

Abstract
A proliferation of mast cells around the small pulmonary blood vessels and the alveolar septae has been noted in models of pulmonary hypertension, and in plexiform lesions of pulmonary arterial hypertension (PAH) in patients. Here, we hypothesize that total mast cell numbers and activation are increased in PAH and that they contribute to vascular remodeling through cellular and soluble proangiogenic effectors. To test this, blood and urine were collected from patients with PAH (N=44), asthma (N=18) and healthy controls (N=29) to quantitate biomarkers of total body mast cell numbers and activation (total and mature tryptase, N-methyl histamine, leukotriene LTE4 and prostaglandin PGD-M). Serum total tryptase was higher in PAH than that in controls suggesting greater numbers of mast cells, but indicators of mast cell activation (mature tryptase, LTE4 and PGD-M) were similar among PAH, asthma, and controls. Immunohistochemistry of lung tissues identified mast cells as primarily perivascular and connective tissue chymase+ type in PAH, rather than mucosal phenotype. Intervention with mast cell inhibitors cromolyn and fexofenadine was performed in 9 patients for 12 weeks to identify the influence of mast cell products on the pathologic proangiogenic environment. Treatment decreased total tryptase and LTE-4 levels over time of treatment. This occurred in parallel to a drop in vascular endothelial growth factor (VEGF) and circulating proangiogenic CD34+CD133+ progenitor cells, which suggests that mast cells may promote vascular remodeling and dysfunction. In support of this, levels of exhaled nitric oxide, a vasodilator that is generally low in PAH, increased at the end of the 12-week mast cell blockade and antihistamine. These results suggest that mast cells might contribute to the pulmonary vascular pathologic processes underlying PAH. More studies are needed to confirm their potential contribution to the disease.
Keywords: mast cells, pulmonary arterial hypertension, tryptase, proangiogenic progenitor cells
Pulmonary arterial hypertension (PAH) is a severe condition clinically defined by elevated mean pulmonary artery pressure. Endothelial and smooth muscle proliferation and dysfunction were the pathological defining features of the panvascular arteriopathic remodeling that ultimately lead to right heart failure and death.[1–3] However, treatment strategies have primarily focused on vasodilator drugs which have limited effect on long-term survival and little or no effect on the vascular remodeling.[4] Goals of new strategies are to halt or reverse the vascular remodeling; thus, investigation of pathways that may fuel the proliferative angiopathy is important for design of novel therapies.[5] In this context, mast cells play a central role in angiogenesis.[6,7] Although derived from bone marrow progenitors (as are all hematopoietic cells), differentiation of mast cells occurs only after recruitment of the circulating progenitors into local tissues where the environment dictates the final long-lived phenotype of the cell. Mast cells are mainly localized in mucosa and connective tissues exposed to the outside world. They are critical for activation of innate and adaptive immunity.[8] However, evidence from cancer biology studies now reveal that mast cells are master regulators of angiogenesis through the production of vascular endothelial growth factor (VEGF) and release of proangiogenic proteases.[6,7] In classic pathologic descriptions written over a century ago, Heath et al. identified mast cells as “plentiful” within plexiform lesions of idiopathic PAH (IPAH); particularly, he noted their presence in the early cellular lesions.[9] Other studies have confirmed the greater numbers of mast cells in experimental models of pulmonary hypertension.[10–12] Despite the known presence of abundant mast cells in the pulmonary vascular lesions, their role in the mechanisms of the pulmonary vascular disease is unclear. In model systems, hypoxia-related vasoconstriction and remodeling of the pulmonary artery wall were ascribed in part to release of factors from mast cells, which degranulate under hypoxic conditions.[13] This led to the speculation that mast cell secretion of a multitude of factors which affect vascular tone or activate the renin-angiotensin system likely contribute to the pathogenesis of pulmonary hypertension.
Classically, mast cells are identified in tissues by their unique positivity for tryptase, a serine proteinase, which has been identified as one of the important proangiogenic factors released by peritumoral mast cells infiltrating cancers that may contribute to neoangiogenesis of malignancies.[14,15] The immature form of tryptase is constitutively released from mast cells, and thus serum total tryptase levels reflect mast cell numbers in the body. When activated, mature tryptase is discharged from mast cells, and this has been used as a marker of mucosal mast cell activation. Interestingly, reports suggest that mast cells in PAH are of the tryptase+/chymase+ “connective tissue” phenotype.[16,17] The serine protease chymase, independent of angiotensin converting enzymes, can lead to localized production of angiotensin II, and the activation of endothelins and matrix metalloproteases, which collectively govern vasomotor tone and neovascularization.[7,18,19] Thus, the perivascular lesional distribution of mast cells in the PAH lung strongly implies a pathophysiological role for mast cell products in driving the lung-specific vascular hypertension and proliferative vascular remodeling. Indeed, in murine models of pulmonary hypertension associated with left heart disease and in monocrotaline-induced rodent pulmonary hypertension, treatment with a mast cell stabilizer or use of mast cell deficient rats attenuated vascular remodeling.[20] Likewise, the early use of mast cell stabilizer cromolyn, or antagonists of the chemokine receptors involved in recruitment of mast cell progenitors, reduce the development of vascular remodeling and right ventricular hypertrophy in hypoxia-induced rodent pulmonary hypertension.[21,22]
The contribution of mast cells in the pathogenesis of human PAH is unknown, but studies identify mast cell progenitors in the circulation of IPAH patients, as well as greater numbers of progenitors and mast cells in the pulmonary arterial lesions.[23] Here, we hypothesize that mast cells contribute to PAH pathogenesis through production of secreted factors which dictate pulmonary hypertension and vascular remodeling. To test this, mast cell numbers, phenotype, and activation in PAH were compared to healthy controls and individuals with asthma, a control disease typified by high numbers and activation of mast cells. To investigate the potential contribution to the pathogenesis of PAH, mast cell degranulation products that promote angiogenesis and the mobilization and recruitment of proangiogenic myeloid progenitors were determined before and after treatment of patients with mast cell blockade cromolyn and the H1 histamine antagonist fexofenadine.
