PubMed] [Google Scholar]
- 11.Rich S, Hart K, Kieras K, Brundage BH. Thromboxane synthetase inhibition in primary pulmonary hypertension. Chest. 1987;91:356–60. doi: 10.1378/chest.91.3.356. [DOI] [PubMed] [Google Scholar]
- 12.Langleben D, Christman BW, Barst RJ, Dias VC, Galie N, et al. Effects of the thromboxane synthetase inhibitor and receptor antagonist terbogrel in patients with primary pulmonary hypertension. Am Heart J. 2002;143:E4. doi: 10.1067/mhj.2002.121806. [DOI] [PubMed] [Google Scholar]
- 13.Hamberg M, Samuelsson B. Detection and isolation of an endoperoxide intermediate in prostaglandin biosynthesis. Proc Natl Acad Sci USA. 1973;70:899–903. doi: 10.1073/pnas.70.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72:2994–8. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP, Versteeg HH. Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Cell Biol. 2004;36:1187–205. doi: 10.1016/j.biocel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Fike CD, Kaplowitz MR, Pfister SL. Arachidonic acid metabolites and an early stage of pulmonary hypertension in chronically hypoxic newborn pigs. Am J Physiol. 2003;284:L316–23. doi: 10.1152/ajplung.00228.2002. [DOI] [PubMed] [Google Scholar]
- 17.Fike CD, Zhang Y, Kaplowitz MR. Thromboxane inhibition reduces an early stage of chronic hypoxia-induced pulmonary hypertension in piglets. J Appl Physiol. 2005;99:670–6. doi: 10.1152/japplphysiol.01337.2004. [DOI] [PubMed] [Google Scholar]
- 18.Fike CD, Aschner JL, Slaughter JC, Kaplowitz MR, Zhang Y, Pfister SL. Pulmonary arterial responses to reactive oxygen species are altered in newborn piglets with chronic hypoxia-induced pulmonary hypertension. Pediatr Res. 2011;70:136–41. doi: 10.1203/PDR.0b013e3182207ce7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorman RR, Johnson RA, Spilman CH, Aiken JW. Inhibition of platelet thromboxane A 2 synthase activity by sodium 5-(3’-pyridinylmethyl)benzofuran-2-carboxylate. Prostaglandins. 1983;26:325–42. doi: 10.1016/0090-6980(83)90099-0. [DOI] [PubMed] [Google Scholar]
- 20.Mohrland JS, VanderLugt JT, Gorman RR, Lakings DB. Thromboxane synthase activity and platelet function after furegrelate administration in man. J Clin Pharmacol. 1989;29:53–8. doi: 10.1002/j.1552-4604.1989.tb03237.x. [DOI] [PubMed] [Google Scholar]
- 21.Mohrland JS, VanderLugt JT, Lakings DB. Multiple dose trial of the thromboxane synthase inhibitor furegrelate in normal subjects. Eur J Clin Pharmacol. 1990;38:485–8. doi: 10.1007/BF02336688. [DOI] [PubMed] [Google Scholar]
- 22.Lakings DB, Friis JM, Lunan CM, VanderLugt JT, Mohrland JS. Pharmacokinetics of furegrelate after oral administration to normal humans. Pharm Res. 1989;6:53–7. doi: 10.1023/a:1015899602741. [DOI] [PubMed] [Google Scholar]
- 23.Wynalda MA, Liggett WF, Fitzpatrick FA. Sodium 5-(3’-pyridinylmethyl)benzofuran-2-carboxylate (U-63557A), a new, selective thromboxane synthase inhibitor: Intravenous and oral pharmacokinetics in dogs and correlation with ex situ thromboxane B2 production. Prostaglandins. 1983;26:311–24. doi: 10.1016/0090-6980(83)90098-9. [DOI] [PubMed] [Google Scholar]
- 24.Hirenallur SD, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, et al. Upregulation of vascular calcium channels in neonatal piglets with hypoxia-induced pulmonary hypertension. Am J Physiol. 2008;295:L915–24. doi: 10.1152/ajplung.90286.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perneby C, Granstrom E, Beck O, Fitzgerald D, Harhen B, Hjemdahl P. Optimization of an enzyme immunoassay for 11-dehydro-thromboxane B 2 in urine: Comparison with GC-MS. Thromb Res. 1999;96:427–36. doi: 10.1016/s0049-3848(99)00126-7. [DOI] [PubMed] [Google Scholar]
- 26.Molthen RC, Karau KL, Dawson CA. Quantitative models of the rat pulmonary arterial tree morphometry applied to hypoxia-induced arterial remodeling. J Appl Physiol. 2004;97:2372–2384. doi: 10.1152/japplphysiol.00454.2004. discussion 2354. [DOI] [PubMed] [Google Scholar]
- 27.Bui KC, Hammerman C, Hirschl R, Snedecor SM, Cheng KJ, Chan L, et al. Plasma prostanoids in neonatal extracorporeal membrane oxygenation.Influence of meconium aspiration. J Thorac Cardiovasc Surg. 1991;101:612–7. [PubMed] [Google Scholar]
- 28.Dobyns EL, Wescott JY, Kennaugh JM, Ross MN, Stenmark KR. Eicosanoids decrease with successful extracorporeal membrane oxygenation therapy in neonatal pulmonary hypertension. Am J Respir Crit Care Med. 1994;149:873–80. doi: 10.1164/ajrccm.149.4.8143049. [DOI] [PubMed] [Google Scholar]
- 29.Pakala R, Benedict CR. Effect of serotonin and thromboxane A2 on endothelial cell proliferation: Effect of specific receptor antagonists. J Lab Clin Med. 1998;131:527–37. doi: 10.1016/s0022-2143(98)90061-0. [DOI] [PubMed] [Google Scholar]
- 30.Pakala R, Willerson JT, Benedict CR. Effect of serotonin, thromboxane A2, and specific receptor antagonists on vascular smooth muscle cell proliferation. Circulation. 1997;96:2280–6. doi: 10.1161/01.cir.96.7.2280. [DOI] [PubMed] [Google Scholar]
- 31.Sachinidis A, Flesch M, Ko Y, Schror K, Bohm M, Dusing R, et al. Thromboxane A2 and vascular smooth muscle cell proliferation. Hypertension. 1995;26:771–80. doi: 10.1161/01.hyp.26.5.771. [DOI] [PubMed] [Google Scholar]
- 32.Fediuk J, Gutsol A, Nolette N, Dakshinamurti S. Thromboxane-induced actin polymerization in hypoxic pulmonary artery is independent of Rho. Am J Physiol Lung Cell Mol. 2012;302:L13–26. doi: 10.1152/ajplung.00016.2011. [DOI] [PubMed] [Google Scholar]