

Abstract
To characterize somatic alterations in colorectal carcinoma, we conducted a genome-scale analysis of 276 samples, analysing exome sequence, DNA copy number, promoter methylation and messenger RNA and microRNA expression. A subset of these samples (97) underwent low-depth-of-coverage whole-genome sequencing. In total, 16% of colorectal carcinomas were found to be hypermutated: three-quarters of these had the expected high microsatellite instability, usually with hypermethylation and MLH1 silencing, and one-quarter had somatic mismatch-repair gene and polymerase ε (POLE) mutations. Excluding the hypermutated cancers, colon and rectum cancers were found to have considerably similar patterns of genomic alteration. Twenty-four genes were significantly mutated, and in addition to the expected APC, TP53, SMAD4, PIK3CA and KRAS mutations, we found frequent mutations in ARID1A, SOX9 and FAM123B. Recurrent copy-number alterations include potentially drug-targetable amplifications of ERBB2 and newly discovered amplification of IGF2. Recurrent chromosomal translocations include the fusion of NAV2 and WNT pathway member TCF7L1. Integrative analyses suggest new markers for aggressive colorectal carcinoma and an important role for MYC-directed transcriptional activation and repression.
Supplementary information
The online version of this article (doi:10.1038/nature11252) contains supplementary material, which is available to authorized users.
Subject terms: Genomics, Chromosome abnormality, DNA sequencing, Colorectal cancer
The Cancer Genome Atlas consortium reports on their genome-wide characterization of somatic alterations in colorectal cancer; in addition to revealing a remarkably consistent pattern of genomic alteration, with 24 genes being significantly mutated, the study identifies new targets for therapeutic intervention and suggests an important role for MYC-directed transcriptional activation and repression.
Supplementary information
The online version of this article (doi:10.1038/nature11252) contains supplementary material, which is available to authorized users.
Mutations associated with colorectal cancer
The Cancer Genome Atlas consortium reports on their genome-wide characterization of somatic alterations in colorectal carcinoma. Combined analysis of exome sequence data, DNA copy number, promoter methylation, messenger RNA and microRNA expression, as well as low-coverage whole-genome sequencing reveal that 16% of these carcinomas have hypermutation. Of these, three-quarters have the expected high microsatellite instability, but the rest have somatic mismatch repair gene mutations. The data reveal a remarkably consistent pattern of genomic alteration, with 24 genes being significantly mutated. As well as identifying new biomarkers for aggressive colorectal carcinoma, the data imply an important role for MYC-directed transcriptional activation and repression.
Supplementary information
The online version of this article (doi:10.1038/nature11252) contains supplementary material, which is available to authorized users.
Main
The Cancer Genome Atlas project plans to profile genomic changes in 20 different cancer types and has so far published results on two cancer types1,2. We now present results from multidimensional analyses of human colorectal carcinoma (CRC).
CRC is an important contributor to cancer mortality and morbidity. The distinction between the colon and the rectum is largely anatomical, but it has both surgical and radiotherapeutic management implications and it may have an impact on prognosis. Most investigators divide CRC biologically into those with microsatellite instability (MSI; located primarily in the right colon and frequently associated with the CpG island methylator phenotype (CIMP) and hyper-mutation) and those that are microsatellite stable but chromosomally unstable.
A rich history of investigations (for a review see ref. 3) has uncovered several critical genes and pathways important in the initiation and progression of CRC (ref. 3). These include the WNT, RAS−MAPK, PI3K, TGF-β, P53 and DNA mismatch-repair pathways. Large-scale sequencing analyses4,5,6 have identified numerous recurrently mutated genes and a recurrent chromosomal translocation. Despite this background, we have not had a fully integrated view of the genetic and genomic changes and their significance for colorectal tumorigenesis. Further insight into these changes may enable deeper understanding of the pathophysiology of CRC and may identify potential therapeutic targets.
Results
Tumour and normal pairs were analysed by different platforms. The specific numbers of samples analysed by each platform are shown in Supplementary Table 1.
Exome-sequence analysis
To define the mutational spectrum, we performed exome capture DNA sequencing on 224 tumour and normal pairs (all mutations are listed in Supplementary Table 2). Sequencing achieved >20-fold coverage of at least 80% of targeted exons. The somatic mutation rates varied considerably among the samples. Some had mutation rates of <1 per 106 bases, whereas a few had mutations rates of >100 per 106. We separated cases (84%) with a mutation rate of <8.24 per 106 (median number of non-silent mutations, 58) and those with mutation rates of >12 per 106 (median number of total mutations, 728), which we designated as hypermutated (Fig. 1).
Figure 1. Mutation frequencies in human CRC.
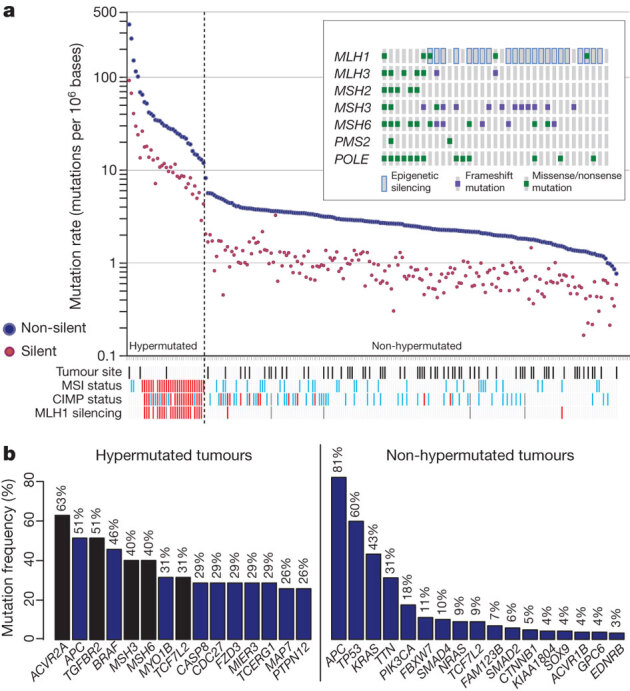
a, Mutation frequencies in each of the tumour samples from 224 patients. Note a clear separation of hypermutated and non-hypermutated samples. Red, MSI high, CIMP high or MLH1 silenced; light blue, MSI low, or CIMP low; black, rectum; white, colon; grey, no data. Inset, mutations in mismatch-repair genes and POLE among the hypermutated samples. The order of the samples is the same as in the main graph. b, Significantly mutated genes in hypermutated and non-hypermutated tumours. Blue bars represent genes identified by the MutSig algorithm and black bars represent genes identified by manual examination of sequence data.