MATERIALS AND METHODS
Blood and urine samples from individuals with PAH, asthma, and healthy controls were collected to measure mast cell products. Clinical data were collected from tests performed as part of standard of care of patients. A mechanistic investigation of cromolyn and fexofenadine to block mast cells activation was performed in a subgroup of PAH patients to determine whether mast cells contribute to the high levels of vasoactive and proangiogenic factors which contribute to the vascular remodeling of PAH. Inclusion criteria included age of at least 18 years, a diagnosis of pulmonary arterial hypertension (PAH), and stability on current PAH medications. Exclusion criteria included participation in other studies, hepatic insufficiency (transaminase levels >4-fold the upper limit of normal, bilirubin >2-fold the upper limit for normal), renal insufficiency (creatinine level, >2.0 mg/dl), pregnancy, breast feeding, lack of use of safe contraception, acute heart failure, known allergy to any of the study drugs, or history of drug or alcohol abuse within the last 12 months. None of the patients enrolled in the mechanistic study had a diagnosis of asthma. All patients provided written informed consent under an IRB-approved protocol.
Mast cell blockade-directed therapy
Individuals received treatment for 12 weeks with a mast cell stabilizer, cromolyn 800 mcg administered by a multidose inhaler with spacer by two puffs, four times a day and an antihistamine, fexofenadine, 180 mg daily. Patients were evaluated at baseline (time 0) four weeks prior to medications, and after Weeks 4, 8, and 12 of treatment. Medications were brought to each visit to check for adherence and dosage. Patients were asked at each visit on accurate use of the inhaler and compliance. Spirometry, lung diffusing capacity, brain natriuretic peptide (BNP), echocardiogram, and 6-minute walk distance were evaluated at each study visit. Blood and urine samples were obtained at each visit for measures of vasoactive and angiogenic factors produced by mast cells, and circulating proangiogenic progenitor cells.
Lung function, diffusing capacity for carbon monoxide and exhaled nitric oxide
Spirometry was performed on each study participant according to American Thoracic Society (ATS) Guidelines using a Viasys Master Screen spirometer (San Diego, Calif., USA). The single breath carbon monoxide diffusing capacity (DLCO) was performed weekly using a Viasys Master Screen analyzer (San Diago, Calif., USA), and measurements were performed at the same time of day on each visit to minimize the effect of diurnal variations.[24] The single-breath DLCO method by ATS standards was performed in duplicate. DLCO was not adjusted for hemoglobin or carboxyhemoglobin. Single-breath on-line measurement of fractional nitric oxide (NO) concentration in expired breath (FENO) was measured using the NIOX (Aerocrine, N.Y., USA).[25] All analyzers were calibrated daily and weekly controls were completed to ensure accuracy.
Assessment of mast cell activation by levels of degranulation products: Serum total and mature tryptase and urinary N-methyl histamine, LTE4, and PGD-M
Tryptase, total and mature, was measured in serum using fluorescent enzyme immunoassay.[26] N-methyl histamine (NMH) was extracted from urine using solid-phase extraction. The elute is analyzed using liquid chromatography-tandem mass spectrometry and quantified using a stable isotope labeled internal standard.[27] LTE4 and PGD-M were measured in urine. After urine acidification, an extraction step using an Empore C-18 solid-phase extraction column (standard density, 6 ml capacity, 3M, St. Paul, Minn., USA) was completed. The eluate was evaporated under a continuous stream of dry nitrogen, was then dissolved in 100 μl methanol, and was finally filtered using a 0.2 μm Spin-X filter (Corning, Corning, N.Y., USA). LTE4 was measured using ultra pressure liquid chromatography/mass spectrometry. PGD-M levels were measured by mass spectrometry as described previously.[28]
Evaluation of enzyme-linked immunosorbent assay for proangiogenic factors
Erythropoietin (Epo), hepatocyte growth factor (HGF), stem cell factor (SCF), and VEGF were measured in plasma using quantikine enzyme-linked immunosorbent assay (ELISA) (R & D system, Minn., USA).
Mast cells numbers and types in lungs of PAH patients
Lung tissues were obtained from explanted PAH and failed donor lungs for investigation of mast cells. Formalin-fixed paraffin embedded tissue sections were stained with human monoclonal tryptase (Promega G3361, dilution 1/1000) and chymase (Abcam, ab2377, dilution 1/100). Pictures were taken of five random fields from each tissue section at 10× using a Leica DM5000B microscope, equipped with a QImaging Retiga SRY digital camera and using Image Pro v.6.2 with Oasis Turboscan module. To quantify the mast cells, the number of positive cells was counted on each picture in a blinded fashion and the average/field was used as a total number.
