

Abstract
Background
Advanced mast cell (MC) disorders are characterized by uncontrolled growth of neoplastic MC in various organs, mediator-related symptoms, and a poor prognosis. Kit mutations supposedly contribute to abnormal growth and drug resistance in these patients.
Methods
We established a novel canine mastocytoma cell line, NI-1, from a patient suffering from MC leukemia.
Results
NI-1 cells were found to form mastocytoma lesions in NOD/SCID IL-2Rgammanull mice and to harbor several homozygous Kit mutations, including missense mutations at nucleotides 107(C→T) and 1187(A→G), a 12-bp duplication (nucleotide 1263), and a 12-bp deletion (nucleotide 1550). NI-1 cells expressed several MC differentiation antigens, including tryptase, Kit, and a functional IgE receptor. Compared to the C2 mastocytoma cell line harboring a Kit exon 11 mutation, NI-1 cells were found to be less responsive against the Kit tyrosine kinase inhibitors (TKI) masitinib and imatinib, but were even more sensitive against proliferation-inhibitory effects of the mammalian target of rapamycin (mTOR) blocker RAD001 and PI3-kinase/mTOR blocker NVP-BEZ235. The Kit-targeting multikinase inhibitors PKC412 and dasatinib were also found to override TKI resistance in NI-1 cells, and produced growth inhibition with reasonable IC50 values (<0.1 μM).
Conclusion
NI-1 may serve as a useful tool to investigate IgE-dependent reactions and mechanisms of abnormal growth and drug resistance in neoplastic MC in advanced mastocytosis.
Keywords: exon 8 mutation, Kit, mastocytoma, resistance, tyrosine kinase inhibitors
Mastocytosis is a heterogeneous group of neoplasms characterized by abnormal growth and accumulation of mast cells (MC) in one or more organ systems (1, 2). Cutaneous and systemic variants of the disease have been described (1). Systemic mastocytosis (SM) can show an indolent or an aggressive clinical course (1, 2). Mast cell leukemia (MCL) is the leukemic variant of SM (1, 2). In typical cases, MC account for ≥20% of all nucleated cells in bone marrow smears and ≥10% of peripheral blood leukocytes (1). Mast cell leukemia and aggressive SM (ASM) are characterized by rapid disease progression and a poor prognosis (2). In MCL, MC are particularly immature and show rapid proliferation in various organs with consecutive organ damage (1). In all disease variants, patients with mastocytosis suffer from mediator-related symptoms, especially when IgE-dependent reactions occur (1, 2).
A number of different conventional drugs and chemotherapy agents have been suggested for the treatment of patients with advanced SM; especially, interferon-alpha (IFNα) and cladribine (2CdA) were found to induce responses in patients with smoldering SM or slowly progressing ASM (3–5). However, in rapidly progressing ASM or MCL, the response against these agents is usually short-lived (3–5).
A number of previous and more recent data suggest that Kit tyrosine kinase inhibitors (TKI) suppress the growth of neoplastic MC (6–8). However, depending on the variant of SM and molecular defects, neoplastic MC may be resistant against TKI. Likewise, the Kit mutation D816V that is found in most patients with SM confers resistance against imatinib (6, 9). Some of the second-generation TKI like dasatinib or PKC412 have been reported to override drug resistance in Kit D816V-transformed cells (6–8). These agents are currently tested in clinical trials in advanced SM (10–12).
Recent data suggest that transforming Kit mutations are also found in canine mastocytomas (8, 13–15). These mutations are detected in Kit exons 8, 9, 11, 12, or 17 (13–15). None of these mutations confer resistance against imatinib or masitinib (16, 17). Therefore, both drugs have been considered for the treatment of canine mastocytomas (16–20). More recently, masitinib has received approval for the treatment of malignant mastocytomas in canines. However, although clinical responses are seen, they are usually short-lived and may be followed by a relapse (19).
The mechanisms of resistance of canine mastocytoma cells against masitinib remain at present unknown. Among others, one possibility might be that more malignant subclones bear or develop additional Kit mutations that confer resistance. One approach to study the mechanism(s) of resistance to masitinib is to establish novel cell line models. We have established a novel canine mastocytoma cell line designated NI-1. This cell line harbors multiple Kit mutations and a functional IgE receptor (IgER) and was found to respond differentially to various TKI.
Materials and methods
Reagents
The TKI bosutinib, dasatinib, imatinib, sorafenib, sunitinib, and nilotinib, the PI3 kinase/mammalian target of rapamycin (mTOR) blocker NVP-BEZ235, everolimus, the ErbB receptor inhibitors lapatinib, erlotinib, and gefitinib, the Aurora kinase inhibitor tozasertib, and the histone deacetylase (HDAC) inhibitor vorinostat were purchased from ChemieTek (Indianapolis, IN, USA), and masitinib and midostaurin from LC Laboratories (Woburn, MA, USA) (Table 1). Stock solutions were prepared by dissolving in dimethylsulfoxide (Merck, Darmstadt, Germany). RPMI 1640 medium and fetal calf serum (FCS) were purchased from PAA Laboratories (Pasching, Austria), Iscove's modified Dulbecco′s medium (IMDM) from Gibco Life Technologies (Gaithersburg, MD, USA), and 3H-thymidine from PerkinElmer (Waltham, MA, USA). A specification of polyclonal and monoclonal antibodies (mAb) used in this study is shown in Table 2.
Table 1.
Specification of drugs used in this study
| Name | Synonym | Supplier | Main target(s) |
|---|---|---|---|
| Bosutinib | SKI-606 | ChemieTek | ABL, SRC |
| Dasatinib | BMS-354825 | ChemieTek | ABL, SRC, KIT, BTK, LYN |
| Erlotinib | OSI-774 | ChemieTek | EGFR, Her2 |
| Gefitinib | ZD1389 | ChemieTek | EGFR, Her2 |
| Imatinib | STI-571 | ChemieTek | ABL, KIT, PDGFR |
| Lapatinib | GW572016 | ChemieTek | EGFR, Her2 |
| Masitinib | AB1010 | LC Laboratories | KIT, PDGFR, LYN |
| Midostaurin | PKC412 | LC Laboratories | FLT3, KIT |
| Nilotinib | AMN107 | ChemieTek | ABL, KIT, PDGFR |
| Sorafenib | BAY 43-9006 | ChemieTek | Raf, VEGFR, KIT |
| Sunitinib | SU11248 | ChemieTek | PDGFR, VEGFR, KIT |
| Everolimus | RAD001 | ChemieTek | mTOR |
| NVP-BEZ235 | BEZ235 | ChemieTek | mTOR, PI3 kinase |
| Tozasertib | VX-680, MK-0457 | ChemieTek | Aurora Kinase A, KIT |
| Vorinostat | SAHA | ChemieTek | HDAC |
BTK, Bruton's tyrosine kinase; EGFR, epidermal growth factor receptor; PDGFR, platelet-derived growth factor receptor; VEGFR, vascular endothelial growth factor receptor; mTOR, mammalian target of rapamycin; PI3 kinase, phosphoinositide 3 kinase; HDAC, histone deacetylase.
