

Abstract
We have shown previously that hypoxia inhibits the growth of distal human pulmonary artery smooth muscle cells (PASMC) isolated under standard normoxic conditions (PASMCnorm). By contrast, a subpopulation of PASMC, isolated through survival selection under hypoxia was found to proliferate in response to hypoxia (PASMChyp). We sought to investigate the role of hypoxia-inducible factor (HIF) in these differential responses and to assess the relationship between HIF, proliferation, apoptosis, and pulmonary vascular remodeling in emphysema. PASMC were derived from lobar resections for lung cancer. Hypoxia induced apoptosis in PASMCnorm (as assessed by TUNEL) and mRNA expression of Bax and Bcl-2, and induced proliferation in PASMChyp (as assessed by 3H-thymidine incorporation). Both observations were mimicked by dimethyloxallyl glycine, a prolyl-hydroxylase inhibitor used to stabilize HIF under normoxia. Pulmonary vascular remodeling was graded in lung samples taken from patients undergoing lung volume reduction surgery for severe heterogenous emphysema. Carbonic anhydrase IX expression in the medial compartment was used as a surrogate of medial hypoxia and HIF stabilization and increased with increasing vascular remodeling. In addition, a mixture of proliferation, assessed by proliferating-cell nuclear antigen, and apoptosis, assessed by active caspase 3 staining, were both higher in more severely remodeled vessels. Hypoxia drives apoptosis and proliferation via HIF in distinct subpopulations of distal PASMC. These differential responses may be important in the pulmonary vascular remodeling seen in emphysema and further support the key role of HIF in hypoxic pulmonary hypertension.
Keywords: hypoxia, hypoxia-inducible factor, pulmonary arterial hypertension
It has been appreciated for some time that vascular remodeling in the pulmonary circulation may complicate a number of hypoxic lung diseases. Although there may be other factors involved in this process, such as inflammation,[1] hypoxia is regarded as a significant contributor. Indeed, hypoxic exposure is used to create animal models of pulmonary hypertension.
Many histological changes develop in the pulmonary circulation in response to chronic hypoxia involving both proximal and distal pulmonary arteries.[2–6] While significant remodeling in the proximal pulmonary arteries occurs in chronic hypoxia, the hemodynamic significance of this remains uncertain. The predominant site of pulmonary vascular resistance is the distal pulmonary circulation and it is here that hypoxia regulates vascular tone. Consequently, it is likely that changes seen here are of greatest pathological significance in the generation of elevated “fixed” pulmonary vascular resistance as seen in chronic hypoxic pulmonary hypertension.
We have recently demonstrated the existence of two distinct subpopulations of pulmonary artery smooth muscle cell (PASMC) present in the distal pulmonary artery in humans: in one, proliferation occurs under normoxic conditions (PASMCnorm) while in the other, proliferation occurs under hypoxic conditions (PASMChyp).[7] This work has led to the hypothesis that alveolar hypoxia may lead to a selective expansion of the PASMChyp population, which contributes to the remodeling of small peripheral pulmonary arteries.
Hypoxia-inducible factor (HIF) is a key regulator of gene expression in response to hypoxia and we sought to characterize its role in the differential apoptotic/proliferative response of PASMCnorm and PASMChyp to hypoxia. We also set out to assess whether such a mixed response could be observed in vivo in patients with emphysema and the relation to HIF stabilization. We showed that hypoxia drives apoptosis in PASMCnorm and proliferation in PASMChyp in vitro and that this effect was mimicked by pharmacological stabilization of HIF. In addition, in lung specimens removed from patients undergoing lung volume reduction surgery for severe heterogenous emphysema we demonstrated both apoptosis and proliferation in adjacent smooth muscle cell (SMC) populations. Furthermore, we demonstrated that the extent of apoptosis and proliferation, as well as HIF activation, correlated with the degree of vascular remodeling. These data suggest that pulmonary vascular remodeling in hypoxic lung disease may involve HIF-dependent expansion of a hypoxia-proliferative subpopulation of SMCs.
MATERIALS AND METHODS
Tissue samples
Samples of lung tissue for the culture of human pulmonary artery smooth muscle cells were obtained from patients undergoing lobectomy. Approval was obtained from the Cambridgeshire and Glenfield Hospital Local Reseach Ethics Committees and all subjects gave informed consent. Only lungs without histological evidence of significant pulmonary vascular disease were used, as judged by a thoracic pathologist. For analysis of peripheral lung sections from patients with emphysema, samples were collected from patients undergoing lung volume reduction surgery (emphysema specimens) and matched to those undergoing lobectomy for cancer (control specimens).