To assess the basis for the considerably different mutation rates, we evaluated MSI7 and mutations in the DNA mismatch-repair pathway8,9,10 genes MLH1, MLH3, MSH2, MSH3, MSH6 and PMS2. Among the 30 hypermutated tumours with a complete data set, 23 (77%) had high levels of MSI (MSI-H). Included in this group were 19 tumours with MLH1 methylation, 17 of which had CIMP. By comparison, the remaining seven hypermutated tumours, including the six with the highest mutation rates, lacked MSI-H, CIMP or MLH1 methylation but usually had somatic mutations in one or more mismatch-repair genes or POLΕ aberrations seen rarely in the non-hypermutated tumours (Fig. 1).
Gene mutations
Overall, we identified 32 somatic recurrently mutated genes (defined by MutSig11 and manual curation) in the hypermutated and non-hypermutated cancers (Fig. 1b). After removal of non-expressed genes, there were 15 and 17 in the hypermutated and non-hypermutated cancers, respectively (Fig. 1b; for a complete list see Supplementary Table 3). Among the non-hypermutated tumours, the eight most frequently mutated genes were APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, TCF7L2 and NRAS. As expected, the mutated KRAS and NRAS genes usually had oncogenic codon 12 and 13 or codon 61 mutations, whereas the remaining genes had inactivating mutations. CTNNB1, SMAD2, FAM123B (also known as WTX) and SOX9 were also mutated frequently. FAM123B is an X-linked negative regulator of WNT signalling12, and virtually all of its mutations were loss of function. Mutations in SOX9, a gene important for cell differentiation in the intestinal stem cell niche13,14, have not been associated previously with human cancer, but all nine mutated alleles in the non-hypermutated CRCs were frameshift or nonsense mutations. Tumour-suppressor genes ATM and ARID1A also had a disproportionately high number of frameshift or nonsense mutations. ARID1A mutations have recently been reported in CRC and many other cancers15,16.
In the hypermutated tumours, ACVR2A, APC, TGFBR2, MSH3, MSH6, SLC9A9 and TCF7L2 were frequent targets of mutation (Fig. 1b), along with mostly BRAF(V600E) mutations. However, two genes that were frequently mutated in the non-hypermutated cancers were significantly less frequently mutated in hypermutated tumours: TP53 (60 versus 20%, P < 0.0001) and APC (81% versus 51%, P = 0.0023; both Fisher’s exact test). Other genes, including TGFBR2, were mutated recurrently in the hypermutated cancers, but not in the non-hypermutated samples. These findings indicate that hypermutated and non-hypermutated tumours progress through different sequences of genetic events.
As expected, hypermutated tumours with MLH1 silencing and MSI-H showed additional differences in the mutational profile. When we specifically examined 28 genes with long mononucleotide repeats in their coding sequences, we found that the rate of frameshift mutation was 3.6-fold higher than the rate of such mutations in hypermutated tumours without MLH1 silencing and 50-fold higher than that in non-hypermethylated tumours (Supplementary Table 2).
Mutation rate and methylation patterns
As mentioned above, patients with colon and rectal tumours are managed differently17, and epidemiology also highlights differences between the two17. An initial integrative analysis of MSI status, somatic copy-number alterations (SCNAs), CIMP status and gene-expression profiles of 132 colonic and 62 rectal tumours enabled us to examine possible biological differences between tumours in the two locations. Among the non-hypermutated tumours, however, the overall patterns of changes in copy number, CIMP, mRNA and miRNA were indistinguishable between colon and rectal carcinomas (Fig. 2). On the basis of this result, we merged the two for all subsequent analyses.
Figure 2. Integrative analysis of genomic changes in 195 CRCs.
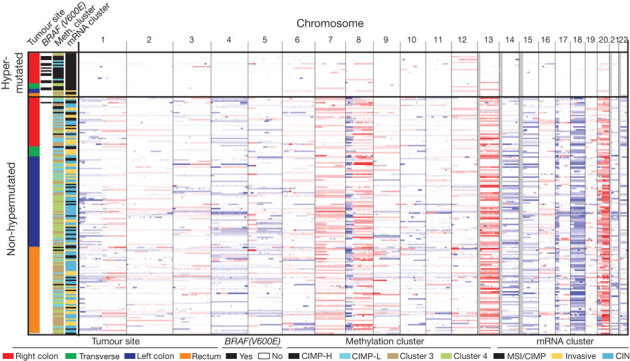
Hypermutated tumours have near-diploid genomes and are highly enriched for hypermethylation, CIMP expression phenotype and BRAF(V600E) mutations. Non-hypermutated tumours originating from different sites are virtually indistinguishable from each other on the basis of their copy-number alteration patterns, DNA methylation or gene-expression patterns. Copy-number changes of the 22 autosomes are shown in shades of red for copy-number gains and shades of blue for copy-number losses.
Unsupervised clustering of the promoter DNA methylation profiles of 236 colorectal tumours identified four subgroups (Supplementary Fig. 1 and Supplementary Methods). Two of the clusters contained tumours with elevated rates of methylation and were classified as CIMP high and CIMP low, as previously described18. The two non-CIMP clusters were predominantly from tumours that were non-hypermutated and derived from different anatomic locations. mRNA expression profiles separated the colorectal tumours into three distinct clusters (Supplementary Fig. 2). One significantly overlapped with CIMP-high tumours (P = 3 × 10−12) and was enriched with hypermutated tumours, and the other two clusters did not correspond with any group in the methylation data. Analysis of miRNA expression by unsupervised clustering (Supplementary Fig. 3) identified no clear distinctions between rectal cancers and non-hypermethylated colon cancers.
