

Abstract
To engage in proliferation, cells need to increase their biomass and replicate their genome. This process presents a substantial bioenergetic challenge: proliferating cells must increase ATP production and acquire or synthesize raw materials, including lipids, proteins and nucleic acids. To do so, proliferating cells actively reprogramme their intracellular metabolism from catabolic mitochondrial oxidative phosphorylation (OXPHOS) to glycolysis and other anabolic pathways. This metabolic reprogramming, which directs nutrient uptake and metabolism during cell activation and proliferation, is under the control of specific signal transduction pathways. The underlying molecular mechanisms of cell metabolism reprogramming and their relevance to physiology and disease are currently under intense study. Several reports have uncovered the mechanisms of metabolic reprogramming that drive high rates of cell proliferation in cancer. Some recent studies have elucidated the physiological role of metabolic reprogramming during T-cell activation, differentiation and trafficking, which are potentially relevant to inflammatory disorders. This review describes the impact of metabolic reprogramming on the pathogenesis of cancer and the physiology of T-cell-mediated immune responses, with an emphasis on the phosphatidyl inositol 3-kinase–serine/threonine kinase–mammalian target of rapamycin pathway and the recently discovered metabolic processes regulated by nuclear factor-κB. These discoveries will hopefully translate into a better understanding of the role of metabolic reprogramming as a key regulator of T-cell-mediated immune responses and offer novel, immune-based therapeutic approaches.
Keywords: glycolysis, mammalian target of rapamycin, migration, nuclear factor-κB, oxidative phosphorylation, T cell
Introduction
For more than 70 years it has been appreciated that cancer cells exhibit an altered metabolism characterized by a high rate of glucose utilization and lactate production despite the presence of sufficient oxygen to oxidize glucose carbon in the mitochondria. Recognition of this unusual metabolic shift in cancer cells stems from experiments performed by the German physiologist Otto Warburg, starting in the 1920s.1,2 The observation that cancer cells generated the majority of their ATP via glycolysis, even when grown in the presence of oxygen, led to the hypothesis that the metabolic shift toward glycolysis observed in cancer cells reflected damage to mitochondrial respiration, which resulted in aerobic glycolysis. Warburg further proposed that this metabolic change toward ‘aerobic glycolysis’ was the root cause of cancer.1,2
Appreciation of the ‘Warburg effect’ as a metabolic signature of tumour cells has grown widely ever since, and it is now well established that the majority of tumours in vivo and transformed cells in vitro exhibit elevated levels of glucose transport and high rates of glycolysis that result in an increase in the production of lactate even in the presence of oxygen.3,4 Indeed, the glycolytic activity of tumours is exploited in clinics by the use of 18fluoro-deoxyglucose positron emission tomography, which detects tumours precisely by virtue of their enhanced ability to take up and metabolize glucose compared with normal tissues.5,6
Warburg’s assumption that aerobic glycolysis is a feature only of cancer cells, however, is now known to be wrong. The Warburg effect is also observed during rapid proliferation of primary cells, and it is viewed as a general feature of anabolic metabolism contributing to cell proliferation. The same reprogramming to aerobic glycolysis is exhibited, for instance, by highly proliferating normal cells, such as activated lymphocytes.7–9
The metabolic requirements of proliferating cells