Table 2.
Specification of antibodies used in this study
| Method | CD # | Name | Company | Clone | Isotype | Fluorochrome |
|---|---|---|---|---|---|---|
| FC | 2 | T11, LFA-2 | BD-Pharmingen | RPA-2.10 | mIgG1 | PE |
| FC | 9 | BA2 | Immunotech | ALB 6 | mIgG1 | FITC |
| FC | 25 | IL-2RA | BD-Pharmingen | 2A3 | mIgG1 | PE |
| FC | 30 | TNFRSF8 | BD-Pharmingen | BerH8 | mIgG1 | PE |
| FC | 31 | PECAM-1 | Miltenyi Biotec | AC128 | mIgG1 | APC |
| FC | 44 | CDW44 | BD-Pharmingen | 515 | mIgG1 | PE |
| FC | 54 | ICAM-1 | Immunotech | 84H10 | mIgG1 | FITC |
| FC | 58 | LFA3 | Immunotech | AICD58 | mIgG2a | FITC |
| FC | 62E | Selectin E | BD-Pharmingen | 68-5H11 | mIgG1 | PE |
| FC | 62L | Selectin L | BD-Pharmingen | DREG-56 | mIgG1 | PE |
| FC | 62P | Selectin P | BD-Pharmingen | AK-4 | mIgG1 | PE |
| FC | 63 | LAMP-3 | Immunotech | CLB-gran12 | mIgG1 | PE |
| FC | 117 | Kit | BD-Pharmingen | 104D2D1 | mIgG1 | PE |
| FC | 162 | Selectin P ligand | BD-Pharmingen | KPL-1 | mIgG1 | PE |
| FC | – | Isotype | BD-Pharmingen | MOPC-21 | mIgG1 | FITC |
| FC | – | Isotype | BD-Pharmingen | MOPC-21 | mIgG1 | PE |
| FC | – | Isotype | BD-Pharmingen | X39 | mIgG2a | FITC |
| ICC | 2 | T11, LFA-2 | Novocastra | AB75 | IgG1 | n.a. |
| ICC | 25 | IL-2RA | Novocastra | 4C9 | IgG2b | n.a. |
| ICC | 34 | – | Novocastra | QBEnd/10 | IgG1 | n.a. |
| ICC | 177 | Kit | Dako | Polyclonal | n.a. | n.a. |
| ICC | – | HDC | Progen | Polyclonal | n.a. | n.a. |
| ICC | – | Tryptase | Dako | AA1 | IgG1 | n.a. |
| ICC | – | Chymase | Chemicon | B7 | IgG1 | n.a. |
FC, flow cytometry; ICC, immunocytochemistry; FITC, fluorescein isothiocyanate; PE, phycoerythrin; APC, allophycocyanin; n.a., not applicable; HDC, histidine decarboxylase.
Establishment of the NI-1 cell line
Mast cells were obtained from the peripheral blood of a 3.5-year-old mixed breed dog. At diagnosis, the patient presented with weight loss, splenomegaly, and lymphadenopathy. Based on blood and bone marrow examinations, the diagnosis of MCL was established. Despite splenectomy, the subject died because of acute hemorrhage from esophagus ulceration. Isolated MC were cultured in RPMI 1640 medium containing 10% FCS and antibiotics at 5% CO2 and 37°C. MC were passaged serially for more than 1 year. Then, cells were single-cell-cloned by limiting dilution. One clone, designated NI-1, was characterized in detail.
Culture of C2 cells and HMC-1 cells
The canine mastocytoma cell line C2 (21) was kindly provided by Dr W. Gold (University of California, San Francisco, CA, USA). The human MCL line HMC-1 (22) was kindly provided by Dr J. H. Butterfield (Mayo Clinic, Rochester, MN, USA). Two subclones were used: HMC-1.1 harboring Kit V560G but not Kit D816V and HMC-1.2 harboring both mutants (6). C2 cells and HMC-1 cells were cultured in IMDM with 10% FCS and antibiotics at 5% CO2 and 37°C. Cells were passaged every 2–3 days and rethawed from an original stock every 6–8 weeks.
Morphology and phenotyping
The morphology of NI-1 cells was evaluated by Wright–Giemsa staining. Immunocytochemistry (ICC) was performed as described (23) using antibodies depicted in Table 2. Flow cytometry was performed according to an established protocol (23) using fluorochrome-labeled mAb (Table 2). IgE receptor expression was analyzed by staining with fluorescein isothiocyanate (FITC)-labeled anti-IgE antibody A40-125F after preloading cells with IgE (Bethyl Laboratories, Montgomery, TX, USA). Antibody reactivity was determined on a FACSCalibur [Becton Dickinson (BD) Biosciences, San Jose, CA, USA]. Electron microscopy was performed essentially as described (24). In brief, NI-1 cells were washed and fixed in OsO4 for 45 min. Then, cells were again washed and incubated with uranyl acetate (5%). After dehydration, cells were embedded in resin and propylene oxide. Embedded cells were cut on an EM UC7 ultramicrotome (Leica, Wetzlar, Germany), transferred to copper grids, and then exposed to uranyl acetate and lead citrate. Sections were photographed using a JEM-1010 transmission electron microscope (JEOL Ltd, Tokyo, Japan).
Sequencing of Kit
The original tumor samples as well as NI-1 cells (before and after xenografting) were examined for Kit mutations by sequencing analysis as described (14, 25). Three large fragments of the Kit cDNA product were amplified, gel-purified using the Qiaex II gel purification kit (Qiagen, Valencia, CA, USA), and sequenced through an automated sequencing technique using fluorescence-labeled dideoxynucleotides with capillary electrophoresis and an ABI sequence analyzer (Applied Biosystems, Foster City, CA, USA).
Western blot experiments
Western blot experiments were performed essentially as described (6, 16) using antibodies against total Kit (Santa Cruz, Santa Cruz, CA, USA) and phosphorylated Kit (Cell Signaling Technology, Danvers, MA, USA). NI-1 cells, HMC-1.2 cells, and cord blood–derived cultured normal MC, generated as reported (26, 27), were examined by Western blotting. Cell lysates were separated in 7.5% SDS polyacrylamide gel electrophoresis, and antibody reactivity was made visible by donkey anti-rabbit IgG and Lumingen PS-3 detection reagent (all from GE Healthcare, Buckinghamshire, UK).
Evaluation of effects of various TKI and other drugs on proliferation of MC
Cells were seeded in 96-well plates (104 cells/well) and incubated with various targeted drugs (37°C, 48 h). In a first screen, drugs were applied at 0.1, 0.5, 1.0, and 2.0 μM. Effective drugs were then examined using additional concentrations. After incubation, 0.5 μCi of 3H-thymidine was added, and thymidine uptake was measured as reported (6). All experiments were performed in triplicate.
