

Abstract
Congenital pulmonary valve stenosis has been associated with the development of massive pulmonary arterial (PA) dilatation. Over time, this dilatation may distort surrounding structures and lead to compression of the left main coronary artery (LMCA) or the left mainstem bronchus. In this report, we describe a patient with a history of chronic thromboembolic pulmonary hypertension (CTEPH) and congenital pulmonic stenosis with massive PA dilatation. He develops exertional chest pain, presenting an unusual differential diagnosis. Novel diagnostic testing was performed to help narrow the differential diagnosis, and the patient responded well to pulmonary vasodilator treatment for progressive pulmonary hypertension.
Keywords: massive pulmonary arterial dilatation, left main coronary artery, chronic thromboembolic pulmonary hypertension
Congenital pulmonary valve stenosis has been associated with the development of massive pulmonary arterial (PA) dilatation. Over time, this dilatation may distort surrounding structures and lead to compression of the left main coronary artery (LMCA) or the left mainstem bronchus.[1] Symptoms from such compression may mimic angina or asthma, respectively. In addition, significant pulmonic stenosis may lead to symptoms of right heart failure including dyspnea, lower extremity edema, and chest pain.
Patients with pulmonary valve stenosis do not typically develop severe pulmonary hypertension. In this report, we describe a patient with a history of chronic thromboembolic pulmonary hypertension (CTEPH) and congenital pulmonic stenosis with massive PA dilatation. The case presents an unusual differential diagnosis, and novel diagnostic testing was performed to help narrow the differential diagnosis.
CASE REPORT
A 55-year-old man with history of pulmonic stenosis (postvalvuloplasty in 2004) and chronic thromboembolic pulmonary hypertension due to antiphospholipid antibody syndrome, presented with two months of progressive chest pain and exertional dyspnea. A CT scan of the chest was remarkable for massive dilatation of the proximal PA (Fig. 1) but did not reveal evidence of acute pulmonary embolism. Our subsequent differential diagnosis included progressive pulmonic stenosis with right ventricular failure, progressive pulmonary hypertension with right ventricular failure, LMCA compression from massive PA dilatation, and left mainstem bronchus compression from PA dilatation. These potential diagnoses were evaluated through a series of diagnostic tests. First, a cardiac MRI revealed a bicuspid pulmonic valve with a peak gradient estimated at 18 mmHg and an impaired right ventricular ejection fraction (27%). Subsequently, a right heart catheterization revealed pulmonary arterial pressure of 93/38 (52), pulmonary capillary wedge pressure of 18 mmHg, right atrial pressure of 18, Qp:Qs = 1:1, and a gradient across the pulmonic valve of 15 mmHg without significant regurgitation. Next, as attention shifted to the potential mechanical complications caused by this massive PA dilatation, pulmonary function testing was performed but did not show obstructive lung disease as would be expected if left main bronchus narrowing contributed significantly to his symptoms. Last, to evaluate the LMCA, a CT angiogram of the coronary arteries was performed (Fig. 2). The CT angiogram showed effacement of the LMCA, but no compression or flow limitation. The most significant finding from these diagnostic tests was severe pulmonary hypertension with elevated pulmonary vascular resistance and without either a significant gradient across the pulmonic valve or physiologic evidence of significant mechanical compression of the surrounding structures. Therefore, we started the patient on bosentan (an endothelin receptor blocker) as his clot burden is distal and not amenable to thromboendarterectomy. Six weeks after the initiation of treatment, the patient had resolution of chest pain, improvement in functional class, a decrease in his BNP to 43 pg/ml, and a decrease in his right ventricular diastolic diameter on transthoracic echocardiogram from 6.2 cm to 5.2 cm. These improvements have sustained over the past year.
Figure 1.
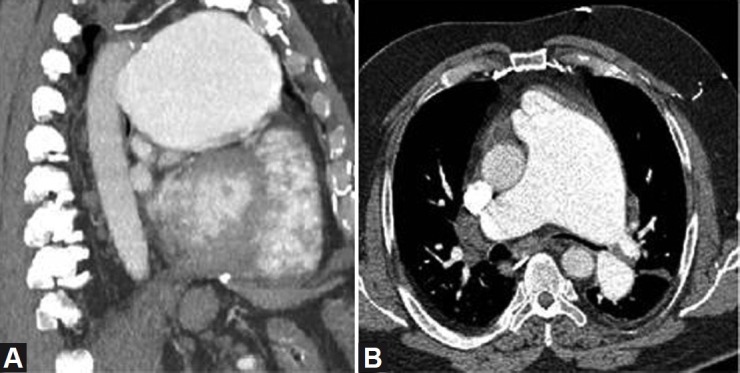
CT images showing massive dilatation of the proximal pulmonary artery on sagittal (A) and axial (B) images, with severe compression of the left mainstem bronchus.
Figure 2.