Culture of human pulmonary artery smooth muscle cells
Peripheral segments of human pulmonary artery were obtained. PASMC were cultured from peripheral arteries (<1–2 mm external diameter) in 20% fetal bovine serum (FBS)/Dulbecco's modified Eagle Medium (DMEM) with smooth muscle growth supplements (bFGF 2ng/ml, EGF 0.5ng/ml, insulin 5μg/ml) (Promocell, Heidelberg, Germany), as previously described.[7] After the first passage, cells were maintained in 10% FBS/DMEM and used between Passages 3-7. The smooth muscle phenotype of isolated cells was confirmed by positive immunoflourescence with antibodies to a smooth muscle actin and smooth muscle specific myosin (Sigma-Aldrich, Poole, UK), as described previously.[8]
Exposure of cells to hypoxia
PASMCs were grown under standard normoxic conditions in a 5% CO2 incubator, before being exposed to hypoxic conditions. Hypoxia was induced by pregassing cell culture medium (DMEM + 25 mM HEPES) with a gas mixture containing 95% N2/5% CO2 for 30 minutes inside a gas-tight isolator which had been flushed with N2. FBS was left to equilibrate in the isolator and was added to the pregassed medium prior to use. Plates were then placed inside specially designed perspex chambers (Bellco Glass Inc., Vineland, N.J.), which were gassed with 95% N2/5% CO2 for 20 minutes. The chambers were kept inside an O2/CO2 incubator (GA156, Leec Limited, Nottingham, UK), preset to provide an atmosphere of 1% O2/5% CO2. The pH (7.35–7.45), PO2 (2–3 kPa) and PCO2 (4–5 kPa) in the medium was checked at the beginning and end of each experiment using a blood gas analyzer (ABL5; Radiometer Ltd., West Sussex, UK). Hypoxic cells were not reoxygenated at any stage during the culture period.
Selection of the hypoxia proliferative subpopulation
PASMC at Passage 1 were plated in 20% FBS/DMEM/smooth muscle growth supplements (as above) and 25 mM HEPES in 96-well plates at an approximate density of 10 cells/well and left overnight to adhere. Plates were then placed under hypoxic conditions as described above, and maintained for up to two to three weeks. Viable, proliferating cells (observed in approximately 5% of the wells) were trypsinised and transferred sequentially to 24 then 6-well plates before passaging into T75 flasks, while maintaining hypoxic conditions throughout.
Proliferation assay
The effects of hypoxia on cell proliferation were quantified by the incorporation of [3H]-thymidine as an index of DNA synthesis, as described previously.[7] Cells were plated at 15×103 cells/well in 10% FBS/DMEM in 24-well plates and left to adhere under normoxic or hypoxic conditions (3kPa). Cells were grown to 60% confluence and then quiesced for 48 hours in normoxic or hypoxic 0.1% FBS/DMEM, unless otherwise specified. At the beginning of the experimental procedure, fresh hypoxic (3kPa) or normoxic medium was added, either alone or with specific pharmacological reagents. [3H]-thymidine (0.5mCi/well) was added after 18 hours for the final six hours. The effect of HIF stabilization was assessed using a prolyl-hydroxylase inhibitor (100 μM and 1mM dimethyloxalyll glycine, DMOG (Alexis, Nottingham, UK)).
Western immunoblotting
Stabilisation of HIF-1α was assessed by Western blotting, following exposure of PASMC to normoxia or hypoxia. Cells were plated in 60 mm dishes and grown to 90% confluence. They were quiesced as in the proliferation assay and then exposed to experimental conditions in 0.1% FBS/DMEM for 24 hours. PASMC were lysed and extracts were boiled in a 1:5 ratio with 5× protein loading buffer for 5 minutes. Samples were loaded onto a 12% gel (HIF-1α) and separated by electrophoresis for one to two hours. The gels were transferred to a nitrocellulose membrane, incubated with blocking buffer, then incubated overnight with a specific mouse antibody to HIF-1α (1:250) (BD Biosciences, CA, USA) at 4°C. Blots were then incubated with an appropriate horseradish-peroxidase-conjugated antibody in a blocking buffer for an hour at room temperature. Blots were developed using enhanced chemiluminesece reagent (Amersham Bioscience, Little Chalfont, UK). Loading controls were performed by incubating the membranes with a specific mouse antibody to HIF-1β (1:200; BD Biosciences, Calif., USA).
Apoptosis assays
A Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labelling (TUNEL) assay was performend to determine the degree of apoptosis. Quantitative polymerase chain reaction (qPCR) for BCl-2 and Bax were also performed. For both assays PASMC were plated into 60 mm dishes at a density of 250×103 cells per plate. Cells were quiesced for 48 hours in 0.1% FBS/DMEM and after the medium was replaced, the cells were treated for 24 hours. At the beginning of the experimental procedure, fresh hypoxic (3 kPa) or normoxic medium was added, either alone or with the addition of DMOG (100 μM) or staurosporine (50 nM) (Sigma-Aldrich).