Evaluation of drug-induced apoptosis in neoplastic MC
To quantify the expression of activated caspase-3 after drug exposure, flow cytometry was performed using an antibody against active caspase-3 (BD Biosciences). Before being stained, HMC-1 cells, C2 cells, and NI-1 cells were cultured in the presence or absence of targeted drugs (37°C; 24 or 48 h). Prior to staining, cells were fixed in formaldehyde (2%) and permeabilized using methanol (100%) at −20°C for 30 min. Expression of active caspase-3 was analyzed on a FACSCalibur (BD Biosciences). To confirm apoptosis in drug-exposed cells, a TUNEL assay was performed using the ‘In situ cell death detection kit-fluorescein’ (Roche, Mannheim, Germany) following the manufacturer's instructions.
Evaluation of IgE-dependent histamine release
Histamine release experiments were performed in NI-1 cells and C2 cells using goat anti-dog anti-IgE antibody A40-125A (0.1–10 μg/ml) (Bethyl Laboratories) or Ca-ionophore A23187 (10 μg/ml) (Sigma-Aldrich, St. Louis, MO). Before being challenged with anti-IgE (37°C, 30 min), cells (1 × 105/ml) were preloaded with dog IgE (Bethyl Laboratories) at 37°C for 2 h. In a separate set of experiments, IgE-preloaded NI-1 cells were incubated in control medium or medium containing masitinib (10 μM), imatinib (10 μM), or midostaurin (0.01–10 μM) at 37°C for 30 min before being exposed to anti-IgE (10 μg/ml) in histamine release buffer (Immunotech, Marseille, France). After incubation, cells were centrifuged at 4°C. Cell-free supernatants were examined for histamine content by radioimmunoassay (Immunotech) (27). Histamine release was expressed as percent of total histamine.
Transplantation of NI-1 cells into NOD/SCID IL-2Rgammanull (NSG) mice
Six-week-old male NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were purchased from Jackson Laboratory (Bar Habor, ME, USA). The study was approved by the ethics committee of the University of Veterinary Medicine Vienna and performed in accordance with guidelines for animal care and protection and protocols approved by the Austrian law (BMWF-66.009/0284-II/10b/2008). NI-1 cells or C2 cells (5 × 106 cells in 0.2 ml) were injected subcutaneously into NSG mice (n = 5). Mice were monitored daily until mice developed palpable tumors. After resection, tumor samples were either frozen in liquid nitrogen or fixed in 10% neutral buffered formalin and embedded in paraffin. In addition, tumor nodules were cut into small pieces and digested in collagenase type II (Worthington Biochemical Corp, Lakewood, NJ, USA) to obtain single-cell suspensions as reported (24, 26).
Statistical evaluation
To determine the significances in differences in proliferation and apoptosis in cells exposed to various drugs, the Student's t-test for dependent samples was applied. Results were considered statistically significant when P was <0.05.
Results
Morphologic and phenotypic characterization of NI-1 cells
In Wright–Giemsa-stained slides, NI-1 cells were found to be immature myeloid cells containing a round nucleus with fine chromatin and one or more nucleoli. The cytoplasm showed numerous vacuoles and a few metachromatic granules (Figure 1A). Electron microscopy confirmed that NI-1 cells are MC progenitors (Figure 1B). As assessed by ICC, NI-1 cells expressed tryptase (Figure 1C, Table 3) and Kit (CD117) (Table 3) as well as histidine decarboxylase (HDC), but did not express CD34 (Table 3). Flow cytometry experiments showed that NI-1 cells also express CD30 (Figure 1D). Expression of Kit was demonstrable in NI-1 cells by ICC (Table 3), flow cytometry (Figure 1E), and Western blotting (Figure 1G). In addition, NI-1 cells were found to express the IgER (Figure 1F). C2 cells were found to display a similar phenotype (Table 3). However, in contrast to NI-1, C2 cells expressed only low levels of IgER and CD30 (Table 3). As determined by radioimmunoassay (RIA), NI-1 cells contained 0.12 ± 0.06 pg histamine per cell, C2 cells 0.3 ± 0.17 pg per cell, HMC-1.1 cells 0.76 ± 0.46 pg per cell, and HMC-1.2 cells 0.4 ± 0.08 pg per cell (Table 3).
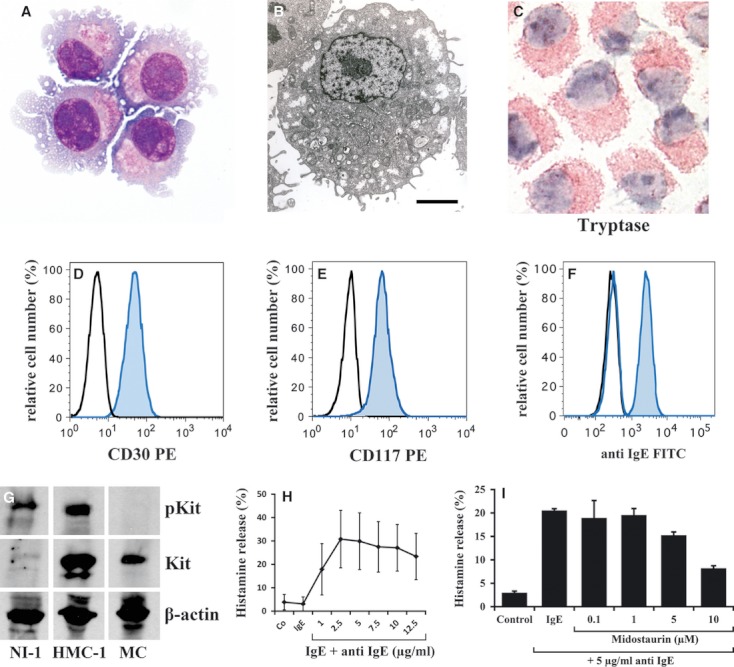
Phenotypic and functional characteristics of NI-1 cells. (A) NI-1 cells were stained by Wright–Giemsa method. As visible, NI-1 cells contained round nuclei with one or more nucleoli, numerous cytoplasmic vacuoles, and several cytoplasmic surface projections. (B) The ultrastructure of NI-1 cells was analyzed by electron microscopy using a JM-1010 transmission electron microscope as described in the text. Electron microscopy confirmed the presence of a mast cell (MC) progenitor, with numerous cytoplasmic surface projections, granule-like structures, and empty containers. Original magnification: ×2000, size bar represents 2 μm. (C) NI-1 cells were stained with an antibody against tryptase and analyzed by indirect immunocytochemistry as described in the text. (D, E) Expression of CD30 and CD117 on NI-1 cells was confirmed by flow cytometry using mAb against CD30 and CD117 (blue histograms). The isotype-matched control antibodies are also shown (black histograms). (F) NI-1 cells were pre-incubated with canine IgE at 4°C for 2 h, then with an FITC-labeled anti-canine anti-IgE antibody (blue histogram) for 30 min. The expression was analyzed by flow cytometry. The black histogram shows unstained NI-1 cells, and the blue open histogram represents NI-1 cells stained with the FITC-labeled anti-IgE antibody without IgE pre-incubation. Antibody reactivity was also controlled by an isotype-matched control antibody (not shown). (G) Western blot analysis of lysates of NI-1 cells, HMC-1.2 cells, and normal cord blood–derived cultured human MCs starved from SCF overnight. Western blotting was performed using an antibody against pKit and an antibody against total Kit. (H) NI-1 cells were preloaded with canine IgE and then incubated with various concentrations (as indicated) of anti-canine anti-IgE antibody at 37°C for 30 min as indicated. Thereafter, NI-1 cells were centrifuged at 4°C, and the supernatants and total suspensions were analyzed for histamine content by RIA. Histamine release results are expressed as percent of total histamine and represent the mean ± SD from four independent experiments. (I) NI-1 cells were pre-incubated with IgE for 2 h and then were washed and incubated with control medium or various concentrations of midostaurin (0.1–10 μM) at 37°C for 30 min, followed by challenge with anti-IgE antibody A40-125 (5 μg/ml) for another 30 min. Then, cells were centrifuged, and histamine content was determined in cell lysates and cell-free supernatants. Histamine release is expressed as percentage of total histamine and represents the mean ± SD of triplicates.