Chromosomal and sub-chromosomal changes
In total, 257 tumours were profiled for SCNAs with Affymetrix SNP 6.0 arrays. Of these tumours, 97 were also analysed by low-depth-of-coverage (low-pass) whole-genome sequencing. As expected, the hypermutated tumours had far fewer SCNAs (Fig. 2). No difference was found between microsatellite-stable and -unstable hypermutated tumours (Supplementary Fig. 4). We used the GISTIC algorithm19 to identify probable gene targets of focal alterations. There were several previously well-defined arm-level changes, including gains of 1q, 7p and q, 8p and q, 12q, 13q, 19q, and 20p and q (ref. 6). (Supplementary Fig. 4 and Supplementary Table 4). Significantly deleted chromosome arms were 18p and q (including SMAD4) in 66% of the tumours and 17p and q (including TP53) in 56%. Other significantly deleted chromosome arms were 1p, 4q, 5q, 8p, 14q, 15q, 20p and 22q.
We identified 28 recurrent deletion peaks (Supplementary Fig. 4 and Supplementary Table 4), including the genes FHIT, RBFOX1 and WWOX with large genomic footprints located in potentially fragile sites of the genome, in near-diploid hypermutated tumours. Other focal deletions involved tumour-suppressor genes such as SMAD4, APC, PTEN and SMAD3. A significant focal deletion of 10p25.2 spanned four genes, including TCF7L2, which was also frequently mutated in our data set. A gene fusion between adjacent genes VTI1A and TCF7L2 through an interstitial deletion was found in 3% of CRCs and is required for survival of CRC cells bearing the translocation4.
There were 17 regions of significant focal amplification (Supplementary Table 4). Some of these were superimposed on broad gains of chromosome arms, and included a peak at 13q12.13 near the peptidase-coding gene USP12 and at ∼500 kb distal to the CRC candidate oncogene CDK8; an adjacent peak at 13q12; a peak containing KLF5 at 13q22.1; and a peak at 20q13.12 adjacent to HNF4A. Peaks on chromosome 8 included 8p12 (which contains the histone methyl-transferase-coding gene WHSC1L1, adjacent to FGFR1) and 8q24 (which contains MYC). An amplicon at 17q21.1, found in 4% of the tumours, contains seven genes, including the tyrosine kinase ERBB2. ERBB2 amplifications have been described in colon, breast and gastro–oesophageal tumours, and breast and gastric cancers bearing these amplifications have been treated effectively with the anti-ERBB2 antibody trastuzumab20,21,22.
One of the most common focal amplifications, found in 7% of the tumours, is the gain of a 100–150-kb region of the chromosome arm 11p15.5. It contains genes encoding insulin (INS), insulin-like growth factor 2 (IGF2) and tyrosine hydroxylase (TH), as well as miR-483, which is embedded within IGF2 (Fig. 3a). We found elevated expression of IGF2 and miR-483 but not of INS and TH (Fig. 3b, c). Immediately adjacent to the amplified region is ASCL2, a transcription factor active in specifying intestinal stem-cell fate23. Although ASCL2 has been implicated as a target of amplification in CRC23,24,25, it was consistently outside the region of amplification and its expression was not correlated with copy-number changes. These observations suggest that IGF2 and miR-483 are candidate functional targets of 11p15.5 amplification. IGF2 overexpression through loss of imprinting has been implicated in the promotion of CRC26,27. MiR-483 may also have a role in CRC pathogenesis28.
Figure 3. Copy-number changes and structural aberrations in CRC.
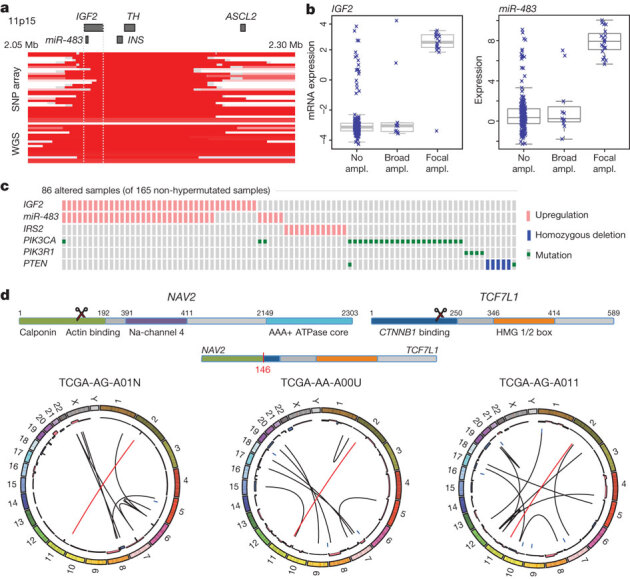
a, Focal amplification of 11p15.5. Segmented DNA copy-number data from single-nucleotide polymorphism (SNP) arrays and low-pass whole-genome sequencing (WGS) are shown. Each row represents a patient; amplified regions are shown in red. b, Correlation of expression levels with copy-number changes for IGF2 and miR-483. c, IGF2 amplification and overexpression are mutually exclusive of alterations in PI3K signalling-related genes. d, Recurrent NAV2–TCF7L2 fusions. The structure of the two genes, locations of the breakpoints leading to the translocation and circular representations of all rearrangements in tumours with a fusion are shown. Red line lines represent the NAV2–TCF7L2 fusions and black lines represent other rearrangements. The inner ring represents copy-number changes (blue denotes loss, pink denotes gain).
A subset of tumours without IGF2 amplification (15%) also had considerably higher levels of IGF2 gene expression (as much as a 100-fold increase), an effect not attributable to methylation changes at the IGF2 promoter. To assess the context of IGF2 amplification/overexpression, we systematically searched for mutually exclusive genomic events using the MEMo method29. We found a pattern of near exclusivity (corrected P < 0.01) of IGF2 overexpression with genomic events known to activate the PI3K pathway (mutations of PIK3CA and PIK3R1 or deletion/mutation of PTEN; Fig. 3c and Supplementary Table 5). The IRS2 gene, encoding a protein linking IGF1R (the receptor for IGF2) with PI3K, is on chromosome 13, which is frequently gained in CRC. The cases with the highest IRS2 expression were mutually exclusive of the cases with IGF2 overexpression (P = 0.04) and also lacked mutations in the PI3K pathway (P = 0.0001; Fig. 3c). These results strongly suggest that the IGF2–IGF1R–IRS2 axis signals to PI3K in CRC and imply that therapeutic targeting of the pathway could act to block PI3K activity in this subset of patients.