For the TUNEL assay, following treatment, the cells were trypsinized and transferred to a poly-L-lysine coated slide at a density of 5,000 cells per slide using a Shandon Cytospin 3 centrifuge (Thermo Fisher Scientific, Mass., USA). The TUNEL assay was performed using the DeadEnd Fluorometric TUNEL System (Promega, Southampton, UK) as described in the manufacturer's instructions. Briefly, formaldehyde fixed cells were rinsed in phosphate-buffered saline (PBS) and permeabilized in 0.2% Triton X-100 solution (5 minutes). The slides were then rinsed twice in PBS (5 minutes, RT (room temperature)) and then immersed in equilibration buffer (5–10 minutes). Recombinant terminal deoxynucleotidyl transferase and nucleotide mix in an equilibration buffer was added to cover the slides and a plastic coverslip applied. Slides were incubated in a humid environment at 37°C for 30 minutes. The reaction was terminated by immersing the slides in 2× sodium chloride/sodium citrate (SSC) (15 minutes, RT). Slides were rinsed three times in PBS (5 minutes, RT). Slides were mounted in Vectashield+DAPI (Vector Laboratories, Calif., USA) to stain the nuclei and examined using a fluorescence microscope (Nikon Eclipse, TE300, Japan). The rate of apoptosis was defined as the proportion of positively labeled nuclei counted in three fields of view (×20 magnification). A positive control was performed by adding ~10 units/ml of DNAse 1 to fixed cells (10 minutes, RT).
For qPCR, DNase-digested total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems) as described in the manufacturer's instructions. qPCR reactions were prepared using the SYBR Green Jumpstart Taq Readymix (Sigma) with the relevant Bcl-2 and Bax sense and antisense primers (Quiagen) and 10 nM fluorescein (Invitrogen). Reactions were amplified on an iCycler (Bio-Rad) using the primers above. The relative expression of target mRNAs was normalized to HPRT (Quiagen) and B2M (Quiagen) using the DDCT method[9] and expressed as the fold-change relative to the control.
Immunohistochemistry
Lung sections were immunostained using antibodies to caspase 3 active (R&D Systems AF835, 2.5 μg/ml), Carbonic Anhydrase IX (Abcam 15086, 1 μg/ml), Proliferating Cell Nuclear Antigen (Novocastra XD943, 0.5 μg/ml), Desmin (Dako M0760, 2.3 μg/ml), Vimentin (Dako M0725, 3.6 μg/ ml) and CD31 (Abcam ab9498, 3.75 μg/ml). Sections were then incubated with a secondary biotinylated antibody, then with the streptavidin-HRP complex (ABC kit, Vector Laboratories). All methods used a chromogenic diaminobenzidine tetrahydrochloride end-point. All tinctoral stains (hematoxylin and eosin, elastic van Gieson (EVG), and picrosirius red) were performed according to standard protocols.[10]
Vascular scoring system
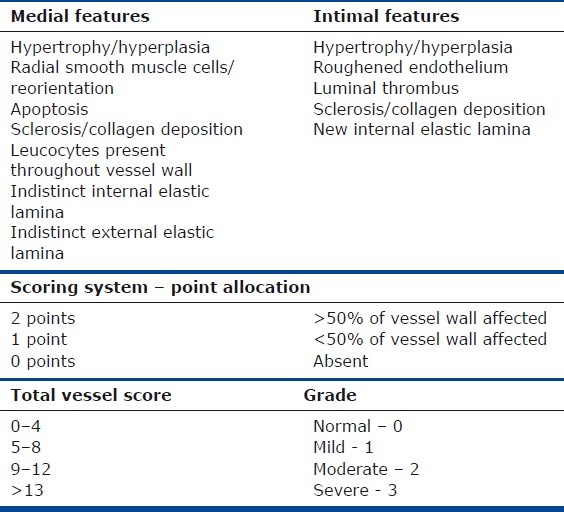
Remodeling of the vessel wall was quantified using Miller's elastic stain (EVG) and picrosirius red. Phenotyping of the cellular component of the vessel wall was studied using CD31 (endothelium), desmin (vascular smooth muscle) and vimentin (mesenchyme) immunohistochemistry. Sections from control/malignancy (n=9) and lung volume reduction surgery (LVRS) (n=10) patients were assessed using a cumulative score grading system (Table 1). All muscular pulmonary arterioles (n=52 control, n=53 emphysema) on every slide were assessed and graded.
Table 1.
Cumulative vessel remodeling grading score
Statistics
Data were presented as mean±standard error unless otherwise stated. Statistical analysis of proliferation and apoptosis assays was performed using a Student's t-test. Differences between the control and emphysema vascular remodeling scores were assessed using a Mann–Whitney test, and presented as median [range]. ANOVA was used to assess caspase 3, PCNA and CA-IX immunostaining in emphysema specimens. All statistical analysis was performed using SPSS v12. Significance was assumed to be at P<0.05.
RESULTS
The hypoxia-proliferative capacity of the PASMChyp subpopulation is preserved despite reoxygenation, indicating a robust phenotype
We have previously demonstrated the differential effects of hypoxia on the proliferation of PASMChyp and PASMCnorm using both [3H]-Thymidine and direct cell counting.[7] In this study, [3H]-Thymidine incorporation alone was used to confirm proliferation in response to hypoxia in PASMChyp (Fig. 1A). When PASMChyp were returned to normoxic culture conditions for 72 hours, quiesced in normoxia for 48 hours and then re-exposed to hypoxia, they maintained their hypoxia-proliferative phenotype (Fig. 1B).
Figure 1.