Table 3.
Characteristics of cell lines
| NI-1 | C2 | HMC-1.1 | HMC-1.2 | NI-1 xeno-TX* | |
|---|---|---|---|---|---|
| Diameter, size (μM) | 11.2 (±0.7) | 14.3 (±0.2) | 17.7 (±0.7) | 14.5 (±0.4) | 12 (±0.6) |
| Histamine (pg/cell) | 0.12 (±0.06) | 0.3 (±0.17) | 0.76 (±0.46) | 0.4 (±0.08) | n.d. |
| Kit mutation status | 2 SiM (Exon 8) | 48-bp ITD (Exon 11) | V560G (Exon 11) | V560G (Exon 11) | 2 SiM (Exon 8) |
| MiM (Exon 8) | D816V (Exon 17) | 2 MiM (Exon 8) | |||
| 12-bp dup (Exon 8) | 12-bp del (Exon 10) | ||||
| Immunocytochemistry | NI-1 | C2 | HMC-1.1 | HMC-1.2 | NI-1 xeno-TX* |
| CD2 | (+) | (+) | (+) | (+) | (+) |
| CD25 | + | + | + | + | n.t. |
| CD34 | − | − | − | − | − |
| CD117/Kit | +++ | +++ | +++ | +++ | +++ |
| HDC | + | + | ++ | + | n.t. |
| Chymase | − | − | − | − | − |
| Tryptase | + | ++ | +++ | +++ | + |
| Flow cytometry | NI-1 | C2 | HMC-1.1 | HMC-1.2 | NI-1 xeno-TX* |
| CD2 | − | − | − | +++ | − |
| CD9 | +/− | − | +++ | +++ | − |
| CD25 | − | − | − | + | − |
| CD30 | +++ | ++ | n.t. | n.t. | n.t. |
| CD44 | +++ | +++ | +++ | +++ | +++ |
| CD54 | + | − | + | +++ | + |
| CD58 | − | +/− | ++ | +++ | − |
| CD62E | + | − | + | + | + |
| CD62L | − | − | − | − | − |
| CD62P | − | − | − | − | − |
| CD63 | − | − | + | +++ | − |
| CD117/Kit | +++ | +++ | +++ | +++ | +++ |
| CD162 | − | − | +++ | +++ | − |
SiM, single point mutation; HDC, histidine decarboxylase; MiM, missense mutation; bp, base pair; dup, duplication; del, deletion; ITD, internal tandem duplication; n.t., not tested; 0–10%: −; 11–20%: +/−; 21–45%: +; 46–70%: ++; 71–100%: +++.
The histamine content in cell lines was determined by RIA. Immunocytochemistry and flow cytometry were performed using anti-leukocyte antibodies as described in the text.
NI-1 xenoTX: NI-1 cells obtained from mast cell tumors developing in NSG mice after xenotransplantation.
NI-1 cells express a functional IgER
As visible in Figure 1H, incubation of IgE-preloaded NI-1 cells with anti-IgE resulted in a dose-dependent release of histamine, suggesting that NI-1 cells express a functional IgER. The Ca-ionophore A23187 also induced histamine liberation in NI-1 cells (data not shown).
NI-1 cells form tumor lesions in NSG mice
To study the tumorigenicity of NI-1 cells, we injected these cells into the skin of NSG mice. Within 3 weeks, NSG mice were found to develop tumor nodules containing neoplastic MC. After resection and isolation, tumor-derived MC could again be cultured and were found to exhibit the same morphology, phenotype, and Kit mutations, compared to the original NI-1 clone (Table 3).
NI-1 cells exhibit a unique profile of Kit mutations
As determined by Western blotting, NI-1 cells contained a constitutively active Kit receptor (Figure 1G). When examining the Kit mutation status, we found that NI-1 cells contain two silent mutations at nucleotide 414 and 507, and two missense mutation sites at nucleotide 107 (C to T) [amino acid 36 (P to L)] as well as at nucleotide 1187 (A to G) [amino acid 396 (Q to R)]. Furthermore, we found a 12-bp duplication mutation at nucleotide 1263 (repeat of AATCCTGACTCA) [amino acid 421–425 (insertion of amino acid QILT)] and a 12-bp shorter isoform of Kit at nucleotide 1550 (deletion of GTAACAGCAAG) [amino acid 517–520 (deletion of amino acid GNSK)]. The primary tumor lesion contained the same Kit mutations as that found in NI-1 cells. C2 cells were also examined for Kit mutations to confirm their identity. As expected, we were able to detect the 48-bp Kit internal tandem duplication (ITD) reported to be expressed in C2 cells (28).
Effects of various antineoplastic drugs on proliferation of NI-1 cells
As assessed by 3H-thymidine uptake, a number of antineoplastic drugs were found to inhibit the proliferation of NI-1 cells (Figure 2A, Table 4). Among these drugs were the Kit kinase blockers midostaurin, dasatinib, sunitinib, tozasertib, imatinib, and sorafenib as well as drugs targeting Kit-downstream signaling molecules, including the mTOR blocker RAD001 and the PI3-kinase/mTOR-targeting drug NVP-BEZ235 (IC50 0.01–0.5 μM). No major growth-inhibitory effects were seen with bosutinib, erlotinib, gefitinib, masitinib, and lapatinib (IC50 > 2 μM) (Table 4). However, the HDAC inhibitor vorinostat was found to block the growth of NI-1 cells (0.1–0.5 μM) (Table 4). The effects of the targeted drugs on caspase 3 activation, and thus their apoptosis-inducing effects on NI-1 cells and other MC lines, are shown in Table 5.