Translocations
To identify new chromosomal translocations, we performed low-pass, paired-end, whole-genome sequencing on 97 tumours with matched normal samples. In each case we achieved sequence coverage of ∼3–4-fold and a corresponding physical coverage of 7.5–10-fold. Despite the low genome coverage, we detected 250 candidate interchromosomal translocation events (range, 0–10 per tumour). Among these events, 212 had one or both breakpoints in an intergenic region, whereas the remaining 38 juxtaposed coding regions of two genes in putative fusion events, of which 18 were predicted to code for in-frame events (Supplementary Table 6). We found three separate cases in which the first two exons of the NAV2 gene on chromosome 11 are joined with the 3′ coding portion of TCF7L1 on chromosome 2 (Supplementary Fig. 5). TCF7L1 encodes TCF3, a member of the TCF/LEF class of transcription factors that heterodimerize with nuclear β-catenin to enable β-catenin-mediated transcriptional regulation. Intriguingly, in all three cases, the predicted structure of the NAV2–TCF7L1 fusion protein lacks the TCF3 β-catenin-binding domain. This translocation is similar to another recurrent translocation identified in CRC, a fusion in which the amino terminus of VTI1A is joined to TCF4, which is encoded by TCF7L2, a homologue of TCF7L1 that is deleted or mutated in 12% of non-hypermutated tumours4. We also observed 21 cases of translocation involving TTC28 located on chromosome 22 (Supplementary Table 6). In all cases the fusions predict inactivation of TTC28, which has been identified as a target of P53 and an inhibitor of tumour cell growth30. Eleven of the 19 (58%) gene–gene translocations were validated by obtaining PCR products or, in some cases, sequencing the junction fragments (Supplementary Fig. 5).
Altered pathways in CRC
Integrated analysis of mutations, copy number and mRNA expression changes in 195 tumours with complete data enriched our understanding of how some well-defined pathways are deregulated. We grouped samples by hypermutation status and identified recurrent alterations in the WNT, MAPK, PI3K, TGF-β and p53 pathways (Fig. 4, Supplementary Fig. 6 and Supplementary Table 1).
Figure 4. Diversity and frequency of genetic changes leading to deregulation of signalling pathways in CRC.
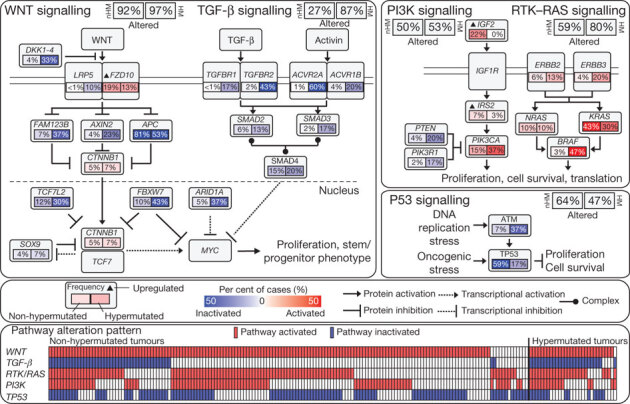
Non-hypermutated (nHM; n = 165) and hypermutated (HM; n = 30) samples with complete data were analysed separately. Alterations are defined by somatic mutations, homozygous deletions, high-level focal amplifications, and, in some cases, by significant up- or downregulation of gene expression (IGF2, FZD10, SMAD4). Alteration frequencies are expressed as a percentage of all cases. Red denotes activated genes and blue denotes inactivated genes. Bottom panel shows for each sample if at least one gene in each of the five pathways described in this figure is altered.
We found that the WNT signalling pathway was altered in 93% of all tumours, including biallelic inactivation of APC (Supplementary Table 7) or activating mutations of CTNNB1 in ∼80% of cases. There were also mutations in SOX9 and mutations and deletions in TCF7L2, as well as the DKK family members and AXIN2, FBXW7 (Supplementary Fig. 7), ARID1A and FAM123B (the latter is a negative regulator of WNT–β-catenin signalling12 found mutated in Wilms’ tumour31). A few mutations in FAM123B have previously been described in CRC32. SOX9 has been suggested to have a role in cancer, but no mutations have previously been described. The WNT receptor frizzled (FZD10) was overexpressed in ∼17% of samples, in some instances at levels of 100× normal. Altogether, we found 16 different altered WNT pathway genes, confirming the importance of this pathway in CRC. Interestingly, many of these alterations were found in tumours that harbour APC mutations, suggesting that multiple lesions affecting the WNT signalling pathway confer selective advantage.
Genetic alterations in the PI3K and RAS–MAPK pathways are common in CRC. In addition to IGF2 and IRS2 overexpression, we found mutually exclusive mutations in PIK3R1 and PIK3CA as well as deletions in PTEN in 2%, 15% and 4% of non-hypermutated tumours, respectively. We found that 55% of non-hypermutated tumours have alterations in KRAS, NRAS or BRAF, with a significant pattern of mutual exclusivity (Supplementary Fig. 6 and Supplementary Table 1). We also evaluated mutations in the erythroblastic leukemia viral oncogene homolog (ERBB) family of receptors because of the translational relevance of such mutations. Mutations or amplifications in one of the four ERBB family genes are present in 22 out of 165 (13%) non-hypermutated and 16 out of 30 (53%) hypermutated cases. Some of the mutations are listed in the COSMIC database33, suggesting a functional role. Intriguingly, recurrent ERBB2(V842I) and ERBB3(V104M) mutations were found in four and two non-hypermutated cases, respectively. Mutations and focal amplifications of ERBB2 (Supplementary Fig. 6) should be evaluated as predictors of response to agents that target those receptors. We observed co-occurrence of alterations involving the RAS and PI3K pathways in one-third of tumours (Fig. 4; P = 0.039, Fisher’s exact test). These results indicate that simultaneous inhibition of the RAS and PI3K pathways may be required to achieve therapeutic benefit.