The Warburg effect is the most widely documented metabolic phenotype exhibited by tumours. As mentioned, however, aerobic glycolysis is not a feature exclusive to cancer cells because highly proliferative activated T cells and stem cells show the same metabolic reprogramming. Explaining the reason why proliferating cells with access to oxygen would deprive themselves of the majority of the ATP that can be produced from glucose metabolism via the oxidative phosphorylation (OXPHOS) pathway in the mitochondria by converting pyruvate into lactate rather than acetyl-CoA has been challenging. Two observations substantiate the importance of the role of the Warburg effect in driving cell proliferation. First, although counterintuitive, the preferential use of glycolysis over OXPHOS for ATP production enables cells to produce ATP at a faster rate. Because the energetic yield per molecule of glucose is much lower for aerobic glycolysis (4 moles ATP/mole glucose) than OXPHOS (36 moles ATP/mole glucose), glucose metabolism appears to be an inefficient metabolic pathway. When glucose is abundant, however, the pace of glycolytic flux guarantees the production of sufficient amounts of ATP to meet the metabolic requirements of proliferating cells (Fig. 1).10 In cancer, several key enzymes of the glycolytic pathway are found to be over-expressed, including hexo-kinase, phospho-fructo-kinase, and pyruvate kinase M2, as well as pyruvate dehydrogenase and lactate dehydrogenase-A, the key limiting enzymes converting the end product of glycolysis, pyruvate, to acetyl-CoA and lactate, respectively. All of these enzymes represent candidate targets for cancer therapy.10–12 Second, rapid glucose metabolism supplies intermediates for biosynthetic pathways, including ribose-5-phosphate and glycine for nucleotide biosynthesis through the pentose-phosphate pathway, and citrate for lipid synthesis through the tricarboxylic acid (TCA) cycle (Fig. 1). Indeed, cells engaged in aerobic glycolysis do not convert all of their pyruvate into lactate. Rather, a measurable fraction of pyruvate is metabolized in the TCA cycle, providing precursors for cataplerotic pathways that consume TCA cycle intermediates to produce fatty acids and amino acids. The discovery that the metabolic enzymes of the TCA cycle, succinate dehydrogenase and fumarate hydratase, function as tumour suppressors and are mutated in human cancers,13–16 thereby favouring the efflux of citrate from mitochondria to the cytosol, underscores the need for tumour cells to increase cataplerotic pathways for biosynthetic reasons. The synthesis of fatty acids requires the active export in the cytosol of the TCA cycle intermediate, citrate, which might otherwise be oxidized in the mitochondria to produce ATP, and the waste product CO2. Exported citrate becomes the substrate of the three enzymes required for fatty acid synthesis, ATP citrate lyase, acetyl-CoA carboxylase, and fatty acid synthase, which are found to be highly expressed and serve oncogenic functions in many human cancers, again representing targets for cancer therapy. Indeed, proliferating cells often display rapid synthesis of fatty acids, cholesterol and isoprenoids, because a large fraction of their membrane lipids are synthesized de novo starting from the carbon sources released downstream of glycolysis, rather than scavenged from extracellular sources.17,18
Figure 1.
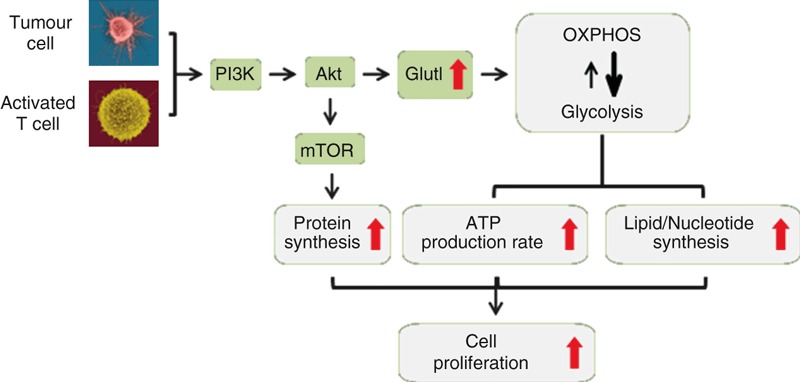
Tumour cells and activated T cells use the same metabolic pathways to support high rates of proliferation. Downstream of growth factor receptors in tumour cells and T-cell receptor (TCR) in activated T cells phosphatidyl inositol 3-kinase (PI3K) -mediated activation of the serine/threonine kinase Akt promotes glucose uptake – through up-regulation of Glut1 – and glycolysis, while oxidative phosphorylation (OXPHOS) is reduced. The preferential use of glycolysis over OXPHOS enables proliferating cells to produce ATP at a faster rate. Rapid glucose metabolism also enables proliferating cells to spare intermediates of the metabolic pathways for the biosynthesis of nucleotides and lipids. Akt also controls mammalian target of rapamycin (mTOR) activation, which promotes protein synthesis. Increased lipid, nucleotide and protein synthesis supports cell proliferation.