Echocardiogram
Two-dimensional echocardiogram and Doppler examinations were performed by an experienced sonographer. Interventricular septal (IVS) thickness in end-diastole, left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), and posterior wall thickness in diastole were measured from the 2D parasternal long-axis image following American Society of Echocardiography (ASE) guidelines. Left ventricular (LV) mass was determined from 2D measurements using the following formula: LV Mass = 1.04((IVS + PW + LVEDD) 3 - LVEDD3) – 13.6g (Devereaux Regression)
LV ejection fraction was determined by visual assessment, and/or apical biplane volumes. LV end-diastolic and end-systolic volumes were calculated from the apical 4- and 2-chamber views using the modified Simpson method. LV fractional shortening was determined from parasternal 2D analysis as (LVEDD-LVESD/LVEDD × 100). Right ventricular (RV) end-diastolic and end-systolic areas were measured in the apical 4-chamber view by tracing the endocardial border of the RV and the tricuspid annular plane. The RV fractional area change was calculated as follows: RV end-diastolic area minus RV end-systolic area divided by RV end-diastolic area × 100. Right atrial volume was measured in the apical 4-chamber view by using the single-plane area-length method. The peak pulmonary artery systolic pressure (PASP) was estimated from the systolic pressure gradient between the RV and the right atrium by the peak continuous-wave Doppler velocity of the TR jet using the modified Bernoulli equation plus estimated right atrial pressure (RAP). RAP was estimated from the subcostal window approach measuring changes in inferior vena caval size and collapsibility as determined by the respiratory sniff test following ASE guidelines. Echo-Doppler was used to estimate the pulmonary vascular resistance (PVR). The highest Doppler continuous wave tricuspid valve peak velocity jet obtained from multiple views (parasternal long axis, parasternal short axis, apical 4-chamber, subcostal, and apical off-axis imaging) was determined as the maximum tricuspid regurgitant velocity (TRV). The pulsed wave Doppler sample was placed in the right ventricular outflow tract (RVOT) at the level of the aortic valve in the parasternal short-axis view just below the pulmonic valve so that pulmonic valve closure could be identified. The Doppler spectrum was traced to determine the time velocity integral of the RVOT. PVR was estimated as follows: PVR (Wood units) = TRVMAX (m/s) / RVOTTVI (cm) × 10 +0.16. Tricuspid annular plane systolic excursion was obtained from the apical 4-chamber RV focused view with M-mode echocardiography across the TV annulus, measuring the distance of longitudinal annular movement from end diastole to end systole toward the apex. With the exception of the TR continuous wave maximum velocity, all echo parameters were measured three times and reported as an average.
Flow cytometry evaluation of proangiogenic progenitor cells
Mononuclear cells (2×106) isolated from peripheral blood and bone marrow were labeled with antihuman CD34-FITC (Becton Dickinson, N.J., USA) and antihuman CD133-PE (Miltenyi Biotec, Auburn, Calif., USA) monoclonal antibodies to quantify CD34+CD133+ cells, as described previously.[29] Nonspecific antibody binding was analyzed in parallel with isotype-matched irrelevant antibodies. Following incubation with antibodies, cell suspensions were washed with PBS/1%BSA/0.02%sodium azide and suspended in FACS flow (Becton Dickinson, N.J., USA). The FACScan flow cytometer (Becton Dickinson, N.J., USA) was used to count 0.5×106 events. Data of at least 0.5×106 events were collected, stored as listmode files, and analyzed using Cell-Quest 3.3 Software (Becton Dickinson, N.J., USA).
Statistical analysis
Descriptive measures for quantitative variables consist of means with appropriately derived standard errors in the form “mean±SE.” Comparisons of PAH, asthmatic, and healthy subjects were performed using ANOVA or t-test when two means were compared. When ANOVA was significant, Tukey was performed for pairwise comparison. Spearman correlation coefficients were used to describe relationships among pairs of quantitative variables in a manner free of the normality assumption. For the mechanistic intervention, outcomes analyzed were all quantitative in nature. The changes in outcomes between visits were assessed using paired t-tests.
RESULTS
Study population
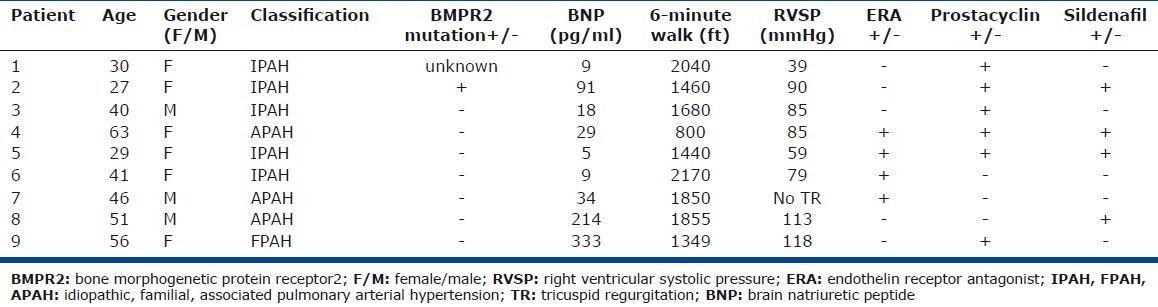
Individuals with PAH (N=44), controls (N=31), and asthma (N=18) provided blood and urine for measurement of mast cell biomarkers in relation to clinical disease. PAH subjects were older (age [years]: PAH [N=44] 45±2; controls [N=31] 36±2; asthma [N=18] 37±3, ANOVA P=0.004). There were more females in the control and PAH groups compared to the asthma group (gender [F/M]: PAH [N=44] 33/11; controls [N=31] 26/5; asthma [N=18] 11/7, ANOVA P=0.03; Table 1). Due to sample limitations, not all assays could be performed in all subjects. The numbers of individuals evaluated for each assay are provided in the text. Separate from the biologic samples of blood and urine, tissue sections of explanted lungs from PAH patients (N=18) undergoing transplant or donor lungs (N=4) not used in transplantation were available for quantification of pulmonary mast cells numbers and phenotype. Nine PAH patients participated in the mast cell blockade study over 16 weeks (Table 2). All participants completed the 16-week study.
Table 1.