The TGF-β signalling pathway is known to be deregulated in CRC and other cancers34. We found genomic alterations in TGFBR1, TGFBR2, ACVR2A, ACVR1B, SMAD2, SMAD3 and SMAD4 in 27% of the non-hypermutated and 87% of the hypermutated tumours. We also evaluated the p53 pathway, finding alterations in TP53 in 59% of non-hypermutated cases (mostly biallelic; Supplementary Table 8) and alterations in ATM, a kinase that phosphorylates and activates P53 after DNA damage, in 7%. Alterations in these two genes showed a trend towards mutual exclusivity (P = 0.016) (Fig. 4, Supplementary Fig. 6 and Supplementary Table 1).
We integrated copy number, gene expression, methylation and pathway data using the PARADIGM software platform35. The analysis showed a number of new characteristics of CRC (Fig. 5a). For example, despite the diversity in anatomical origin or mutation levels, nearly 100% of these tumours have changes in MYC transcriptional targets, both those promoted by and those inhibited by MYC. These findings are consistent with patterns deduced from genetic alterations (Fig. 4) and suggest an important role for MYC in CRC. The analysis also identified several gene networks altered across all tumour samples and those with differential alterations in hypermutated versus non-hypermutated samples (Supplementary Table 7, Supplementary Data on the Cancer Genome Atlas publication webpage).
Figure 5. Integrative analyses of multiple data sets.
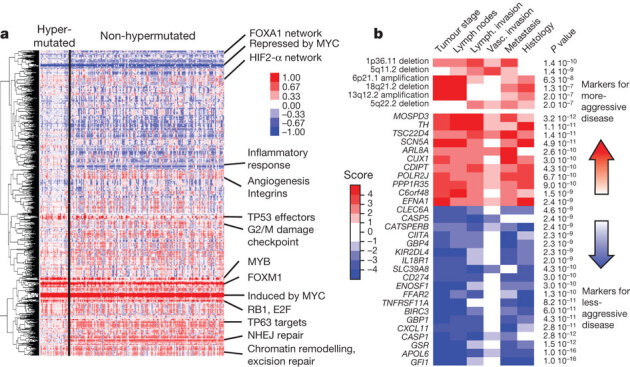
a, Clustering of genes and pathways affected in colon and rectum tumours deduced by PARADIGM analysis. Blue denotes under-expressed relative to normal and red denotes overexpressed relative to normal. Some of the pathways deduced by this method are shown on the right. NHEJ, non-homologous end joining. b, Gene-expression signatures and SCNAs associated with tumour aggression. Molecular signatures (rows) that show a statistically significant association with tumour aggressiveness according to selected clinical assays (columns) are shown in colour, with red indicating markers of tumour aggressiveness and blue indicating the markers of less-aggressive tumours. Significance is based on the combined P value from the weighted Fisher’s method, corrected for multiple testing. Colour intensity and score is in accordance with the strength of an individual clinical–molecular association, and is proportional to log10(P), where P is the P value for that association. To limit the vertical extent of the figure, gene-expression signatures are restricted to a combined P value of P < 10−9 and SCNAs to P < 10−7, and features are shown only if they are also significant in the subset of non-MSI-H samples (the analysis was performed separately on the full data as well as on the MSI-H and non-MSI-H subgroups).
Because most of the tumours used in this study were derived from a prospective collection, survival data are not available. However, the tumours can be classified as aggressive or non-aggressive on the basis of tumour stage, lymph node status, distant metastasis and vascular invasion at the time of surgery. We found numerous molecular signatures associated with tumour aggressiveness, a subset of which is shown in Fig. 5b. They include specific focal amplifications and deletions, and altered gene-expression levels, including those of SCN5A (ref. 36), a reported regulator of colon cancer invasion (see Supplementary Tables 10 and 11 for a full list). Association with tumour aggressiveness is also observed in altered expression of miRNAs and specific somatic mutations (APC, TP53, PIK3CA, BRAF and FBXW7; Supplementary Fig. 8b). Mutations in FBXW7 (38 cases) and distant metastasis (32 cases) never co-occurred (P = 0.0019). Interestingly, a number of genomic regions have multiple molecular associations with tumour aggressiveness that manifest as clinically related genomic hotspots. Examples of this are the region 20q13.12, which includes a focal amplification and multiple genes correlating with tumour aggression, and the region 22q12.3, containing APOL6 (ref. 37) (Supplementary Figures 8 and 9).
Discussion
This comprehensive integrative analysis of 224 colorectal tumour and normal pairs provides a number of insights into the biology of CRC and identifies potential therapeutic targets. To identify possible biological differences in colon and rectum tumours, we found, in the non-hypermutated tumours irrespective of their anatomical origin, the same type of copy number, expression profile, DNA methylation and miRNA changes. Over 94% had a mutation in one or more members of the WNT signalling pathway, predominantly in APC. However, there were some differences between tumours from the right colon and all other sites. Hypermethylation was more common in the right colon, and three-quarters of hypermutated samples came from the same site, although not all of them had MSI (Fig. 2). Why most of the hypermutated samples came from the right colon and why there are two classes of tumours at this site is not known. The origins of the colon from embryonic midgut and hindgut may provide an explanation. As the survival rate of patients with high MSI-related cancers is better and these cancers are hypermutated, mutation rate may be a better prognostic indicator.
Whole-exome sequencing and integrative analysis of genomic data provided further insights into the pathways that are dysregulated in CRC. We found that 93% of non-hypermutated and 97% of hypermutated cases had a deregulated WNT signalling pathway. New findings included recurrent mutations in FAM123B, ARID1A and SOX9 and very high levels of overexpression of the WNT ligand receptor gene FZD10. To our knowledge, SOX9 has not previously been described as frequently mutated in any human cancer. SOX9 is transcriptionally repressed by WNT signalling, and the SOX9 protein has been shown to facilitate β-catenin degradation38. ARID1A is frequently mutated in gynaecological cancers and has been shown to suppress MYC transcription39. Activation of WNT signalling and inactivation of the TGF-β signalling pathway are known to result in activation of MYC. Our mutational and integrative analyses emphasize the critical role of MYC in CRC. We also compared our results with other large-scale analyses6 and found many similarities and few differences in mutated genes (Supplementary Table 3).