Cataplerosis may cause a deleterious imbalance at the level of the TCA cycle as a consequence of the withdrawal of intermediates for biosynthetic reactions. Glutamine metabolism, however, plays a vital role in the compensation of such potential imbalance in the mitochondria. Glutamine conversion to α-ketoglutarate and entry into the TCA cycle generates oxalaocetate, effectively replacing the metabolites that are removed from the cycle in cataplerotic reactions for the biosynthesis of fatty acids, nucleotides and proteins. This process, termed anaplerosis, allows proliferating cells to maintain the integrity of the TCA cycle function in the mitochondria while withdrawing intermediates for biosynthetic reactions. Furthermore, partial oxidation of glutamine to lactate (glutaminolysis) uses the cytosolic malic enzyme and provides cells with NADPH for the reductive reactions of fatty acid and nucleotide biosynthesis. The discovery of activating mutations in isocitrate dehydrogenase and glutaminase in human cancers further supports the evidence that tumour cells actively increase anaplerotic pathways for biosynthetic needs.19–22
The metabolic demands of T-lymphocyte activation
T cells switch between highly proliferative states (i.e. developing thymocytes and antigen-activated T cells) and quiescent states (i.e. naive, memory and anergic T cells). These fates are regulated by signals that, once delivered through T-cell receptors (TCR) and cytokine receptors, induce the activation of different intracellular metabolic pathways.7–9
Resting T cells are dependent on interleukin-7 for survival and derive most of their ATP from OXPHOS. This catabolic metabolism supports their low rate of energy metabolism, which is required to maintain housekeeping functions, such as ion transport and membrane integrity. This same form of metabolism also keeps resting T cells from engaging in cell proliferation. In the absence of extrinsic signals that are necessary to sustain homeostatic processes, resting T cells cannot take up enough extracellular nutrients to maintain themselves. The resulting bioenergetic decline causes apoptosis or metabolic crisis.23,24
Upon stimulation, T cells undergo conversion from a quiescent state to one of active growth. Anabolic processes such as protein and lipid synthesis are increased, whereas catabolic processes including β-oxidation are actively suppressed. One hallmark of this metabolic conversion is that, despite adequate oxygen to support complete oxidation of glucose, cellular ATP production switches from OXPHOS to high-throughput glycolysis,9 the same switch to aerobic glycolysis already discussed for cancer cells,10 which also supports energy production and biosynthetic programmes during T-cell activation (Fig. 1). Indeed, mitogenic stimulation of thymocytes or naive T cells induces an almost 20-fold increase in glucose uptake within 1 hr.25 Productive T-cell activation is known to require two signals: an antigen-specific signal induced by the TCR and a co-stimulatory signal delivered by the surface receptor CD28. These two signals promote not only the release of interleukin-2 but also the induction of glycolytic metabolism.26 A similar co-stimulus is required for full activation of B and natural killer cells, in which it is initiated upon engagement of CD19 and DAP10, respectively.27,28 Activation of TCR along with CD28 co-stimulation triggers the activity of phosphatidylinositol 3′-kinase (PI3K), which leads to the activation of the serine-threonine kinase Akt. This kinase promotes glucose metabolism by stimulating the localization of the glucose transporter Glut1 to the plasma membrane of T cells, so facilitating increased glucose uptake. In addition, Akt stimulates the activity of hexokinase and phosphofructokinase, two rate-limiting enzymes of the glycolytic pathway (Fig. 1). Interestingly, the effects of Akt on glucose metabolism in lymphocytes are antagonized by the inhibitory receptor cytotoxic T-lymphocyte antigen-4, suggesting that antagonists of T-cell activation may function in part by disrupting glucose metabolism.9,24,26,29
T-cell activation is not only accompanied by increased glycolytic metabolism, but also by high rates of protein synthesis that support cell growth and effector functions. Downstream of TCR and CD28, Akt controls the activation state of the mammalian Target of Rapamycin (mTOR) a key regulator of protein synthesis in T cells (Figs 1 and 2).7,8 The mTOR modulates the rate of protein synthesis by regulating both the availability of amino acids and the process of cap-dependent translation. Phosphorylation of components of the translational machinery (i.e. the translation inhibitor 4E-BP1, the translation initiation factor EIF2B, and the ribosomal p70 S6 kinase) by mTOR promotes the initiation of cap-dependent translation.30 Rapamycin, a potent inhibitor of mTOR, suppresses cap-dependent protein translation. The importance of the mTOR pathway for T-cell activation is underscored by the fact that rapamycin causes immunosuppression via the induction of a G1 cell-cycle arrest in proliferating T lymphocytes.7
The role of mTOR in T-cell differentiation
Besides its well-established role in T-cell activation and proliferation, mTOR has recently been shown to serve a crucial function in the induction of anergy, in determining the differentiation fate of CD4+ T cells into inflammatory and regulatory subsets, in the development of CD8+ memory T cells and in the regulation of T-cell migration patterns (Fig. 2).7,8
Figure 2.