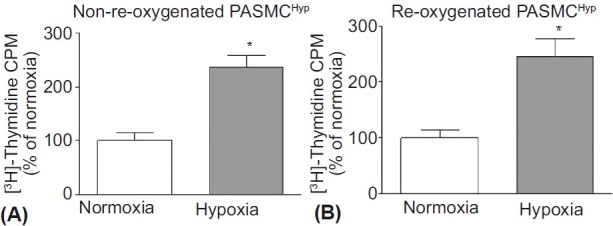
The effect of reoxygenation on the hypoxia-induced proliferative capacity of PASMChyp. DNA synthesis measured by [3H]-Thymidine incorporation (expressed as % of normoxia) is increased in hypoxia whether cells were maintained in hypoxia (A) or returned to normoxia (B) for 72 hours prior to the experiment. n=4; * P<0.05 vs.normoxia.
The PASMChyp and PASMCnorm phenotype can be differentiated by the response to pharmacological stabilisation of HIF
We chose to examine whether the differential response of these sub-populations of PASMC to hypoxia was HIF dependent. Dimethyloxallyl glycine (DMOG) stabilizes HIF- 1α by inhibiting its hydroxylation by prolyl-hydroxylases in the presence of oxygen, thereby preventing its targeting by pVHL for proteosomal degradation. Using Western blot analysis, the stabilization of HIF-1α was examined at different concentrations of DMOG (10 μM, 100 μM and 1 mM), with the effect of 100 μM appearing to match most closely the effect of hypoxia (Fig. 2A). The effects of DMOG on HIF-1α stabilization were similar in both PASMChyp and PASMCnorm (data not shown).
Figure 2.
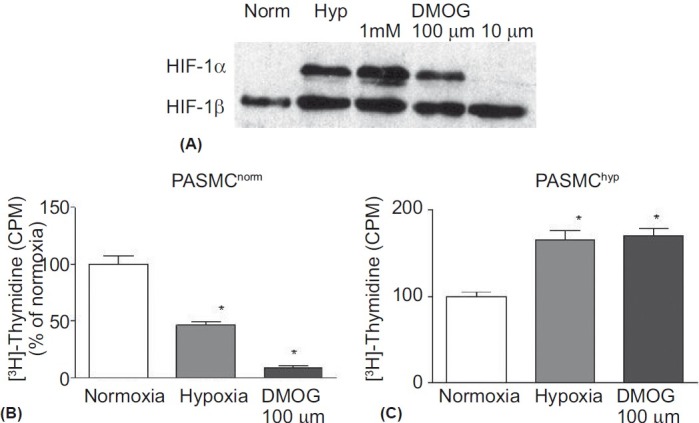
(A) Immunoblot of HIF-1α and HIF-1β (loading control) in PASMCnorm. Cells were treated for 4 hours in either hypoxic medium (Hyp) or normoxic medium (Norm) with or without DMOG at concentrations of 10 μM, 100 μM and 1 mM. Hypoxia and DMOG at the two higher concentrations stabilized HIF-1α and the DMOG dose of 100 μM was chosen as being closest to the physiological level of hypoxia. Both hypoxia and DMOG (100 μM) inhibited [3H]-Thymidine incorporation in PASMCnorm (B) and increased [3H]-Thymidine incorporation in PASMChyp (C). n=3; *P<0.05 vs. normoxia.
Using [3H]-Thymidine incorporation as a surrogate for proliferation, both hypoxia and DMOG (100 μM) had similar effects in PASMChyp (proliferation) and PASMCnorm (inhibition), suggesting that the differential effects of hypoxia on these two subpopulations was HIF-dependent (Fig. 2B and C). At the higher concentration of DMOG (1 mM), there was an inhibitory effect in both populations, suggesting either that this higher concentration was toxic or reflected higher levels of HIF stabilization more akin to anoxia. Conditioned medium from PASMChyp exposed to hypoxia did not alter proliferation in PASMCnorm, suggesting no role for hypoxia-induced autocrine growth factor release (data not shown).
Hypoxia and DMOG induce apoptosis in the PASMCnorm subpopulation
Having demonstrated previously (by cell counting[7]) that the increase in [3H]-Thymidine incorporation seen in PASMChyp reflected cellular proliferation, we proceeded to investigate whether the inhibition of [3H]-Thymidine by hypoxia and DMOG in PASMCnorm occurred as a result of apoptosis. Apoptosis was examined using TUNEL in parallel with Bcl-2 and Bax mRNA expression (Fig. 3). Although the overall rates of apoptosis were low, both hypoxia and DMOG (100 μM) increased the rate of PASMCnorm apoptosis as assessed by TUNEL (Fig. 3A). Bcl-2 expression decreased significantly in response to DMOG, but showed only a nonsignificant trend to decrease in hypoxia (Fig. 3B). Bax expression increased in hypoxia and showed a nonsignificant trend to increase in response to DMOG (100 μM; Fig. 3C). In all assays cells treated with staurosporin (50 nM) showed elevated levels of apoptosis compared to control conditions (Fig. 3).
Figure 3.