Our integrated analysis revealed a diverse set of changes in TCF/LEF-encoding genes, suggesting additional roles for TCF/LEF factors in CRC beyond being passive partners for β-catenin.
Our data suggest a number of therapeutic approaches to CRC. Included are WNT-signalling inhibitors and small-molecule β-catenin inhibitors, which are showing initial promise40,41,42. We find that several proteins in the RTK–RAS and PI3K pathways, including IGF2, IGFR, ERBB2, ERBB3, MEK, AKT and MTOR could be targets for inhibition.
Our analyses show that non-hypermutated adenocarcinomas of the colon and rectum are not distinguishable at the genomic level. However, tumours from the right/ascending colon were more likely to be hypermethylated and to have elevated mutation rates than were other CRCs. As has been recognized previously, activation of the WNT signalling pathway and inactivation of the TGF-β signalling pathway, resulting in increased activity of MYC, are nearly ubiquitous events in CRC. Genomic aberrations frequently target the MAPK and PI3K pathways but less frequently target receptor tyrosine kinases. In conclusion, the data presented here provide a useful resource for understanding this deadly disease and identifying possibilities for treating it in a targeted way.
Methods Summary
Tumour and normal samples were processed by either of two biospecimen core resources, and aliquots of purified nucleic acids were shipped to the genome characterization and sequencing centres (Supplementary Methods). The biospecimen core resources provided sample sets in several different batches. To assess any batch effects we examined the mRNA expression, miRNA expression and DNA methylation data sets using a combination of cluster analysis, enhanced principal component analysis and analysis of variance (Supplementary Methods). Although some differences among batches were detected, we did not correct them computationally because the differences were generally modest and because some of them may reflect biological phenomena (Supplementary Methods).
We used Affymetrix SNP 6.0 microarrays to detect copy-number alterations. A subset of samples was subjected to low-pass (2–5×) whole-genome sequencing (Illumina HiSeq), in part for detection of SCNA and chromosomal translocations43,44. Gene-expression profiles were generated using Agilent microarrays and RNA-Seq. DNA methylation data were obtained using Illumina Infinium (HumanMethylation27) arrays. DNA sequencing of coding regions was performed by exome capture followed by sequencing on the SOLiD or Illumina HiSeq platforms. Details of the analytical methods used are described in Supplementary Methods.
All of the primary sequence files are deposited in dbGap and all other data are deposited at the Data Coordinating Center (DCC) for public access (http://cancergenome.nih.gov/). Data matrices and supporting data can be found at http://tcga-data.nci.nih.gov/docs/publications/coadread_2012/. The data can also be explored through the ISB Regulome Explorer (http://explorer.cancerregulome.org/), Next Generation Clustered Heat Maps (http://bioinformatics.mdanderson.org/main/TCGA/Supplements/NGCHM-CRC) and the cBio Cancer Genomics Portal (http://cbioportal.org). Descriptions of the data can be found at https://wiki.nci.nih.gov/x/j5dXAg and in Supplementary Methods.
Supplementary information
This file contains the legends for Supplementary Tables 1-12, Supplementary Tables 1-9 and Supplementary Data files 1-2, Supplementary Methods, which include 17 Figures and 2 Tables (see Contents for details) and Supplementary Figures 1-9. (PDF 8117 kb)
This file contains Supplementary Table 2. (ZIP 25458 kb)
This file contains Supplementary Tables 1 and 3-12 (ZIP 2809 kb)
Acknowledgements
This work was supported by the following grants from the National Institutes of Health: U24CA143799, U24CA143835, U24CA143840, U24CA143843, U24CA143845, U24CA143848, U24CA143858, U24CA143866, U24CA143867, U24CA143882, U24CA143883, U24CA144025, U54HG003067, U54HG003079 and U54HG003273.
PowerPoint slides
Author Contributions
The Cancer Genome Atlas research network contributed collectively to this study. Biospecimens were provided by the tissue source sites and processed by the Biospecimen Core Resource. Data generation and analyses were performed by the genome-sequencing centers, cancer genome-characterization centers and genome data analysis centers. All data were released through the Data Coordinating Center. Project activities were coordinated by the National Cancer Institute and National Human Genome Research Institute project teams. Project leaders were R.K. and D.A.W. Writing team, T.A., A.J.B., T.A.C., L.D., A.H., S.R.H., R.K., P.W.L., M.M., N.S., I.S., J.M.S., J.T., V.T. and D.A.W.; mutations, M.S.L., L.R.T., D.A.W. and G.G.; copy-number and structural aberrations, A.H.R., A.J.B., A.H. and P.-C.C.; DNA methylation, T.H.; expression, J.T.A.; miRNA, G.R., A.C.; pathways, C.J.C., L.D., T.G., S.N., J.D.R., C.S., N.S., J.M.S. and V.T.
Accession codes
Data deposits
dbGaP accession numbers have been provided in Supplementary Table 1.
Competing interests
The author declare no competing financial interests.
Footnotes
(Participants are arranged by area of contribution, below, and by institution, above.)