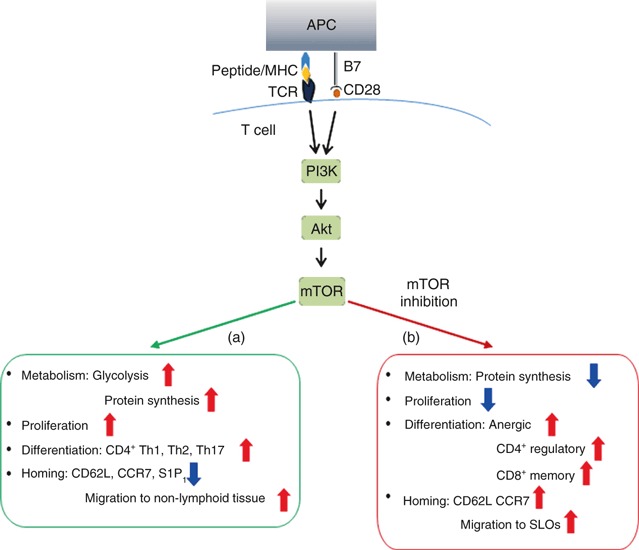
The mammalian target of rapamycin (mTOR) pathway regulates T-cell activation, differentiation and migration. (a) Downstream of T-cell receptor (TCR) and CD28, phosphatidyl inositol 3-kinase (PI3K) leads to activation of the serine/threonine kinase Akt, which subsequently controls mTOR activity. This signalling cascade promotes the glucose metabolism and protein synthesis necessary for T-cell activation, proliferation and differentiation into CD4+ T helper type 1 (Th1), Th2 and Th17 subsets. The PI3K–Akt–mTOR axis also promotes down-regulation of secondary lymphoid organ (SLO) homing receptors CD62L, CCR7 and S1P1. Antigen experienced T cells, therefore, home to their respective non-lymphoid tissues. (b) Inhibition of mTOR via rapamycin or genetic deletion reduces protein synthesis and T-cell proliferation, and promotes differentiation toward anergic, CD4+ regulatory T (Treg) cell and CD8+ memory cell subsets. Rapamycin-mediated inhibition of mTOR causes T effector cells to re-express CD62L and CCR7 and home to secondary lymphoid organs.
It is well established that lack of co-stimulation via CD28 during the primary exposure to antigen results in suboptimal activation of the Akt-mTOR pathway, leading to T-cell anergy.31 Recent data, however, have reinforced this notion by showing that the anergic status in T cells is critically the result of a failure of the Akt–mTOR pathway, in the absence of an appropriate co-stimulus, to up-regulate nutrient transporters and activate glycolytic pathways. Furthermore, interfering with leucine, glucose and energy metabolism via the use of N-acetyl-leucine amide, 2-deoxyglucose and 5-aminoimidazole-4-carboxamide ribonucleoside was sufficient to induce anergy even in the presence of an appropriate co-stimulatory signal.32
Delgoffe et al.33 have shown that mTOR-deficient CD4+ T cells fail to differentiate into T helper type 1 (Th1), Th2 and Th17 subsets upon activation. Rather they differentiate towards a Foxp3+ regulatory (Treg) phenotype. These results are in agreement with other studies showing that rapamycin prevents the differentiation of Th17 cells while favouring the development of Treg cells.34,35 Altogether these observations highlight the importance of the mTOR pathway in the differentiation of mature CD4+ helper T cells.