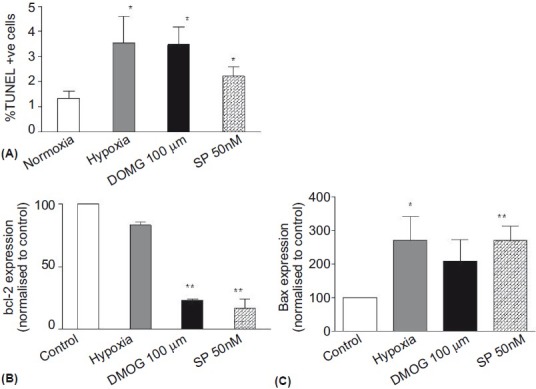
Apoptosis of PASMCnorm population assessed by TUNEL (A) and Bcl-2 and Bax (B and C). Hypoxia, DMOG (100 μM) and staurosporine (SP) (50 nM) induced apoptosis when assessed by TUNEL. Hypoxia and SP increased Bax expression and DMOG and SP decreased Bcl-2 expression. n=3; * P<0.05; ** P<0.01 compared with control.
Pulmonary arteries in emphysematous lung show increased remodeling and carbonic anhydrase IX expression
The distribution of vascular scores for control (n=9) and emphysema lungs (n=10) are shown in Figure 4. More extensive medial and intimal remodeling was seen in the pulmonary arteries in the emphysema lungs compared with controls (P<0.001). In the majority of emphysema specimens there was intimal hypertrophy and medial hyperplasia in the arteriolar walls. Reorientation of the matrix fibers within the media (to adopt a radial alignment) was observed in the LVRS samples, which resulted in PASMC invasion of the intima. These fibers stained positive for desmin and vimentin stained negative for CD31, suggesting a mesenchymal origin for these cells and matrix hyperplasia.
Figure 4.
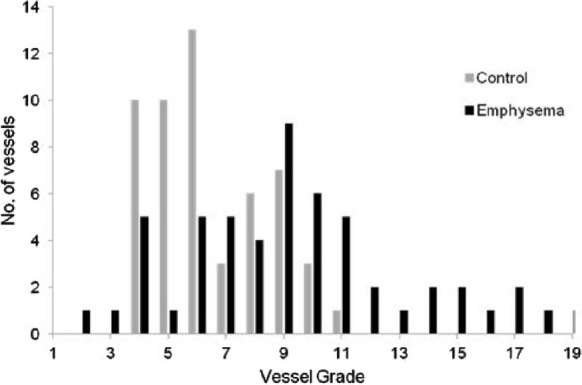
Distribution of remodeling scores in control (malignancy) and lung volume reduction surgery (LVRS) specimens. Median (range) scores were LVRS 4 (1-9) LVRS vs. control 3 (1-6) for intimal score (P=0.006); LVRS 5 (1-11) vs. control 3 (0-7) for medial score (P<0.001); and LVRS 9 (2-20) vs. 6 (4-11) for total score (P<0.001).
Apoptosis of medial cells, confirmed by active caspase 3, was widely distributed in arteriolar vessels in emphysema specimens, with the apoptotic cells nesting within areas of the vessel wall showing matrix hyperplasia (Fig. 5A and B). The amount of apoptosis as assessed by active caspase 3 and proliferation assessed by an increase in PCNA staining in vessels increased with higher remodeling scores (Fig. 5C, P<0.001). Furthermore, in the vessels with greatest remodeling there were higher levels of carbonic anhydrase expression, suggesting an association with HIF stabilization (Fig. 5D and E, P<0.001).
Figure 5.
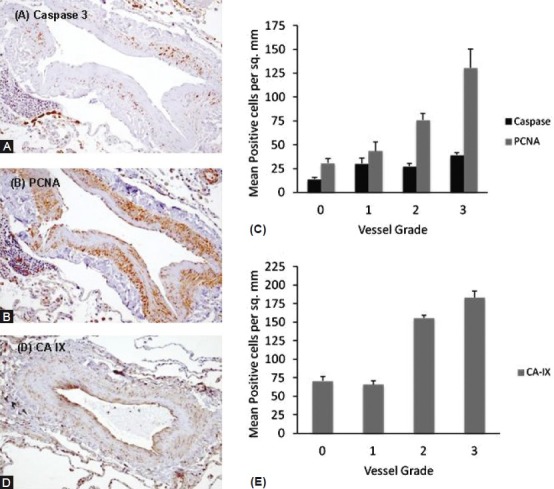
Representative immunohisto-chemistry of active Caspase 3 (A) and proliferating cell nuclear antigen (PCNA) (B) of muscular pulmonary arterioles taken from emphysema specimens. Active Caspase 3 and PCNA increased with vessel grade (C) (ANOVA, P<0.001). Representative immunohistochemistry of carbonic anhydrase IX (CA-IX) expression (D), which increased with vessel grade (E) (ANOVA, P<0.001).