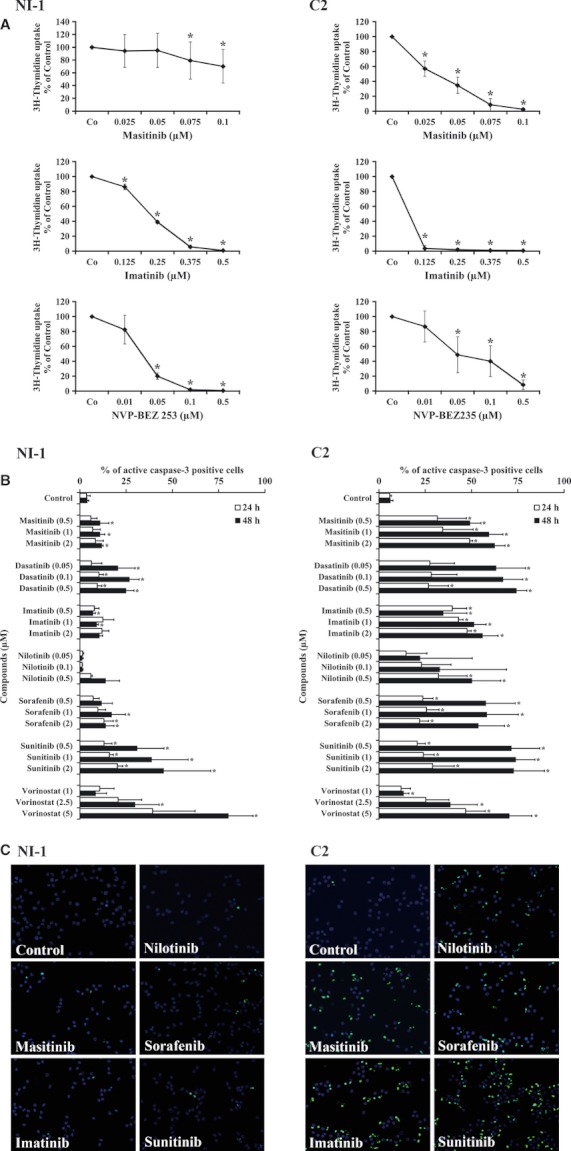
Effects of various drugs on proliferation and apoptosis of NI-1 cells and C2 cells. (A) NI-1 cells (left panels) and C2 cells (right panels) were incubated in control medium or with various concentrations of targeted drugs (masitinib, imatinib, NVP-BEZ235, as indicated) at 37°C for 48 h. Then, 3H-thymidine uptake was measured as described in the text. Results show the percent of control and represent the mean ± SD of at least three independent experiments. Asterisk: P < 0.05. (B) NI-1 cells and C2 cells were incubated in control medium or in medium containing various concentrations of targeted drugs (as indicated) at 37°C for 24 h (open bars) or 48 h (black bars). Then, cells were recovered, and the expression of activated caspase-3 was assessed by flow cytometry. Results show the percentage of active caspase-3-positive cells and represent the mean ± SD of three independent experiments. Asterisk: P < 0.05. (C) TUNEL assay experiments using NI-1 cells, C2 cells, and various targeted drugs. Cells were incubated in control medium or in medium containing masitinib (2 μM), imatinib (2 μM), nilotinib (2 μM), sorafenib (2 μM), and sunitinib (2 μM) as indicated (37°C, 24 h). The TUNEL assay was performed as described in the text. As visible, the targeted drugs applied induced apoptosis in C2 cells but not in NI-1 cells, confirming results obtained by flow cytometry with an antibody against active caspase-3.
Table 4.
Effects of various drugs on proliferation of mast cell lines
| IC50 (μM) | ||||
|---|---|---|---|---|
| NI-1 | C2 | HMC-1.1 | HMC-1.2 | |
| Bosutinib | >2 | >2 | >2 | 0.5–1 |
| Dasatinib | <0.003 | 0.003–0.006 | 0.0015–0.003 | 1–2 |
| Erlotinib | >2 | >2 | >2 | >2 |
| Everolimus | <0.1 | >2 | >2 | >2 |
| Gefitinib | >2 | >2 | >2 | >2 |
| Imatinib | 0.125–0.25 | 0.025–0.05 | 0.01–0.025 | >2 |
| Lapatinib | >2 | >2 | >2 | >2 |
| Masitinib | 0.1–0.2 | 0.025–0.05 | 0.005–0.01 | >2 |
| Midostaurin | <0.1 | <0.1 | 0.1–0.25 | 0.1–0.25 |
| Nilotinib | 0.2–0.3 | 0.1–0.2 | 0.025–0.05 | 1–2 |
| NVP-BEZ235 | 0.01–0.05 | 0.01–0.05 | 0.05–0.075 | 0.01–0.05 |
| Sunitinib | 0.006–0.013 | 0.006–0.013 | 0.003–0.006 | >2 |
| Sorafenib | 0.1–0.25 | 0.005–0.1 | 0.01–0.015 | >2 |
| Tozasertib | <0.1 | >2 | >2 | <0.1 |
| Vorinostat | 0.1–0.5 | 0.1–0.5 | 0.5–1 | 0.5–1 |
Cells were incubated with various concentrations of targeted drugs at 37°C for 48 h. Then, 3H-thymidine uptake was measured. The mean IC50 values from at least three independent experiments are shown.
Table 5.
Effects of targeted drugs on caspase 3 activation in neoplastic mast cells
| ED50 (μM) | ||||
|---|---|---|---|---|
| NI-1 | C2 | HMC-1.1 | HMC-1.2 | |
| Bosutinib | >5 | >5 | >5 | >5 |
| Dasatinib | >0.5 | <0.05 | <0.05 | >2 |
| Erlotinib | >5 | >5 | >5 | >5 |
| Everolimus | >5 | >5 | >5 | >5 |
| Gefitinib | >2 | >2 | >2 | >2 |
| Imatinib | >2 | 0.5–1 | 0.01–0.05 | >2 |
| Lapatinib | >2 | >2 | >2 | >2 |
| Masitinib | >2 | 0.5–1 | 0.05–0.1 | >5 |
| Midostaurin | >1 | 0.5–1 | 0.25–0.5 | 0.5–1 |
| Nilotinib | >0.5 | 0.1–0.5 | <0.05 | >2 |
| NVP-BEZ235 | >5 | >5 | 0.05–0.1 | >5 |
| Sunitinib | >2 | <0.5 | <0.05 | >5 |
| Sorafenib | >2 | <0.5 | 0.05–0.1 | >5 |
| Tozasertib | >2 | >2 | 2.5–5 | >5 |
| Vorinostat | 2.5–5 | 2.5–5 | 2.5–5 | >5 |
Cells were incubated with various concentrations of targeted drugs at 37°C for 48 h. Then, the percentage of caspase-3+ cells was determined by flow cytometry. The mean ED50 values from three independent experiments are shown.