With regard to CD8+ T cells, recent reports indicate that inhibition of the mTOR pathway via rapamycin promotes the differentiation of highly functional memory CD8+ T cells.36,37
Hence, mTOR signals orchestrate the commitment to distinct differentiation pathways by CD4+ and CD8+ T lymphocytes. Inhibition of mTOR in naive CD4+ T cells promotes the generation of regulatory T cells whereas inhibition of mTOR in naive CD8+ T cells promotes the generation of long-lived memory T cells. Although regulatory and memory T cells are functionally diverse, their metabolic demands are similar and less than those of effector CD4+ and CD8+ T cells.
The leptin–mTOR metabolic axis in the control of T-cell-mediated immunity
Leptin is a cytokine-like hormone that controls food intake, metabolism and T-cell function. The effects of leptin on adaptive immune responses have been extensively investigated in the context of CD4+ T cells where it potentiates pro-inflammatory Th1 immune responses while constraining Treg-cell-mediated immunosuppression.38 Freshly isolated human Treg cells produce high levels of leptin and constitutively express the leptin receptor (ObR) on their surface, and the leptin pathway acts as a negative signal for their proliferation. In vitro antibody-mediated leptin neutralization during anti-CD3 and anti-CD28 stimulation, however, reverses the anergic status of Treg cells and leads to proliferation. These findings are in agreement with the increased rates of Treg proliferation found in leptin-deficient and ObR-deficient mice.39 The anergic status of Treg cells depends on the constitutively elevated activity of the mTOR pathway downstream of leptin signals. A transient inhibition of mTOR with rapamycin before TCR stimulation reverses the anergic status of Treg cells, which become highly proliferative even in the absence of interleukin-2. In vivo transient inhibition of mTOR with rapamycin ameliorates the onset and progression of autoimmune encephalomyelitis. These effects are associated with an increased frequency of Treg cells that precedes the peak of the disease in mice pre-treated with rapamycin.40 These observations may explain why individuals with low levels of leptin are prone to infections but show reduced incidence of autoimmunity. Furthermore, they suggest a way forward to manipulate Treg cells through control of metabolism to achieve high levels of proliferation and ultimately promote immunosuppression.
Is there a metabolic control of T-cell migration?
Early studies claimed a role for oxidative and glucose metabolism in the control of polymorphonuclear leucocyte (PMN) migration. Ischaemia is a recurrent feature of pathological events, such as myocardial infarction, leading to severe reduction of the cellular oxygen supply and consequently of the intracellular oxidative metabolism. The rapid recruitment of PMN in the ischaemic tissues is one of the most important causes of ischaemia/reperfusion injury. Exposure of PMN to hypoxia enhances the expression of the β2 integrins, lymphocyte function-associated antigen-1 (LFA-1) and macrophage-1 antigen (Mac-1), which increase PMN adhesiveness for intercellular adhesion molecule-1, the major endothelial ligand for β2 integrins.41,42
Mutations in the glucose-6-phosphate transporter cause glycogen storage disease type Ib as a result of disrupted glucose homeostasis. Patients with glycogen storage disease type Ib show immunodeficiency that is at least partly the result of an impairment of mobility and chemotaxis by PMN.43
Some recent evidence suggests that the metabolic status of T cells can influence their homing patterns. Expression of the adhesion molecule CD62L (also known as l-selectin) and the chemokine receptors CC-chemokine receptor 7 (CCR7) and sphingosine-1-phosphate receptor 1 (S1P1) on the surface of naive T cells facilitates their trafficking to secondary lymphoid organs. Upon TCR engagement, the PI3K–Akt–mTOR ‘metabolic’ axis promotes the down-regulation of CD62L, CCR7 and S1P1, and prompts effector T cells expressing adhesion molecules (such as very late antigen 4 and ligands for P-selectin and E-selectin) and chemokine receptors (such as CXCR3 and CCR5) to traffic to the sites of inflammation.44 Rapamycin-mediated inhibition of mTOR causes effector T cells to re-express CD62L and CCR7, and home to secondary lymphoid organs where they are trapped away from the target cells in the periphery.44,45 Therefore, rapamycin can promote immunosuppression also by redirecting effector T cells from peripheral tissues to secondary lymphoid organs (Fig. 2).