DISCUSSION
We have demonstrated previously that two distinct smooth muscle cell subtypes can be derived from the distal pulmonary artery – one that grows well under standard normoxic culture conditions (PASMCnorm) and another where growth is selected for by hypoxia (~3 kPa).[7] The present study provides further characterization of these cell types and shows that the hypoxia-selected cells display a robust and enduring phenotype, maintaining their proliferative response to hypoxia even after freeze-thawing and a period of sustained reoxygenation. This response was HIF dependent as competitive inhibition of prolyl hydroxylases by dimethyloxallylglycine (DMOG) mimicked both growth inhibition in the PASMCnorm and growth stimulation in PASMChyp. We also show that hypoxia induces apoptosis in PASMCnorm via a HIF-dependent mechanism. Following this observation, we show the presence and colocalization of both apoptotic and proliferating smooth muscle cells in the pulmonary arteries of emphysematous lung and that the extent of HIF activation increases with worsening severity of pulmonary vascular remodeling. These results suggest that distinct smooth muscle cell populations present in the distal resistance pulmonary arteries may contribute differentially to vascular remodeling in hypoxic lung disease through apoptosis and proliferation and through HIF-transcribed pathways.
Hypoxic pulmonary vascular remodeling
Perhaps the commonest hypoxic lung disease that leads to pulmonary vascular remodeling is chronic obstructive pulmonary disease (COPD). In this condition there are clearly many other influences which may contribute to the vascular remodeling process, such as cigarette smoke and inflammation. Importantly, intimal thickening and an increase in the number of muscularized pulmonary arteries is found in patients with mild COPD and in healthy smokers even in the absence of airflow obstruction or systemic hypoxemia.[11–14] However, in patients with severe COPD,[15,16] the same histological changes are seen as those described in hypoxic animal models[4,5] and native highlanders, suggesting that alveolar hypoxia is a significant contributor to vascular remodeling.
Distinct smooth muscle cell phenotypes, with different proliferative and matrix-producing responses, have also been observed in the proximal bovine pulmonary artery.[17] These cells appear to respond differently during the development of hypoxia-induced pulmonary hypertension, with proliferation being restricted to cells that are negative for the cytoskeletal protein, metavinculin.[18] The neomuscularization seen in previously nonmuscularized arteries has been proposed to arise from the differentiation and proliferation of pericytes and intermediate cells or the recruitment of other cell types, either from the lung (interstitial fibroblasts) or the circulation (mesenchymal precursor cells).[6]
However, we and others have reported major functional differences between PASMCs isolated from the proximal and distal pulmonary circulation.[8,19] In addition, we have shown that two PASMC populations can be isolated from the distal pulmonary artery. One is selected for when freshly isolated cells are grown under “normal” culture conditions and undergo apoptosis on exposure to hypoxia; the other are cells isolated by a process of hypoxic selection when PASMCs are plated at early passage at low density and grown under hypoxic conditions for one to two weeks.[7] These cells subsequently proliferate in hypoxia. It is possible that sparse cells may undergo a functional switch to proliferate more readily under hypoxic conditions, but we show that this phenotype is maintained after freeze/thawing and reoxygenation, making this appear less likely.
Studies of the hypoxic neonatal calf have shown a mixed phenotype of cells in the distal pulmonary circulation.[20] Scattered “myofibroblast” cells expressing lower levels of α-SM-actin, as well as other cells expressing haematopoeitic (CD45), leukocytic/monocytic (CD11b, CD14) and stem-cell (cKit) markers were seen within the pulmonary artery media, and when cells were isolated from the distal pulmonary artery, two populations appeared. The first population was more slow-growing and was phenotypically characteristic of the SMC isolated from normotensive calves. The second appeared later in culture, but was subsequently much faster-growing, had a rhomboidal shape and expressed progenitor cell markers (CD34, CD73 and cKit) and was phenotypically and functionally distinct when compared with fibroblasts. These cells appeared to be mesenchymal in origin, constitutively expressing type I procollagen and with time in culture expressed the myofibroblast marker, α-SM-actin. The proliferation of the SMC population was inhibited by hypoxia as in our study, whereas the rhomboidal population was markedly stimulated by hypoxia.
While some similarities exist between Frid et al.[20] and our own study, there are significant differences in methodology. Our cells were obtained from macroscopically normal lung and the two sub-populations were isolated under normoxic and hypoxic conditions, whereas the cells in the study by Frid et al.[20] were isolated from pulmonary hypertensive calves and both populations were derived under normoxia. Both our cell populations had similar morphology unlike the populations derived by Frid et al.[20] which appeared to be progenitor like in origin. Nonetheless, both studies have led to the hypothesis that alveolar hypoxia may lead to a selective expansion of a subpopulation of cells, which in vitro exhibit increased proliferation under hypoxic conditions and that these cells may contribute to the remodeling of small peripheral pulmonary arteries. It is perhaps more important to note in our study that hypoxia also induced apoptosis in the normally resident PASMCnorm population, which further supports the hypothesis that these cells do not seem likely to contribute to remodeling.
The role of hypoxia-inducible factors
The data from the present study suggest that hypoxia, acting via a hypoxia-inducible factor (HIF)-dependent pathway, may be important in pulmonary vascular remodeling. HIF consists of α and β subunits. Under conditions of normoxia, the α subunit is hydroxylated by prolyl hydroxylases and subsequently targeted by von Hippel Lindau protein (pVHL) for proteosomal degradation. In hypoxia, HIF translocates to the nucleus as a heterodimer with the β subunit where it acts as a master regulator of the transcriptional response to hypoxia. The α subunit has three known isoforms – 1, 2 and 3.