Characteristics of all PAH population
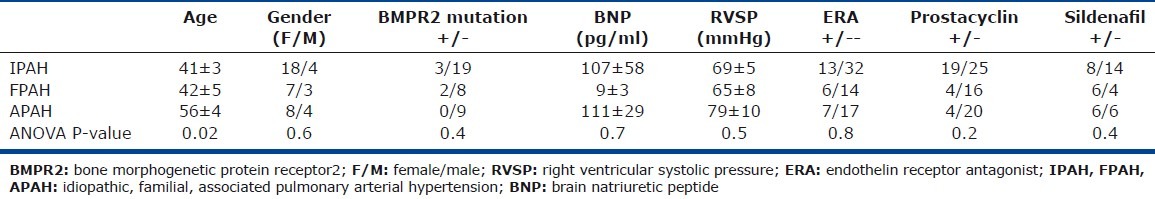
Table 2.
Mast cell-blockade study sample characteristics
Mast cells in pulmonary hypertension
Lung sections were stained for tryptase and chymase to localize, characterize, and quantitate the mast cells in the lungs. There was a higher number of tryptase+ mast cells in lung from patients with PAH compared to controls (mast cells per high power field: PAH [N=12] 56±7; control [N=3] 28±5; t-test P=0.01). As previously shown in human PAH lung[16,17] mast cells were concentrated around vascular lesions and had a tryptase+/chymase+ proteinase phenotype, suggesting a “connective tissue” mast cell phenotype (Fig. 1). In line with the histological findings, serum total tryptase levels were higher in PAH patients compared to controls (total tryptase [ng/ml]: PAH [N=44] 5.7±0.7; controls [N=29] 3.1±0.4; asthma [N=18] 3.8±0.8; ANOVA P=0.01 and Tukey P<0.05 for PAH compared to controls; (Fig. 2). In subclass analyses, idiopathic PAH (IPAH) patients had higher tryptase levels compared to familial PAH (FPAH) (total tryptase [ng/ml]: IPAH [N=33] 6.3±0.8; FPAH [N=10] 4.1±1.4; P=0.03]. There was a minor but significant correlation between total serum tryptase levels and BNP in PAH patients (Spearman R=0.5, P=0.006; Fig. 2) In contrast, levels of mature tryptase were low in all subjects with no difference between PAH and controls (mature tryptase <1 for all samples). Similarly, urinary N-methyl histamine was not different among PAH, controls, and asthma (urinary N-methyl histamine [ug/g creatinine]: PAH [N=9] 108±26; controls [N=5] 88±8; asthma [N=3] 98±26; ANOVA P=0.8 and all tukey P>0.05]. Urinary LTE4 and PGD-M levels, which are rapidly produced by IgE-sensitized mucosal mast cells activated via specific allergen(s), were also similar among the groups (LTE4 [pg/mg creatinine]: PAH [N=7] 42±7; asthma [N=16] 131±65; controls [N=15] 60±9; ANOVA P=0.4; PGD-M [ng/mg creatinine]: PAH [N=7] 2.8±0.3; asthma [N=16] 2.2±0.3; controls [N=14] 2.4±0.3; ANOVA P=0.5]. Likewise, IgE levels among PAH, asthmatic and control individuals were similar (IgE [IU/ml]: PAH [N=40] 108±43, asthma [N=18] 147±53; controls [N=29] 50±17; ANOVA P=0.3 and all Tukey P>0.05]. Altogether, these findings indicate that PAH lungs contain greater numbers of the nonallergic, connective tissue phenotype of mast cells, but not activated mucosal mast cells.
Figure 1.
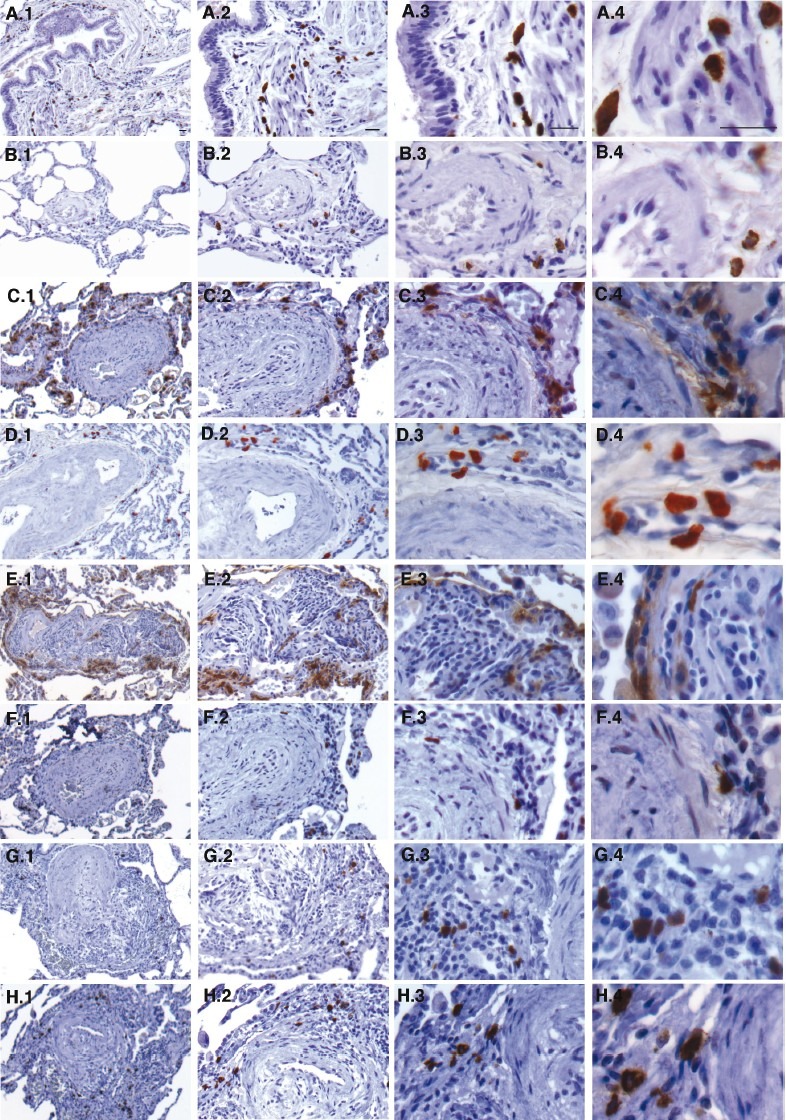
Increased numbers of connective tissue type mast cells in PAH lungs. (A–H) Tryptase and chymase immunostaining of explanted control and PAH lungs. (A–E) Tryptase stained lung tissue sections. (F–H) Chymase stained lung tissue sections. Explanted PAH and failed donor (control) lung paraffin embedded tissue sections were immunostained for tryptase and chymase to identify and characterize mast cell phenotype (brown cells). Mast cell numbers were increased in the lungs of patients with PAH