The direct effect of metabolism on the ability of T cells to migrate into and exit from their homing sites, however, has never been investigated. This is surprising considering that the well-established control of T-cell migration by TCR and co-stimulators46–48 implies that metabolic changes induced by these receptors can influence the efficiency and topography of T-cell trafficking. The metabolic machinery is also likely to directly affect and be affected by T-cell migratory events, as T cells continuously re-circulate between different microenvironments (e.g. blood, lymphoid tissues and peripheral tissues) in which they are exposed to different nutrient availability and oxygen tension, and must adapt their metabolic pathways to effectively mediate immune responses. Here, we have highlighted the role of the mTOR pathway as a common denominator of these events. This field is mostly unexplored at present, and represents a fascinating area for future research, to gain a better understanding of the physiology of T-cell trafficking and the pathological mechanisms leading to T-cell-mediated inflammation.
The control of cell metabolism by nuclear factor-κB transcription factors
Transcription factors of the nuclear factor-κB (NF-κB)/Rel family control immunity, inflammation and cell survival.49 Much less is known about the control of metabolic circuitries by NF-κB in physiological contexts and disease. A recent study identified a link between p53, NF-κB and glycolysis.50 In this study, NF-κB activity was found to be enhanced in p53-deficient primary cells. Activation of NF-κB, by loss of p53, caused an increase in the rate of aerobic glycolysis via up-regulation of glucose transporter Glut3, a mechanism that was suggested to be the basis for the increased Ras-induced transformation of p53-deficient cells.
Mauro et al.51 have identified an additional metabolic function for NF-κB as a physiological regulator of mitochondrial respiration, and have established that this function of NF-κB suppresses the metabolic reprogramming to aerobic glycolysis in cells and prevents necrosis upon nutrient starvation. This metabolic function of NF-κB involves the p53-dependent up-regulation of mitochondrial Synthesis of Cytochrome c Oxidase 2 (SCO2), which increases OXPHOS and reduces glycolytic flux in cells.
However, NF-κB may control metabolism also via additional, p53-independent mechanisms. Future studies will unveil these additional metabolic functions of NF-κB and determine their significance in physiological and pathogenic contexts. Although much has been discovered about the role of metabolism reprogramming in lymphocyte functions, whether the metabolic activities regulated by NF-κB are relevant to its control of the immune response is presently not known.
Conclusions
Changes in metabolism and bioenergetics have emerged as a common feature in cancer. The switch to aerobic glycolysis, particularly, benefits both growth and survival of tumours in vivo. On the other hand, this metabolic switch renders cancer cells ‘addicted’ to glucose. The inhibition of glycolysis is therefore likely to hit cancers much harder than normal tissues. The inhibitors of glycolysis, 3-bromopyruvate and 2-deoxyglucose, for instance, have been shown to cause the preferential killing of cancer cells.52 Additionally, drugs targeting key metabolic control points important for aerobic glycolysis, such as pyruvate kinase-M2 or lactate dehydrogenase-A, warrant investigation as potential cancer therapeutics.10
In addition, drugs developed for the treatment of metabolic diseases have shown efficacy in the cure of cancer. A number of retrospective clinical studies have established that the widely used type 2 diabetes drug, metformin, and the more potent metformin-related compound, phenformin, provide benefits in cancer prevention as well as improved outcomes when used with other cancer therapies.53,54 These evidences support the view that cancer and metabolic diseases, such as obesity, diabetes and atherosclerosis, share a similar metabolic signature. Interestingly in this regard, different metabolic diseases often develop in the same patient: obese individuals, for instance, have higher risk to develop type 2 diabetes, and insulin-resistance is frequently associated with cardiovascular disease such as atherosclerosis and hypertension, and with cancer.55–57 Pro-inflammatory T cells have been shown to play major roles in the pathogenesis of all these metabolic diseases and in cancer.57–62 Identifying the molecular links between metabolism control and T-cell-mediated immune responses may therefore lead us to a better understanding of the pathogenesis of metabolic diseases and the development of novel, effective therapeutic strategies.
Acknowledgments
C.M. is supported by the British Heart Foundation. F.M.M-B. is supported by the British Heart Foundation, the Medical Research Council of the UK and the Gates Foundation.