Effects of targeted drugs on other MC lines and comparison to NI-1 cells
C2 cells were found to respond clearly better to certain Kit kinase inhibitors (imatinib, masitinib) than NI-1 cells (Figure 2A). However, other kinase inhibitors including the PI3-kinase blocker NVP-BEZ235 and the mTOR blocker everolimus showed an even more potent antiproliferative effect on NI-1 cells compared to effects produced in C2 cells (Figure 2A, Table 4). We also compared drug responses in HMC-1 cells. As expected, HMC-1.1 cells were found to be more sensitive against several Kit TKI including imatinib, nilotinib, and masitinib than HMC-1.2 cells (Table 4). In addition, we found that sorafenib and sunitinib are potent inhibitors of growth of HMC-1.1 cells, whereas these TKI did not inhibit the growth of HMC-1.2 cells at pharmacologically meaningful drug concentrations (Table 4). All in all, the drug-response profile of NI-1 resembles more that of HMC-1.2 cells than that of HMC-1.1 cells, whereas the resistance profile of C2 resembles that of HMC-1.1 cells.
Effects of various antineoplastic drugs on apoptosis in NI-1 cells and C2 cells
As assessed by flow cytometry, only sunitinib and vorinostat produced substantial caspase 3 cleavage in NI-1 cells (Figure 2B, Table 5). Less pronounced effects on caspase cleavage were seen with bosutinib and dasatinib. The other drugs tested, including masitinib (up to 2 μM) and imatinib (up to 2 μM) (Figure 2B, Table 5) as well as NVP-BEZ235 (up to 5 μM) and everolimus (up to 5 μM) (Table 5), did not induce apoptosis in these cells. By contrast, in C2 cells, various targeted drugs including masitinib, imatinib, nilotinib, dasatinib, sunitinib, sorafenib, and vorinostat were found to induce caspase cleavage at relatively low concentrations (Figure 2B, Tables 4 and 5). Apoptosis-inducing drug effects on NI-1 and C2 cells were confirmed by TUNEL assay (Figure 2C). We also compared responses to targeted drugs in HMC-1.1 and HMC-1.2 cells. Confirming previous data, HMC-1.1 cells were found to respond better to imatinib, nilotinib, dasatinib, and masitinib compared to HMC-1.2 cells (Table 5). In addition, we found that HMC-1.1 cells are more sensitive against sorafenib, tozasertib, and sunitinib compared to HMC-1.2 cells (Table 5). The other drugs tested showed comparable apoptosis-inducing effects on HMC-1.1 and HMC-1.2 cells (Table 5).
Effects of various TKI on IgE-dependent histamine release in NI-1 cells
Patients with mastocytosis often suffer IgE-dependent mediator-related symptoms. We have recently shown that midostaurin blocks IgE-dependent histamine release in human MC. In the present study, midostaurin was found to inhibit IgE-mediated secretion of histamine in NI-1 cells in a dose-dependent manner (Figure 1I), whereas no effects were seen with masitinib or imatinib (not shown).
Discussion
Mastocytomas are among the most frequent life-threatening neoplasms in dogs (29). During the past few years, Kit has been identified as a major molecular target in neoplastic MC in humans and canines (6–12). In both species, Kit-targeting TKI have been used to counteract the growth of neoplastic MC (6–12, 16–18). However, resistances against TKI have been described (10, 12, 18). Although the exact mechanisms remain unknown, several observations suggest that mutations in Kit and other genes may be involved. To better define the mechanisms of drug resistance in canine MC disorders, it seems important to create novel robust cell line models. We have established a novel canine MC line, NI-1, which harbors several Kit mutations and exhibits relative resistance against masitinib and other Kit-targeting TKI.
NI-1 cells were generated from a canine patient suffering from MCL. The origin of NI-1 from a MC progenitor was confirmed by morphologic studies, histamine content, electron microscopy, and immunophenotyping. An intriguing observation was that NI-1 cells express CD30, a marker antigen that is preferentially expressed in advanced mastocytosis (30). In line with this observation, NI-1 cells were found to form mastocytoma lesions in immunodeficient mice. Compared to C2 cells, NI-1 cells appeared to be a more immature and more rapidly growing tumor cell line in vitro and in vivo, which may be explained by the fact that NI-1 cells were generated from an extremely immature MC tumor.
Only a few well-characterized canine mastocytoma cell lines have been described so far (20, 31–33). In some of these cell lines, Kit-activating mutations have been identified. C2 cells express a Kit-activating ITD in exon 11 (29). However, this mutation does not confer resistance against imatinib or masitinib (16, 17). In NI-1 cells, several other Kit mutations, including missense mutations at nucleotides 107(C→T) and 1187(A→G), a 12-bp deletion at nucleotide 1550, and a 12-bp duplication at nucleotide 1263, were detectable. The latter mutation has been described to be a Kit-activating defect (15). Correspondingly, NI-1 cells were found to express a constitutively activated (phosphorylated) Kit receptor. Whether the 12-bp deletion at nucleotide 1550 causes resistance against various TKI such as masitinib and imatinib remains unknown.
Resistance against masitinib and other similar TKI is an important issues in the management and therapy of advanced canine MC tumors (13–15, 18–20). In fact, these patients have a poor prognosis. We asked whether our new cell line model can be employed to screen for targeted drugs that can overcome resistance against masitinib in canine MC tumors. Indeed, we were able to identify several drugs that effectively block the growth of NI-1 cells. Among these drugs were the PI3-kinase/mTOR blocker NVP-BEZ235, everolimus, and the TKI dasatinib, midostaurin, and sunitinib. These drugs produced growth inhibition (thymidine uptake) in NI-1 cells with an IC50 below 0.1 μM. In case of NVP-BEZ235 and everolimus, NI-1 cells were found to be even more sensitive cells compared to C2 cells. One explanation for this result is that the PI3-kinase/mTOR pathway plays a particular role for malignant Kit-triggered growth of NI-1 cells. A similar role of the PI3-kinase/mTOR pathway has been proposed for human neoplastic MC expressing Kit D816V (34). In fact, it has been described that HMC-1.2 cells exhibiting Kit D816V are more sensitive against the growth-inhibitory effect of rapamycin compared to HMC-1.1 cells (34). In the present study, this observation could be confirmed using the mTOR blocker everolimus and the mTOR/PI3-kinase blocker NVP-BEZ235. Both drugs exhibited more potent effects on the growth of HMC-1.2 cells than on HMC-1.1 cells in our experiments.
We also examined the effects of the mTOR inhibitors and other targeted drugs on survival and apoptosis in NI-1 cells and other MC lines. As expected, most TKI exerting growth-inhibitory effects also produced major apoptosis-inducing effects. However, as expected, the mTOR blockers, which may primarily act on neoplastic MC via cell cycle inhibition, did not show major pro-apoptotic effects on NI-1 cells.