compared to controls (P=0.01) and localized predominantly to perivascular regions as opposed to submucosal regions as in control lungs. Mast cells in PAH lungs were tryptase+ and chymase+ consistent with a connective tissue phenotype as opposed to primarily tryptase+ in control lungs. Panels A–B – Control lungs stained for tryptase: (A) Mast cells are seen in the submucosal regions of the airways of control lungs. (B) Mast cells around a blood vessel in a control lung are less compared to PAH lungs. Panels C–H – PAH lungs: (C–E) Tryptase+ mast cells in PAH are in the perivascular adventitia and increased in number. (F–H) Mast cells in the perivascular regions are chymase+. In panels E–G where plexiform lesions are noted, mast cells are seen within the lesions. Magnification: (1) 5×, (2) 10×, (3) 20×, (4) 40×. Scale bar: 25μm.
Figure 2.
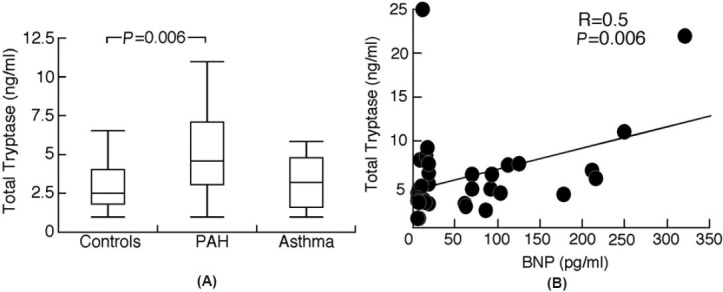
(A) Serum total tryptase levels are higher in PAH than controls. Total serum tryptase levels are higher in PAH compared to controls indicating greater total numbers of mast cells in PAH. Box plots indicate median values, upper and lower quartiles. (B) Serum total tryptase levels correlate with brain natriuretic peptide (BNP). Total tryptase levels are related to disease severity assessed by BNP.
Mast cell blockade in PAH
Given the elevated numbers of mast cells in PAH lungs and the relationship of tryptase to BNP, a subgroup of PAH patients was placed on mast cell stabilizer cromolyn and antihistamine fexofenadine to evaluate effects on the PAH proangiogenic milieu. Mature tryptase did not change over time; however, total serum tryptase dropped from baseline to Weeks 4 and 12 (tryptase [ng/ml]: baseline 5.2±0.5; Week 4 4.3±0.4; Week 12 4.2±0.4; all P<0.05). Likewise, PGD-M did not change significantly, however LTE4 appeared to be lower with treatment at Week 8 (LTE4 [pg/mg creatinine]: baseline 63±17; Week 8 41±11; P=0.04; Fig. 3). To assess the potential contribution of mast cells for angiogenesis, we measured circulating angiogenic factors, including VEGF, HGF, EPO, SCF. VEGF, a potent proangiogenic factor secreted by mast cells, decreased early after four weeks of treatment (VEGF [pg/ ml]: baseline 400±117; Week 4 335±112; P=0.03; Fig. 3). Other angiogenic factors did not vary over the time of the study (all P>0.05; Table 3). VEGF dictates angiogenesis in part by induction of myeloid progenitor cell mobilization into the circulation and subsequent recruitment to local vascular bed. Consistent with the temporal drop of VEGF at Week 4, circulating CD34+CD133+ proangiogenic progenitor myeloid cells consequently decreased by Week 4 of treatment (CD34+CD133+ cells (%): baseline 0.11±0.03; Week 4 0.08±0.03; week 12 0.07±0.02, all P<0.05; Fig. 3). Furthermore, numbers of CD34+CD133+ cells were related to the levels of serum VEGF across the time of the study (Spearman R=0.4, P=0.02) but not with the other angiogenic factors (all P>0.05).
Figure 3.