Contributor Information
The Cancer Genome Atlas Network:
Donna M. Muzny, Matthew N. Bainbridge, Kyle Chang, Huyen H. Dinh, Jennifer A. Drummond, Gerald Fowler, Christie L. Kovar, Lora R. Lewis, Margaret B. Morgan, Irene F. Newsham, Jeffrey G. Reid, Jireh Santibanez, Eve Shinbrot, Lisa R. Trevino, Yuan-Qing Wu, Min Wang, Preethi Gunaratne, Lawrence A. Donehower, Chad J. Creighton, David A. Wheeler, Richard A. Gibbs, Michael S. Lawrence, Douglas Voet, Rui Jing, Kristian Cibulskis, Andrey Sivachenko, Petar Stojanov, Aaron McKenna, Eric S. Lander, Stacey Gabriel, Gad Getz, Li Ding, Robert S. Fulton, Daniel C. Koboldt, Todd Wylie, Jason Walker, David J. Dooling, Lucinda Fulton, Kim D. Delehaunty, Catrina C. Fronick, Ryan Demeter, Elaine R. Mardis, Richard K. Wilson, Andy Chu, Hye-Jung E. Chun, Andrew J. Mungall, Erin Pleasance, A. Gordon Robertson, Dominik Stoll, Miruna Balasundaram, Inanc Birol, Yaron S. N. Butterfield, Eric Chuah, Robin J. N. Coope, Noreen Dhalla, Ranabir Guin, Carrie Hirst, Martin Hirst, Robert A. Holt, Darlene Lee, Haiyan I. Li, Michael Mayo, Richard A. Moore, Jacqueline E. Schein, Jared R. Slobodan, Angela Tam, Nina Thiessen, Richard Varhol, Thomas Zeng, Yongjun Zhao, Steven J. M. Jones, Marco A. Marra, Adam J. Bass, Alex H. Ramos, Gordon Saksena, Andrew D. Cherniack, Stephen E. Schumacher, Barbara Tabak, Scott L. Carter, Nam H. Pho, Huy Nguyen, Robert C. Onofrio, Andrew Crenshaw, Kristin Ardlie, Rameen Beroukhim, Wendy Winckler, Gad Getz, Matthew Meyerson, Alexei Protopopov, Juinhua Zhang, Angela Hadjipanayis, Eunjung Lee, Ruibin Xi, Lixing Yang, Xiaojia Ren, Hailei Zhang, Narayanan Sathiamoorthy, Sachet Shukla, Peng-Chieh Chen, Psalm Haseley, Yonghong Xiao, Semin Lee, Jonathan Seidman, Lynda Chin, Peter J. Park, Raju Kucherlapati, J. Todd Auman, Katherine A. Hoadley, Ying Du, Matthew D. Wilkerson, Yan Shi, Christina Liquori, Shaowu Meng, Ling Li, Yidi J. Turman, Michael D. Topal, Donghui Tan, Scot Waring, Elizabeth Buda, Jesse Walsh, Corbin D. Jones, Piotr A. Mieczkowski, Darshan Singh, Junyuan Wu, Anisha Gulabani, Peter Dolina, Tom Bodenheimer, Alan P. Hoyle, Janae V. Simons, Matthew Soloway, Lisle E. Mose, Stuart R. Jefferys, Saianand Balu, Brian D. O’Connor, Jan F. Prins, Derek Y. Chiang, D. Neil Hayes, Charles M. Perou, Toshinori Hinoue, Daniel J. Weisenberger, Dennis T. Maglinte, Fei Pan, Benjamin P. Berman, David J. Van Den Berg, Hui Shen, Timothy Triche Jr, Stephen B. Baylin, Peter W. Laird, Gad Getz, Michael Noble, Doug Voet, Gordon Saksena, Nils Gehlenborg, Daniel DiCara, Juinhua Zhang, Hailei Zhang, Chang-Jiun Wu, Spring Yingchun Liu, Sachet Shukla, Michael S. Lawrence, Lihua Zhou, Andrey Sivachenko, Pei Lin, Petar Stojanov, Rui Jing, Richard W. Park, Marc-Danie Nazaire, Jim Robinson, Helga Thorvaldsdottir, Jill Mesirov, Peter J. Park, Lynda Chin, Vesteinn Thorsson, Sheila M. Reynolds, Brady Bernard, Richard Kreisberg, Jake Lin, Lisa Iype, Ryan Bressler, Timo Erkkilä, Madhumati Gundapuneni, Yuexin Liu, Adam Norberg, Tom Robinson, Da Yang, Wei Zhang, Ilya Shmulevich, Jorma J. de Ronde, Nikolaus Schultz, Ethan Cerami, Giovanni Ciriello, Arthur P. Goldberg, Benjamin Gross, Anders Jacobsen, Jianjiong Gao, Bogumil Kaczkowski, Rileen Sinha, B. Arman Aksoy, Yevgeniy Antipin, Boris Reva, Ronglai Shen, Barry S. Taylor, Timothy A. Chan, Marc Ladanyi, Chris Sander, Rehan Akbani, Nianxiang Zhang, Bradley M. Broom, Tod Casasent, Anna Unruh, Chris Wakefield, Stanley R. Hamilton, R. Craig Cason, Keith A. Baggerly, John N. Weinstein, David Haussler, Christopher C. Benz, Joshua M. Stuart, Stephen C. Benz, J. Zachary Sanborn, Charles J. Vaske, Jingchun Zhu, Christopher Szeto, Gary K. Scott, Christina Yau, Sam Ng, Ted Goldstein, Kyle Ellrott, Eric Collisson, Aaron E. Cozen, Daniel Zerbino, Christopher Wilks, Brian Craft, Paul Spellman, Robert Penny, Troy Shelton, Martha Hatfield, Scott Morris, Peggy Yena, Candace Shelton, Mark Sherman, Joseph Paulauskis, Julie M. Gastier-Foster, Jay Bowen, Nilsa C. Ramirez, Aaron Black, Robert Pyatt, Lisa Wise, Peter White, Monica Bertagnolli, Jen Brown, Timothy A. Chan, Gerald C. Chu, Christine Czerwinski, Fred Denstman, Rajiv Dhir, Arnulf Dörner, Charles S. Fuchs, Jose G. Guillem, Mary Iacocca, Hartmut Juhl, Andrew Kaufman, Bernard Kohl III, Xuan Van Le, Maria C. Mariano, Elizabeth N. Medina, Michael Meyers, Garrett M. Nash, Phillip B. Paty, Nicholas Petrelli, Brenda Rabeno, William G. Richards, David Solit, Pat Swanson, Larissa Temple, Joel E. Tepper, Richard Thorp, Efsevia Vakiani, Martin R. Weiser, Joseph E. Willis, Gary Witkin, Zhaoshi Zeng, Michael J. Zinner, Carsten Zornig, Mark A. Jensen, Robert Sfeir, Ari B. Kahn, Anna L. Chu, Prachi Kothiyal, Zhining Wang, Eric E. Snyder, Joan Pontius, Todd D. Pihl, Brenda Ayala, Mark Backus, Jessica Walton, Jon Whitmore, Julien Baboud, Dominique L. Berton, Matthew C. Nicholls, Deepak Srinivasan, Rohini Raman, Stanley Girshik, Peter A. Kigonya, Shelley Alonso, Rashmi N. Sanbhadti, Sean P. Barletta, John M. Greene, David A. Pot, Kenna R. Mills Shaw, Laura A. L. Dillon, Ken Buetow, Tanja Davidsen, John A. Demchok, Greg Eley, Martin Ferguson, Peter Fielding, Carl Schaefer, Margi Sheth, Liming Yang, Mark S. Guyer, Bradley A. Ozenberger, Jacqueline D. Palchik, Jane Peterson, Heidi J. Sofia, and Elizabeth Thomson.