In a substantial number of patients with (advanced) SM, mediator-related symptoms occur and represent a clinical problem (35–40). In several of these cases, IgE-dependent reactions can be documented (35–40). It has also been described that Kit-dependent and IgER-dependent pathways may act in concert to provoke mediator release in MC (41). However, so far, no MC lines expressing a functional IgER as well as relevant Kit mutations are available (32, 33). NI-1 cells were found to express the IgER on their surface. In addition, we were able to show that cross-linking of the IgER on NI-1 cells is followed by histamine release. Finally, we were able to show that midostaurin inhibits IgE-dependent secretion of histamine in NI-1 cells, which is in line with the observation that this TKI also blocks IgE-mediated histamine release in human MC and basophils (27).
In summary, we have established a novel canine MC line that may serve as a valuable tool for studying various aspects of neoplastic MC in advanced canine MC tumors. Specifically, this novel cell line should assist in exploring the mechanisms of abnormal growth and drug resistance in canine MC tumors, IgE-dependent activation of neoplastic MC, and the effects of various novel targeted drugs.
Acknowledgments
We would like to thank Elisabeth Wieser, Denise Klein, and Gabriele Stefanzl for skillful technical assistance. Supported by: Von Fircks-Fonds; a Mastocytosis Research Grant of the Clinic for Internal Medicine and Infectious Diseases, University of Veterinary Medicine Vienna; and the Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich – FWF grants #P21173-B13, #SFB-F4611.
Author contributions
E.H. cultured the cells and performed drug effect experiments; E.H. and M.W. collected samples, analyzed data, and wrote the paper; E.H., B.P., H.H., T.R., S.C-R., K.S., and W.F.P. did sample preparation and/or molecular analysis; T.T and V.Y-G. did sequencing of the kit oncogene and karyotype analysis; T.R. conducted tumorigenicity studies in NSG mice; L.K. performed electron microscopic analysis; and P.V. designed the study, analyzed the data, and wrote the paper.
Conflict of interest
P.V. received a research grant from Novartis and from BMS and is a consultant with Novartis for a TKI trial in aggressive mastocytosis but has no other conflicts of interest relevant to this manuscript. The remaining authors declare no conflict of interests relevant to this manuscript.
References
- Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603–625. doi: 10.1016/s0145-2126(01)00038-8. [DOI] [PubMed] [Google Scholar]
- Metcalfe DD. Mast cells and mastocytosis. Blood. 2008;112:946–956. doi: 10.1182/blood-2007-11-078097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluin-Nelemans HC, Jansen JH, Breukelman H, Wolthers BG, Kluin PM, Kroon HM, et al. Response to interferon alfa-2b in a patient with systemic mastocytosis. N Engl J Med. 1992;326:619–623. doi: 10.1056/NEJM199202273260907. [DOI] [PubMed] [Google Scholar]
- Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, Van‘t Wout JW, Verhoef G, Gerrits WB, et al. Cladribine therapy for systemic mastocytosis. Blood. 2003;102:4270–4276. doi: 10.1182/blood-2003-05-1699. [DOI] [PubMed] [Google Scholar]
- Bohm A, Sonneck K, Gleixner KV, Schuch K, Pickl WF, Blatt K, et al. In vitro and in vivo growth-inhibitory effects of cladribine on neoplastic mast cells exhibiting the imatinib-resistant KIT mutation D816V. Exp Hematol. 2010;38:744–755. doi: 10.1016/j.exphem.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Bohm A, et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006;107:752–759. doi: 10.1182/blood-2005-07-3022. [DOI] [PubMed] [Google Scholar]
- Gleixner KV, Mayerhofer M, Sonneck K, Gruze A, Samorapoompichit P, Baumgartner C, et al. Synergistic growth-inhibitory effects of two tyrosine kinase inhibitors, dasatinib and PKC412, on neoplastic mast cells expressing the D816V-mutated oncogenic variant of KIT. Haematologica. 2007;92:1451–1459. doi: 10.3324/haematol.11339. [DOI] [PubMed] [Google Scholar]
- Hadzijusufovic E, Peter B, Rebuzzi L, Baumgartner C, Gleixner KV, Gruze A, et al. Growth-inhibitory effects of four tyrosine kinase inhibitors on neoplastic feline mast cells exhibiting a Kit exon 8 ITD mutation. Vet Immunol Immunopathol. 2009;132:243–250. doi: 10.1016/j.vetimm.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin C, Brockow K, D'Ambrosio C, Kirshenbaum AS, Ma Y, Longley BJ, et al. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol. 2003;31:686–692. doi: 10.1016/s0301-472x(03)00112-7. [DOI] [PubMed] [Google Scholar]
- Gotlib J, Berube C, Growney JD, Chen CC, George TI, Williams C, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood. 2005;106:2865–2870. doi: 10.1182/blood-2005-04-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotlib J. KIT mutations in mastocytosis and their potential as therapeutic targets. Immunol Allergy Clin North Am. 2006;26:575–592. doi: 10.1016/j.iac.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Verstovsek S, Tefferi A, Cortes J, O'Brien S, Garcia-Manero G, Pardanani A, et al. Phase II study of dasatinib in Philadelphia chromosome-negative acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res. 2008;14:3906–3915. doi: 10.1158/1078-0432.CCR-08-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London CA, Galli SJ, Yuuki T, Hu ZQ, Helfand SC, Geissler EN. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp Hematol. 1999;27:689–697. doi: 10.1016/s0301-472x(98)00075-7. [DOI] [PubMed] [Google Scholar]
- Zemke D, Yamini B, Yuzbasiyan-Gurkan V. Mutations in the juxtamembrane domain of c-KIT are associated with higher grade mast cell tumors in dogs. Vet Pathol. 2002;39:529–535. doi: 10.1354/vp.39-5-529. [DOI] [PubMed] [Google Scholar]
- Letard S, Yang Y, Hanssens K, Palmerini F, Leventhal PS, Guery S, et al. Gain-of-function mutations in the extracellular domain of KIT are common in canine mast cell tumors. Mol Cancer Res. 2008;6:1137–1145. doi: 10.1158/1541-7786.MCR-08-0067. [DOI] [PubMed] [Google Scholar]