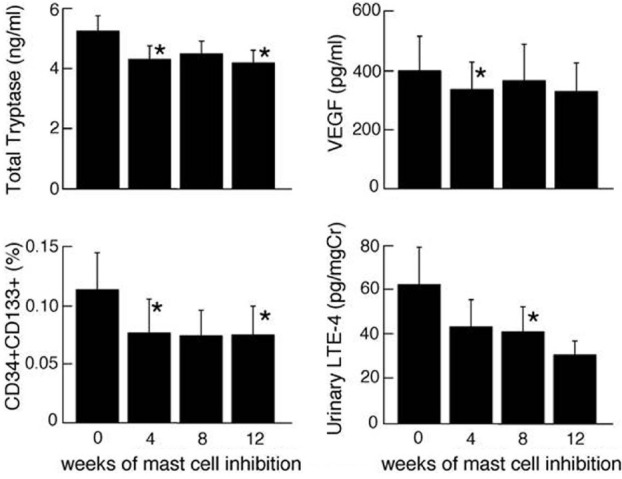
Mast cell blockade for 12 weeks with cromolyn and fexofenadine. Mast cell activation and proangiogenic biomarkers were measured at baseline Week 0 (prior to start of therapy). Four weeks later medications were started, and patients evaluated after 4, 8, and 12 weeks of therapy. Total tryptase and urinary LTE4 dropped with mast cell blockade therapy. Proangiogenic CD34+CD133+ myeloid progenitors and VEGF also decreased with mast cell blockade. Asterisk significant changes compared to baseline (all P<0.05).
Table 3.
Proangiogenic factors with mast cell blockade

Exhaled NO was evaluated over the time of mast cell blockade to detect potential positive vascular functional effects with treatment. NO increased significantly at the end of the study from Weeks 8 to 12 of therapy (NO [ppb]: Week 8 15±2; Week 12 17±2; P=0.02; Fig. 4). Overall, NO was inversely related to VEGF (Spearman R=-0.6; P<0.0001; Fig. 4) and CD34+CD133+ cells (Spearman R=-0.4; P=0.02).
Figure 4.
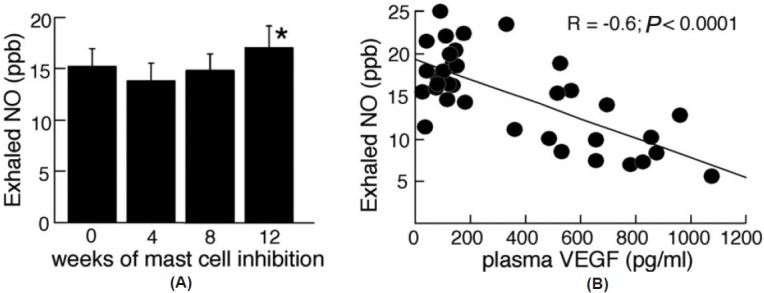
(A) Nitric oxide increases with mast cell blockade. Exhaled NO measured over the course of the study suggests improved pulmonary vascular health near the end of the 12-week therapy. The asterisk represents significant change from week 8 to week 12 (P<0.05). (B) NO levels are inversely correlated to VEGF. The inverse relationship suggests that the decrease of VEGF, which occurs with mast cell blockade, was associated with increasing levels of NO. Points are derived from all time points in the study.
Effects of mast cell blockade on clinical parameters
Although the study was aimed to determine effects of mast cell blockade on proangiogenic factors and cells, clinical evaluation of patients were evaluated for potential benefits and/or adverse effects. Lung functions (FEV1, FVC and FEV1/FVC, DLCO) were similar across the time of the interventional study (all P>0.05); likewise, BNP and 6-Minute Walk Distance did not change (all P>0.1). The tricuspid annular plane systolic excursion (TAPSE) decreased from baseline to Week 12 of the study (TAPSE [cm]: baseline 2±0.2; Week 12 1.8±0.1; P=0.03) as did left ventricular (LV) mass average (LV mass average: baseline 214±17; Week 12 171±18; P=0.02). Serum total tryptase levels were inversely related to clinical parameters, including the 6-Minute Walk Distance (Spearman R=-0.4; P=0.006) and TAPSE (Spearman R=-0.5; P=0.04). Similarly, LTE-4 was correlated with disease severity over time (6-Min Walk: Spearman R=-0.5; P=0.004, BNP: Spearman R=0.5; P=0.005 and RAP: Spearman R=0.5; P=0.05; Table 3). In addition, 6-Minute Walk was inversely related to several proangiogenic factors, including Epo, HGF, VEGF, and SCF (Epo: Spearman R=-0.6; P<0.0001; HGF: Spearman R=-0.6; P=0.0001; VEGF: Spearman R=-0.5; P=0.004; SCF: Spearman R=-0.6; P=0.006). HGF was also related to right atrial pressure and TAPSE (right atrial pressure: Spearman R=0.7; P=0.005 and TAPSE: Spearman R=-0.6; P=0.03).
DISCUSSION
Although mast cells have long been described as abundant in the lungs of patients with PAH,[9] their contribution to the pathophysiology of the disease is unclear. In this report, mast cells are again shown to be abundantly present in PAH. In addition, the mast cell is identified as connective tissue type that expresses both tryptase and chymase proteases.[16,17] The small mechanistic intervention with cromolyn to block mast cell degranulation and fexofenadine to block mast cell histamine vasoactive effects led to a decrease of total serum tryptase and urinary LTE4 in parallel to a drop in proangiogenic CD34+CD133+ cells and mast cell secreted proangiogenic VEGF. These findings suggest that mast cells may contribute to the vascular pathophysiology.