References
- 1.The Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fearon ER. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 4.Bass AJ, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A–TCF7L2 fusion. Nature Genet. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 6.Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 7.Umar A, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aaltonen LA, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 9.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 10.Parsons R, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 11.Dooley AL, et al. Nuclear factor I/B is an oncogene in small cell lung cancer. Genes Dev. 2011;25:1470–1475. doi: 10.1101/gad.2046711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Major MB, et al. Wilms tumor suppressor WTX negatively regulates WNT/β-catenin signaling. Science. 2007;316:1043–1046. doi: 10.1126/science/1141515. [DOI] [PubMed] [Google Scholar]
- 13.Mori-Akiyama Y, et al. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology. 2007;133:539–546. doi: 10.1053/j.gastro.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Bastide P, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol. 2007;178:635–648. doi: 10.1083/jcb.200704152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012;33:100–103. doi: 10.1002/humu.21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 17.Minsky BD. Unique considerations in the patient with rectal cancer. Semin. Oncol. 2011;38:542–551. doi: 10.1053/j.seminoncol.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Hinoue T, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–282. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl Acad. Sci. USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camps J, et al. Integrative genomics reveals mechanisms of copy number alterations responsible for transcriptional deregulation in colorectal cancer. Genes Chromosom. Cancer. 2009;48:1002–1017. doi: 10.1002/gcc.20699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varley JM, Swallow JE, Brammar WJ, Whittaker JL, Walker RA. Alterations to either c-erbB-2(neu) or c-myc proto-oncogenes in breast carcinomas correlate with poor short-term prognosis. Oncogene. 1987;1:423–430. [PubMed] [Google Scholar]
- 22.Yokota J, et al. Amplification of c-erbB-2 oncogene in human adenocarcinomas in vivo. Lancet. 1986;327:765–767. doi: 10.1016/s0140-6736(86)91782-4. [DOI] [PubMed] [Google Scholar]
- 23.van der Flier LG, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–912. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 24.Jubb AM, Hoeflich KP, Haverty PM, Wang J, Koeppen H. Ascl2 and 11p15.5 amplification in colorectal cancer. Gut. 2011;60:1606–1607. doi: 10.1136/gut.2010.231746. [DOI] [PubMed] [Google Scholar]
- 25.Stange DE, et al. Expression of an ASCL2 related stem cell signature and IGF2 in colorectal cancer liver metastases with 11p15.5 gain. Gut. 2010;59:1236–1244. doi: 10.1136/gut.2009.195701. [DOI] [PubMed] [Google Scholar]
- 26.Cui H, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa H, et al. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc. Natl Acad. Sci. USA. 2001;98:591–596. doi: 10.1073/pnas.011528698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veronese A, et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010;70:3140–3149. doi: 10.1158/0008-5472.CAN-09-4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brady CA, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivera MN, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315:642–645. doi: 10.1126/science.1137509. [DOI] [PubMed] [Google Scholar]
- 32.Scheel SK, et al. Mutations in the WTX-gene are found in some high-grade microsatellite instable (MSI-H) colorectal cancers. BMC Cancer. 2010;10:413. doi: 10.1186/1471-2407-10-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forbes, S. A. et al. The catalogue of somatic mutations in cancer (COSMIC). Curr. Protoc. Hum. Genet. Ch. 10, Unit 10.11. (2008) [DOI] [PMC free article] [PubMed]
- 34.Massagué J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 35.Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.House CD, et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010;70:6957–6967. doi: 10.1158/0008-5472.CAN-10-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Lu H, Jiang Z, Pastuszyn A, Hu CA. Apolipoprotein l6, a novel proapoptotic Bcl-2 homology 3-only protein, induces mitochondria-mediated apoptosis in cancer cells. Mol. Cancer Res. 2005;3:21–31. [PubMed] [Google Scholar]
- 38.Topol L, Chen W, Song H, Day TF, Yang Y. Sox9 inhibits Wnt signaling by promoting β-catenin phosphorylation in the nucleus. J. Biol. Chem. 2009;284:3323–3333. doi: 10.1074/jbc.M808048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagl NG, Jr, Zweitzig DR, Thimmapaya B, Beck GR, Jr, Moran E. The c-myc gene is a direct target of mammalian SWI/SNF-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 2006;66:1289–1293. doi: 10.1158/0008-5472.CAN-05-3427. [DOI] [PubMed] [Google Scholar]
- 40.Chen B, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ewan K, et al. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010;70:5963–5973. doi: 10.1158/0008-5472.CAN-10-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sack U, et al. S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol. Biol. Cell. 2011;22:3344–3354. doi: 10.1091/mbc.E10-09-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xi R, et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc. Natl Acad. Sci. USA. 2011;108:E1128–E1136. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains the legends for Supplementary Tables 1-12, Supplementary Tables 1-9 and Supplementary Data files 1-2, Supplementary Methods, which include 17 Figures and 2 Tables (see Contents for details) and Supplementary Figures 1-9. (PDF 8117 kb)
This file contains Supplementary Table 2. (ZIP 25458 kb)
This file contains Supplementary Tables 1 and 3-12 (ZIP 2809 kb)
Data Availability Statement
Data deposits
dbGaP accession numbers have been provided in Supplementary Table 1.