- Gleixner KV, Rebuzzi L, Mayerhofer M, Gruze A, Hadzijusufovic E, Sonneck K, et al. Synergistic antiproliferative effects of KIT tyrosine kinase inhibitors on neoplastic canine mast cells. Exp Hematol. 2007;35:1510–1521. doi: 10.1016/j.exphem.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Dubreuil P, Letard S, Ciufolini M, Gros L, Humbert M, Casteran N, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE. 2009;4:e7258. doi: 10.1371/journal.pone.0007258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn KA, Ogilvie G, Rusk T, Devauchelle P, Leblanc A, Legendre A, et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J Vet Intern Med. 2008;22:1301–1309. doi: 10.1111/j.1939-1676.2008.0190.x. [DOI] [PubMed] [Google Scholar]
- Hahn KA, Legendre AM, Shaw NG, Phillips B, Ogilvie GK, Prescott DM, et al. Evaluation of 12- and 24-month survival rates after treatment with masitinib in dogs with nonresectable mast cell tumors. Am J Vet Res. 2010;71:1354–1361. doi: 10.2460/ajvr.71.11.1354. [DOI] [PubMed] [Google Scholar]
- London CA. Tyrosine kinase inhibitors in veterinary medicine. Top Companion Anim Med. 2009;24:106–112. doi: 10.1053/j.tcam.2009.02.002. [DOI] [PubMed] [Google Scholar]
- DeVinney R, Gold WM. Establishment of two dog mastocytoma cell lines in continuous culture. Am J Respir Cell Mol Biol. 1990;3:413–420. doi: 10.1165/ajrcmb/3.5.413. [DOI] [PubMed] [Google Scholar]
- Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988;12:345–355. doi: 10.1016/0145-2126(88)90050-1. [DOI] [PubMed] [Google Scholar]
- Rebuzzi L, Willmann M, Sonneck K, Gleixner KV, Florian S, Kondo R, et al. Detection of vascular endothelial growth factor (VEGF) and VEGF receptors Flt-1 and KDR in canine mastocytoma cells. Vet Immunol Immunopathol. 2007;115:320–333. doi: 10.1016/j.vetimm.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Samorapoompichit P, Kiener HP, Schernthaner GH, Jordan JH, Agis H, Wimazal F, et al. Detection of tryptase in cytoplasmic granules of basophils in patients with chronic myeloid leukemia and other myeloid neoplasms. Blood. 2001;98:2580–2583. doi: 10.1182/blood.v98.8.2580. [DOI] [PubMed] [Google Scholar]
- Webster JD, Kiupel M, Yuzbasiyan-Gurkan V. Evaluation of the kinase domain of c-KIT in canine cutaneous mast cell tumors. BMC Cancer. 2006;6:85. doi: 10.1186/1471-2407-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valent P, Ashman LK, Hinterberger W, Eckersberger F, Majdic O, Lechner K, et al. Mast cell typing: demonstration of a distinct hematopoietic cell type and evidence for immunophenotypic relationship to mononuclear phagocytes. Blood. 1989;73:1778–1785. [PubMed] [Google Scholar]
- Krauth MT, Mirkina I, Herrmann H, Baumgartner C, Kneidinger M, Valent P. Midostaurin (PKC412) inhibits immunoglobulin E-dependent activation and mediator release in human blood basophils and mast cells. Clin Exp Allergy. 2009;39:1711–1720. doi: 10.1111/j.1365-2222.2009.03353.x. [DOI] [PubMed] [Google Scholar]
- Ma Y, Longley BJ, Wang X, Blount JL, Langley K, Caughey GH. Clustering of activating mutations in c-KIT's juxtamembrane coding region in canine mast cell neoplasms. J Invest Dermatol. 1999;112:165–170. doi: 10.1046/j.1523-1747.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- London CA, Seguin B. Mast cell tumors in the dog. Vet Clin North Am Small Anim Pract. 2003;33:473–489. doi: 10.1016/s0195-5616(03)00003-2. [DOI] [PubMed] [Google Scholar]
- Sotlar K, Cerny-Reiterer S, Petat-Dutter K, Hessel H, Berezowska S, Mullauer L, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585–595. doi: 10.1038/modpathol.2010.224. [DOI] [PubMed] [Google Scholar]
- Ishiguro T, Kadosawa T, Mori K, Takagi S, Okumura M, Fujinaga T. Establishment and characterization of a new canine mast cell tumor cell line. J Vet Med Sci. 2001;63:1031–1034. doi: 10.1292/jvms.63.1031. [DOI] [PubMed] [Google Scholar]
- Amagai Y, Tanaka A, Ohmori K, Matsuda H. Establishment of a novel high-affinity IgE receptor-positive canine mast cell line with wild-type c-kit receptors. Biochem Biophys Res Commun. 2008;366:857–861. doi: 10.1016/j.bbrc.2007.12.053. [DOI] [PubMed] [Google Scholar]
- Lin TY, Thomas R, Tsai PC, Breen M, London CA. Generation and characterization of novel canine malignant mast cell line CL1. Vet Immunol Immunopathol. 2009;127:114–124. doi: 10.1016/j.vetimm.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabillot-Carre M, Lepelletier Y, Humbert M, de Sepuvelda P, Hamouda NB, Zappulla JP, et al. Rapamycin inhibits growth and survival of D816V-mutated c-kit mast cells. Blood. 2006;108:1065–1072. doi: 10.1182/blood-2005-06-2433. [DOI] [PubMed] [Google Scholar]
- Valent P, Akin C, Escribano L, Fodinger M, Hartmann K, Brockow K, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest. 2007;37:435–453. doi: 10.1111/j.1365-2362.2007.01807.x. [DOI] [PubMed] [Google Scholar]
- Niedoszytko M, Oude Elberink JN, Bruinenberg M, Nedoszytko B, de Monchy JG, te Meerman GJ, et al. Gene expression profile, pathways, and transcriptional system regulation in indolent systemic mastocytosis. Allergy. 2011;66:229–237. doi: 10.1111/j.1398-9995.2010.02477.x. [DOI] [PubMed] [Google Scholar]
- Niedoszytko M, de Monchy J, van Doormaal JJ, Jassem E, Oude Elberink JN. Mastocytosis and insect venom allergy: diagnosis, safety and efficacy of venom immunotherapy. Allergy. 2009;64:1237–1245. doi: 10.1111/j.1398-9995.2009.02118.x. [DOI] [PubMed] [Google Scholar]
- Niedoszytko M, Bruinenberg M, van Doormaal JJ, de Monchy JG, Nedoszytko B, Koppelman GH, et al. Gene expression analysis predicts insect venom anaphylaxis in indolent systemic mastocytosis. Allergy. 2011;66:648–657. doi: 10.1111/j.1398-9995.2010.02521.x. [DOI] [PubMed] [Google Scholar]
- Matito A, Bartolome-Zavala B, Alvarez-Twose I, Sanchez-Matas I, Escribano L. IgE-mediated anaphylaxis to Hippobosca equina in a patient with systemic mastocytosis. Allergy. 2010;65:1058–1059. doi: 10.1111/j.1398-9995.2009.02270.x. [DOI] [PubMed] [Google Scholar]
- Bonadonna P, Zanotti R, Pagani M, Caruso B, Perbellini O, Colarossi S, et al. How much specific is the association between hymenoptera venom allergy and mastocytosis? Allergy. 2009;64:1379–1382. doi: 10.1111/j.1398-9995.2009.02108.x. [DOI] [PubMed] [Google Scholar]
- Hundley TR, Gilfillan AM, Tkaczyk C, Andrade MV, Metcalfe DD, Beaven MA. Kit and FcepsilonRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood. 2004;104:2410–2417. doi: 10.1182/blood-2004-02-0631. [DOI] [PubMed] [Google Scholar]