HIF-1α ± mice display less muscularization of the distal pulmonary arteries on exposure to chronic hypoxia (three weeks at 10% O2) as well as significantly less right ventricular hypertrophy compared with wild-type littermates.[21] This has also been replicated in HIF-2α +/- mice in association with decreased circulating endothelin-1 (ET-1) and catecholamine levels.[22] Furthermore, an activating HIF-2α mutation has been associated with autosomal dominant erythrocytosis and pulmonary arterial hypertension in man.[23] Finally, mice homozygous for a mutation in the pVHL gene, mimicking the human form of Chuvash polycythaemia, developed spontaneous pulmonary hypertension and vascular remodeling, which was suppressed by HIF-2α, but not HIF-1α, heterozygosity.[24] A role for HIF-3α in pulmonary vascular cells has not yet been established.
We have shown that the downstream response to HIF stabilization in hypoxia can differentiate two phenotypically distinct populations of smooth muscle cell. In one population, HIF stabilization results in apoptosis and in the other, proliferation. Both processes were seen in areas of lung affected by emphysema, therefore likely to be hypoxic, and both apoptosis and proliferation increased in prevalence with worsening remodeling. Furthermore, evidence of HIF stabilization, quantified by carbonic anhydrase IX expression, was greater in the more severely remodeled vessels, suggesting an association between HIF-dependent transcription, proliferation/apoptosis and remodeling in vivo. Expression profiling of laser-microdissected murine intrapulmonary arteries has underlined the importance of HIF in hypoxic pulmonary vascular remodeling by demonstrating that 15 out of 21 upregulated genes after 24 hours of hypoxia carried potential hypoxia response elements.[25]
Although hypoxia may well be the dominant stimulus for HIF-dependent cellular responses, other stimuli may also be involved, both in vitro and in vivo. HIF has been implicated in nonhypoxic pulmonary arterial hypertension with immunohistological evidence of HIF-1α and β and VEGF/VEGFR2 (regulated by HIF) overexpression in plexiform lesions of patients with idiopathic pulmonary arterial hypertension.[26] In COPD, pulmonary vascular remodeling can be identified in patients with mild lung disease, suggesting a direct vascular effect from cigarette smoking or inflammation, independent from hypoxia.[1] Indeed, studies in proximal PASMCs have shown that growth factors may induce higher levels of HIF-1α than hypoxia itself and that they act in synergism with hypoxia to increase HIF-1α.[27,28]
To our knowledge the apoptotic effect of hypoxia on distal PASMC has not been described before. HIF induction in both tumor and nontumor cell lines under conditions of anoxia has been shown to induce apoptosis via BNIP3, a member of the Bcl-2 family;[29] and bovine aortic smooth muscle cells also undergo apoptosis via a HIF-dependent mechanism.[30] Thus, there appears to be consensus that HIF is a key orchestrator of PASMC behavior, either controlled by hypoxia or growth factors, but that its effects are context-specific, both with regard to the cell type and the severity of hypoxia.
Although debate continues over the importance of alveolar hypoxia vs inflammation in driving vascular changes in chronic lung disease, patients with germline homozygosity for a hypomorphic VHL allele, which results in defective HIF degradation show an exaggerated acute hypoxic pulmonary vasoconstrictor response on a background of increased pulmonary vascular tone. Despite the fact that the precise relationship between hypoxic pulmonary vasoconstriction and chronic pulmonary vascular remodeling has not being established, this observation suggests an important role for hypoxia acting through HIF in pulmonary vascular homeostasis.[31]
CONCLUSION
We have demonstrated that the predominant smooth muscle cell present in the distal pulmonary artery media undergoes apoptosis when subjected to hypoxia, but that a more sparse resident cell population with similar morphology can be isolated from normal distal pulmonary artery media, which displays hypoxia-mediated proliferation. Both these processes are active in the media of vessels undergoing remodeling in hypoxic lung and appear to be under HIF-dependent control both in vitro and in vivo. HIF downregulation, induced either pharmacologically or through treatment with oxygen or growth factor inhibition, may be an important target for the treatment of pulmonary vascular remodeling in hypoxic lung diseases.
ACKNOWLEDGMENT
Dr. Luke Howard was supported by a grant (no. FS/04/057/17496) from the British Heart Foundation.
Footnotes
Source of Support: British Heart Foundation, Grant no. FS/04/057/17496
Conflict of Interest: None declared.