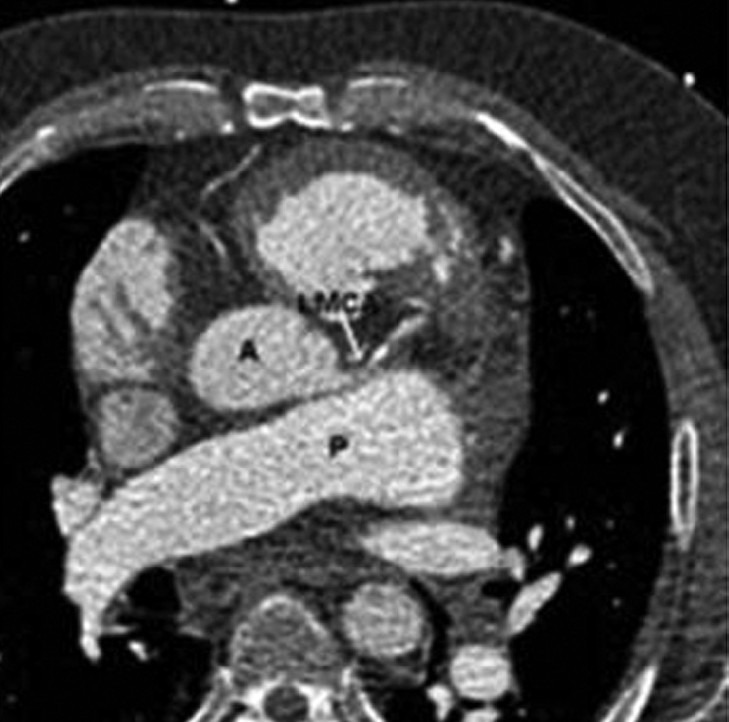
CT angiography of the coronary arteries shows the enlarged pulmonary artery (PA) effacing, but not compressing, the left main coronary artery (LMCA) as it leaves the aortic root (A).
DISCUSSION
Our case has several unique aspects. First, an isolated bicuspid pulmonic valve in the absence of other congenital defects is exceedingly rare.[2] While it is associated with maternal rubella infection,[3] our patient had no knowledge of any complication during his mother's pregnancy. As in patients with bicuspid aortic valves, a bicuspid pulmonic valve tends to calcify over time, leading to significant stenosis.[4] Because of the very low resistance and high compliance of the main pulmonary artery, the “jet” of blood created by pulmonic stenosis can lead to remarkable dilatation of the proximal pulmonary artery (compared with the proximal aorta in patients with aortic stenosis). In our patient, massive enlargement of the PA led to narrowing of the left mainstem bronchus as well as effacement of the LMCA. Because both the size of the proximal pulmonary artery and the extent of pulmonary hypertension are associated with the development of LMCA compression syndrome, and our patient has dramatic PA enlargement and pulmonary hypertension, we felt that he was at significant risk of LMCA compression syndrome.[5] He was reluctant to undergo left heart catheterization, having just had a right heart catheterization. Therefore, CT angiography of the coronary arteries was performed to evaluate for LMCA compression syndrome. This is a novel use of this diagnostic tool and helped us to narrow the patient's differential diagnosis. In conclusion, we present a patient with massive pulmonary artery dilatation and concurrent CTEPH, presenting a unique and difficult diagnostic challenge which led to the initiation of pulmonary vasodilator treatment and subsequent resolution of his presenting symptoms.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Hungate RG, Newman B, Meza MP. 51. Left mainstem bronchial narrowing: a vascular compression syndrome? Evaluation by magnetic resonance imaging. Pediatr Radiol. 1999;28:527–32. doi: 10.1007/s002470050404. [DOI] [PubMed] [Google Scholar]
- 2.Jashari R, Van Hoeck B, Goffin Y, Vanderkelen A. A The incidence of congenital bicuspid or bileaflet and quadricuspid or quadrileaflet arterial valves in 3,861 donor hearts in the European Homograft Bank. J Heart Valve Dis. 2009;18:337–44. [PubMed] [Google Scholar]
- 3.Stoermer J, Galal O, Arafa R, Rupprath G, Galal I, Neifer B. A rare combination: persistent ductus arteriosus and pulmonary stenoses.Is there a correlation with rubella embryopathy. Klin Padiatr. 1989;201:28–32. doi: 10.1055/s-2007-1025271. [DOI] [PubMed] [Google Scholar]
- 4.Magnoni M, Turri C, Roghi A, Merlanti B, Maseri A. An inverted location of the bicuspid valve disease: A variant of a variant. Circulation. 2011;124:e513–5. doi: 10.1161/CIRCULATIONAHA.111.055285. [DOI] [PubMed] [Google Scholar]
- 5.Mesquita SM, Castro CR, Ikari NM, Oliveira SA, Lopes AA. Likelihood of left main coronary artery compression basaed on pulm trunk diameter in patients with pulm hypertension. Am J Med. 2004;116:369–74. doi: 10.1016/j.amjmed.2003.11.015. [DOI] [PubMed] [Google Scholar]