Disclosures
The authors declare no conflict of interest.
References
- 1.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269. [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 4.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatzivassiliou G, Zhao F, Bauer DE, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 6.Jadvar H, Alavi A, Gambhir SS. 18F-FDG uptake in lung, breast, and colon cancers: molecular biology correlates and disease characterization. J Nucl Med. 2009;50:1820. doi: 10.2967/jnumed.108.054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Rios F, Sanchez-Arago M, Garcia-Garcia E, et al. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- 8.Peter C, Waldmann H, Cobbold SP. MTOR signalling and metabolic regulation of T cell differentiation. Curr Opin Immunol. 2010;22:655. doi: 10.1016/j.coi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 13.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 16.Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 17.Frezza C, Zheng L, Folger O, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 18.Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer. 2009;100:1369. doi: 10.1038/sj.bjc.6605007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 24.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 25.Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem. 1994;269:31484. [PubMed] [Google Scholar]
- 26.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 27.Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol. 2011;23:178. doi: 10.1016/j.coi.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol. 2003;33:2223. doi: 10.1002/eji.200324048. [DOI] [PubMed] [Google Scholar]
- 30.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wells AD. New insights into the molecular basis of T cell anergy: anergy factors, avoidance sensors, and epigenetic imprinting. J Immunol. 2009;182:7331. doi: 10.4049/jimmunol.0803917. [DOI] [PubMed] [Google Scholar]
- 32.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol. 2009;183:6095. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 35.Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol. 2007;7:1819. doi: 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4:371. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 39.De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, La Cava A, Matarese G. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26:241. doi: 10.1016/j.immuni.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Procaccini C, De Rosa V, Galgani M, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madjdpour C, Jewell UR, Kneller S, et al. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284:L360. doi: 10.1152/ajplung.00158.2002. [DOI] [PubMed] [Google Scholar]
- 42.Montoya MC, Luscinskas FW, del Pozo MA, Aragones J, de Landazuri MO. Reduced intracellular oxidative metabolism promotes firm adhesion of human polymorphonuclear leukocytes to vascular endothelium under flow conditions. Eur J Immunol. 1997;27:1942. doi: 10.1002/eji.1830270818. [DOI] [PubMed] [Google Scholar]
- 43.Chen LY, Shieh JJ, Lin B, et al. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum Mol Genet. 2003;12:2547. doi: 10.1093/hmg/ddg263. [DOI] [PubMed] [Google Scholar]
- 44.Sinclair LV, Finlay D, Feijoo C, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9:513. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. 2011;11:109. doi: 10.1038/nri2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cernuda-Morollon E, Millan J, Shipman M, Marelli-Berg FM, Ridley AJ. Rac activation by the T-cell receptor inhibits T cell migration. PLoS ONE. 2010;5:e12393. doi: 10.1371/journal.pone.0012393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jarmin SJ, David R, Ma L, et al. T cell receptor-induced phosphoinositide-3-kinase p110delta activity is required for T cell localization to antigenic tissue in mice. J Clin Invest. 2008;118:1154. doi: 10.1172/JCI33267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider H, Downey J, Smith A, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313:1972. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 49.Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 50.Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- 51.Mauro C, Leow SC, Anso E, et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011;13:1272. doi: 10.1038/ncb2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613. [PubMed] [Google Scholar]
- 53.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cuzick J, DeCensi A, Arun B, et al. Preventive therapy for breast cancer: a consensus statement. Lancet Oncol. 2011;12:496. doi: 10.1016/S1470-2045(11)70030-4. [DOI] [PubMed] [Google Scholar]
- 55.Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-β and NF-κB. Nat Med. 2011;17:883. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 57.Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 59.Lahoute C, Herbin O, Mallat Z, Tedgui A. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat Rev Cardiol. 2011;8:348. doi: 10.1038/nrcardio.2011.62. [DOI] [PubMed] [Google Scholar]
- 60.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 61.Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10:203. doi: 10.1016/j.coph.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schiffrin EL. T lymphocytes: a role in hypertension? Curr Opin Nephrol Hypertens. 2010;19:181. doi: 10.1097/MNH.0b013e3283360a2e. [DOI] [PubMed] [Google Scholar]