Mast cell blockade and antihistamine therapy did not translate into clinical improvement. This lack of clinical improvement is most likely multifactorial, possibly due to limited sample size and/or short duration of therapy. The mechanistic intervention included nine patients who were treated for only 12 weeks and were not powered to detect clinical improvement. Furthermore, cromolyn is a weak mast cell blocker in humans; a more potent mast cell inhibition might have provided an indication of clinical benefit. C–kit inhibitors which affect mast cells, such as imatininb, are being studied in pulmonary hypertension and have shown promising results.[30] Mast cells secrete vasomotor mediators and factors that promote angiogenesis and vascular remodeling. The patients enrolled in the study had advanced PAH and almost certainly had severe remodeling, which might also explain the lack of clinical response, and indeed even worsening clinical status by some echocardiographic parameters. In fact, our findings suggest worsening cardiac function based on TAPSE. This seems to indicate that mast cells blockade affect the myocardium independently of its effect on the vasculature. Banasova et al. used cromolyn in a murine hypoxia model of pulmonary hypertension and found that if given during the very early phase of hypoxia exposure, pulmonary hypertension, and vascular remodeling were reduced;[22] if provided late in the model, beneficial responses were not observed.[22] In another study using a rodent model of flow associated pulmonary hypertension, early treatment with cromolyn attenuated vascular remodeling, shown by reduced pulmonary artery wall thickness, muscularization, and wall/lumen ratio.[31] However, the effects on the pulmonary vascular bed were not associated with positive effects on RV hemodynamics; in fact there was no improvement in pulmonary arterial pressures and RV hypertrophy.[31] Similarly, Gambaryan et al. used a CXCR4 antagonist and a CXCR7 antagonist to inhibit recruitment of progenitors, including mast cell progenitor in a murine model of hypoxia-induced PH.[21] They showed that when the drugs are used early on before development of PH (preventive strategy), they prevented vascular remodeling, PH, and the perivascular accumulation of c-kit+ progenitors. However, when used to treat established PH, the drugs did not abrogate the vascular remodeling, RV hypertrophy, and increased pulmonary artery pressures.[21] These findings and others[10] suggest that mast cell targeted therapy might work best if early in the course of the disease, and that the primary effect may be attenuation of vascular remodeling and/or vasomotor tone.
In this study, exhaled NO increased at the end of the treatment period. Several studies have shown that NO levels are low in PAH[32,33] and mechanistically related to dysfunctional vascular endothelium.[34–36] Furthermore, treatments that decrease pulmonary artery pressures are associated with increase of exhaled NO.[32,33] Thus, although there was only a slight increase of NO at the end of the study and no significant change in pulmonary artery pressures, the rise in NO suggests a possible vascular effect. Unfortunately, the lack of a control group limits firm conclusions regarding the significance of changes in NO. On the other hand, studies in asthma, which is characterized by high levels of exhaled NO due to airway expression of the inducible NO synthase,[37] demonstrate that NO levels are generally unaffected by cromolyn.[38] Other biological markers changed with therapy, but changes were not consistent. While increase in NO was noted at the end of the study, LTE-4 dropped at Week 8 and VEGF at Week 4. These variable effects could be due to several limitations, including the small sample size, poor bioavailability of drug, and consistency of dose delivery to the lung vascular compartment. Finally, the time of treatment may have been inadequate, i.e., a longer exposure to cromolyn might be required for consistent effects to be apparent.
Overall, our findings support prior reports that point to mast cells in the vascular processes leading to PAH.[9,10,17,20–23] Further studies are needed to determine if mast cell blockade and/or more potent mast cell targeted therapies early in the course of the disease can impact the angiopathic processes and improve patients’ outcome.
ACKNOWLEDGMENTS
We thank B. Savasky and J. Hanson for excellent technical assistance; D. Hatala, Dr. J. Drazba, and Dr. A. J. Peterson in the Lerner Research Institute Digital Imaging Core; C. Shemo and S. O‘Bryant in the Lerner Research Institute Flow Cytometry Core for technical advice and excellent assistant with instrument operation; and M. Koo for study coordination. We also thank Dr. M. Aldred for BMPR2 analysis and Drs. R. Dweik, C. Jennings and O. Minai for help in patient recruitment.
Footnotes
Source of Support: This work was supported by the Cleveland Clinic Research Programs Council. Kewal Asosingh is a scholar of the international society for advancement of cytometry.
Conflict of Interest: None declared.
REFERENCES
- 1.Farber HW, Loscalzo J. Pulmonary arterial hypertension. The New England journal of medicine. 2004;351:1655–65. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, McLaughlin VV. The 4th World Symposium on Pulmonary Hypertension.Introduction. J Am Coll Cardiol. 2009;54(1 Suppl):S1–2. doi: 10.1016/j.jacc.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–31. doi: 10.1161/CIRCULATIONAHA.104.503540. [DOI] [PubMed] [Google Scholar]
- 4.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–63. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 5.Ghofrani HA, Barst RJ, Benza RL, Champion HC, Fagan KA, Grimminger F, et al. Future perspectives for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S108–17. doi: 10.1016/j.jacc.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theoharides TC. Mast cells and pancreatic cancer. N Engl J Med. 2008;358:1860–1. doi: 10.1056/NEJMcibr0801519. [DOI] [PubMed] [Google Scholar]