REFERENCES
- 1.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodeling in smokers and patients with mild COPD. Eur Respir J. 2002;19:632–8. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 2.Arias-Stella J, Saldana M. The Terminal Portion of the Pulmonary Arterial Tree in People Native to High Altitudes. Circulation. 1963;28:915–25. doi: 10.1161/01.cir.28.5.915. [DOI] [PubMed] [Google Scholar]
- 3.Heath D, Smith P, Rios Dalenz J, Williams D, Harris P. Small pulmonary arteries in some natives of La Paz, Bolivia. Thorax. 1981;36:599–604. doi: 10.1136/thx.36.8.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hislop A, Reid L. New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol. 1976;57:542–54. [PMC free article] [PubMed] [Google Scholar]
- 5.Meyrick B, Reid L. The effect of continued hypoxia on rat pulmonary arterial circulation.An ultrastructural study. Lab Invest. 1978;38:188–200. [PubMed] [Google Scholar]
- 6.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: Cellular and molecular mechanisms. Circ Res. 2006;99:675–91. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 7.Yang X, Sheares KK, Davie N, Upton PD, Taylor GW, Horsley J, et al. Hypoxic induction of cox-2 regulates proliferation of human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2002;27:688–96. doi: 10.1165/rcmb.2002-0067OC. [DOI] [PubMed] [Google Scholar]
- 8.Wharton J, Davie N, Upton PD, Yacoub MH, Polak JM, Morrell NW. Prostacyclin analogues differentially inhibit growth of distal and proximal human pulmonary artery smooth muscle cells. Circulation. 2000;102:3130–6. doi: 10.1161/01.cir.102.25.3130. [DOI] [PubMed] [Google Scholar]
- 9.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 10.Bancroft JD, Gamble M. 6th ed. Philadelphia: Churchill Livingstone; 2007. Theory and Practice of Histological Techniques. [Google Scholar]
- 11.Barbera JA, Riverola A, Roca J, Ramirez J, Wagner PD, Ros D, et al. Pulmonary vascular abnormalities and ventilation-perfusion relationships in mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994;149:423–9. doi: 10.1164/ajrccm.149.2.8306040. [DOI] [PubMed] [Google Scholar]
- 12.Hale KA, Niewoehner DE, Cosio MG. Morphologic changes in the muscular pulmonary arteries: Relationship to cigarette smoking, airway disease, and emphysema. Am Rev Respir Dis. 1980;122:273–8. doi: 10.1164/arrd.1980.122.2.273. [DOI] [PubMed] [Google Scholar]
- 13.Magee F, Wright JL, Wiggs BR, Pare PD, Hogg JC. Pulmonary vascular structure and function in chronic obstructive pulmonary disease. Thorax. 1988;43:183–9. doi: 10.1136/thx.43.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright JL, Lawson L, Pare PD, Hooper RO, Peretz DI, Nelems JM, et al. The structure and function of the pulmonary vasculature in mild chronic obstructive pulmonary disease. The effect of oxygen and exercise. Am Rev Respir Dis. 1983;128:702–7. doi: 10.1164/arrd.1983.128.4.702. [DOI] [PubMed] [Google Scholar]
- 15.Wright JL, Petty T, Thurlbeck WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trial. Lung. 1992;170:109–24. doi: 10.1007/BF00175982. [DOI] [PubMed] [Google Scholar]
- 16.Wilkinson M, Langhorne CA, Heath D, Barer GR, Howard P. A pathophysiological study of 10 cases of hypoxic cor pulmonale. Q J Med. 1988;66:65–85. [PubMed] [Google Scholar]
- 17.Frid MG, Moiseeva EP, Stenmark KR. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ Res. 1994;75:669–81. doi: 10.1161/01.res.75.4.669. [DOI] [PubMed] [Google Scholar]
- 18.Wohrley JD, Frid MG, Moiseeva EP, Orton EC, Belknap JK, Stenmark KR. Hypoxia selectively induces proliferation in a specific subpopulation of smooth muscle cells in the bovine neonatal pulmonary arterial media. J Clin Invest. 1995;96:273–81. doi: 10.1172/JCI118031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–63. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 20.Frid MG, Li M, Gnanasekharan M, Burke DL, Fragoso M, Strassheim D, et al. Sustained hypoxia leads to the emergence of cells with enhanced growth, migratory, and promitogenic potentials within the distal pulmonary artery wall. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1059–72. doi: 10.1152/ajplung.90611.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–6. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. 2003;111:1519–27. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112:919–21. doi: 10.1182/blood-2008-04-153718. [DOI] [PubMed] [Google Scholar]
- 24.Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest. 2010;120:827–39. doi: 10.1172/JCI36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwapiszewska G, Wilhelm J, Wolff S, Laumanns I, Koenig IR, Ziegler A, et al. Expression profiling of laser-microdissected intrapulmonary arteries in hypoxia-induced pulmonary hypertension. Respir Res. 2005;6:109. doi: 10.1186/1465-9921-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195:367–74. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 27.Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem. 2000;275:26765–71. doi: 10.1074/jbc.M003325200. [DOI] [PubMed] [Google Scholar]
- 28.Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible factor-1alpha promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H2528–34. doi: 10.1152/ajpheart.01077.2005. [DOI] [PubMed] [Google Scholar]
- 29.Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–76. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- 30.Gao W, Ferguson G, Connell P, Walshe T, Murphy R, Birney YA, et al. High glucose concentrations alter hypoxia-induced control of vascular smooth muscle cell growth via a HIF-1alpha-dependent pathway. J Mol Cell Cardiol. 2007;42:609–19. doi: 10.1016/j.yjmcc.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]