- 7.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes & development. 1999;13:1382–97. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiological reviews. 1997;77:1033–79. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 9.Heath D, Yacoub M. Lung mast cells in plexogenic pulmonary arteriopathy. J Clin Pathol. 1991;44:1003–6. doi: 10.1136/jcp.44.12.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahal BK, Kosanovic D, Kaulen C, Cornitescu T, Savai R, Hoffmann J, et al. Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir Res. 2011;12:60. doi: 10.1186/1465-9921-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker A, McMurtry IF, Alexander AF, Reeves JT, Grover RF. Lung mast cell density and distribution in chronically hypoxic animals. J Appl Physiol. 1977;42:174–8. doi: 10.1152/jappl.1977.42.2.174. [DOI] [PubMed] [Google Scholar]
- 12.van Albada ME, Bartelds B, Wijnberg H, Mohaupt S, Dickinson MG, Schoemaker RG, et al. Gene expression profile in flow-associated pulmonary arterial hypertension with neointimal lesions. Am J Physiol Lung Cell Mol Physiol. 2010;298:L483–91. doi: 10.1152/ajplung.00106.2009. [DOI] [PubMed] [Google Scholar]
- 13.Nadziejko CE, Loud AV, Kikkawa Y. Effect of alveolar hypoxia on pulmonary mast cells in vivo. The American review of respiratory disease. 1989;140:743–8. doi: 10.1164/ajrccm/140.3.743. [DOI] [PubMed] [Google Scholar]
- 14.Blair RJ, Meng H, Marchese MJ, Ren S, Schwartz LB, Tonnesen MG, et al. Human mast cells stimulate vascular tube formation.Tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribatti D, Vacca A, Nico B, Crivellato E, Roncali L, Dammacco F. The role of mast cells in tumour angiogenesis. Br J Haematol. 2001;115:514–21. doi: 10.1046/j.1365-2141.2001.03202.x. [DOI] [PubMed] [Google Scholar]
- 16.Mitani Y, Ueda M, Maruyama K, Shimpo H, Kojima A, Matsumura M, et al. Mast cell chymase in pulmonary hypertension. Thorax. 1999;54:88–90. doi: 10.1136/thx.54.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamada H, Terai M, Kimura H, Hirano K, Oana S, Niimi H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am J Respir Crit Care Med. 1999;160:1303–8. doi: 10.1164/ajrccm.160.4.9810058. [DOI] [PubMed] [Google Scholar]
- 18.Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochimica et biophysica acta. 2009;1796:19–26. doi: 10.1016/j.bbcan.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishi K, Jin D, Takai S, Muramatsu M, Katayama H, Tamai H, et al. Role of chymase-dependent angiotensin II formation in monocrotaline-induced pulmonary hypertensive rats. Pediatr Res. 2006;60:77–82. doi: 10.1203/01.pdr.0000219431.45075.d9. [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann J, Yin J, Kukucka M, Yin N, Saarikko I, Sterner-Kock A, et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J. 2011;37:1400–10. doi: 10.1183/09031936.00043310. [DOI] [PubMed] [Google Scholar]
- 21.Gambaryan N, Perros F, Montani D, Cohen-Kaminsky S, Mazmanian M, Renaud JF, et al. Targeting of c-kit+ haematopoietic progenitor cells prevents hypoxic pulmonary hypertension. Eur Respir J. 2011;37:1392–9. doi: 10.1183/09031936.00045710. [DOI] [PubMed] [Google Scholar]
- 22.Banasova A, Maxova H, Hampl V, Vízek M, Povýsilová V, Novotná J, et al. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration. 2008;76:102–7. doi: 10.1159/000121410. [DOI] [PubMed] [Google Scholar]
- 23.Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:116–23. doi: 10.1164/rccm.201006-0905OC. [DOI] [PubMed] [Google Scholar]
- 24.Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26:720–35. doi: 10.1183/09031936.05.00034905. [DOI] [PubMed] [Google Scholar]
- 25.ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am J Respir Crit Care Med. 2005;171:912–30. doi: 10.1164/rccm.200406-710ST. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz LB, Bradford TR, Rouse C, Irani AM, Rasp G, Van der Zwan JK, et al. Development of a new, more sensitive immunoassay for human tryptase: Use in systemic anaphylaxis. J Clin Immunol. 1994;14:190–204. doi: 10.1007/BF01533368. [DOI] [PubMed] [Google Scholar]
- 27.Martens-Lobenhoffer J, Neumann HJ. Determination of 1-methylhistamine and 1-methylimidazoleacetic acid in human urine as a tool for the diagnosis of mastocytosis. J Chromatogr B Biomed Sci Appl. 1999;721:135–40. doi: 10.1016/s0378-4347(98)00481-2. [DOI] [PubMed] [Google Scholar]
- 28.Awad JA, Morrow JD, Roberts LJ., 2nd Simplification of the mass spectrometric assay for the major urinary metabolite of prostaglandin D2. J Chromatogr. 1993;617:124–8. doi: 10.1016/0378-4347(93)80430-c. [DOI] [PubMed] [Google Scholar]
- 29.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, Farver C, et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172:615–27. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182:1171–7. doi: 10.1164/rccm.201001-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartelds B, van Loon RL, Mohaupt S, Wijnberg H, Dickinson MG, Boersma B, et al. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest. 2012;141:651–60. doi: 10.1378/chest.11-0663. [DOI] [PubMed] [Google Scholar]
- 32.Ozkan M, Dweik RA, Laskowski D, Arroliga AC, Erzurum SC. High levels of nitric oxide in individuals with pulmonary hypertension receiving epoprostenol therapy. Lung. 2001;179:233–43. doi: 10.1007/s004080000064. [DOI] [PubMed] [Google Scholar]
- 33.Machado RF, Londhe Nerkar MV, Dweik RA, Hammel J, Janocha A, Pyle J, et al. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Med. 2004;37:1010–7. doi: 10.1016/j.freeradbiomed.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. Faseb J. 2004;18:1746–8. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 36.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. New Eng J Med. 1995;333:214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 37.Guo FH, Comhair SA, Zheng S, Dweik RA, Eissa NT, Thomassen MJ, et al. Molecular mechanisms of increased nitric oxide (NO) in asthma: Evidence for transcriptional and post-translational regulation of NO synthesis. J Immunol. 2000;164:5970–80. doi: 10.4049/jimmunol.164.11.5970. [DOI] [PubMed] [Google Scholar]
- 38.Cowan DC, Hewitt RS, Cowan JO, Palmay R, Williamson A, Lucas SJ, et al. Exercise-induced wheeze: Fraction of exhaled nitric oxide-directed management. Respirology. 2010;15:683–90. doi: 10.1111/j.1440-1843.2010.01740.x. [DOI] [PubMed] [Google Scholar]