

Abstract
Copy number variations (CNV) within the genome are extremely abundant. In this closeup, Canales and Walz discuss how CNV are associated with normal variation, genomic disorders, genome evolution, adaptive traits and how the use of a novel screen described by Ermakova et al in this issue that is designed to identify human disease-relevant phenotypes associated with CNV in the mouse can help elucidating susceptibility or predisposition to diseases loci.
Keywords: CNV susceptibility, gene dosage, phenotypic variability
It has been 20 years since it was first reported that a genetic, autosomal dominant neurodegenerative disorder was not caused by small alterations of the coding deoxyribonucleic acid (DNA) sequence, but by genomic rearrangements and gene dosage effects (Lupski et al, 1991). Specifically, J. R. Lupski and co-workers identified a duplication of a 3 cM interval on human chromosome 17p12 in members of multiple families presenting with the Charcot–Marie–Tooth neuropathy type A (CMT1A) disorder. Shortly after, the gene coding for peripheral myelin protein 22 (PMP22), a component of myelin, was identified as the dosage sensitive one within the duplicated ∼1.4 megabases (Mb) region responsible for the clinical presentation of the disease (Valentijn et al, 1992). Interestingly, the reduced gene dosage of PMP22 is the most common genetic cause of hereditary neuropathy with liability to pressure palsies (HNPP).
Besides CMT1A and HNPP, we now know many examples of genomic disorders caused by reciprocal microdeletion or microduplication of specific genomic regions (or copy number variations, CNV), i.e. DiGeorge syndrome (22q11.2 deletion) and 22q11.2 duplication syndrome, Williams–Beuren syndrome (7q11.23 deletion) and 7q11.23 duplication syndrome, Smith–Magenis (17p11.2 deletion) and Potocki–Lupski (17p11.2 duplication) syndromes, as well as Miller–Dieker lissencephaly (17p13.3 deletion) and (17p13.3) duplication syndromes, among several others. For some of these syndromes, a positive correlation between an excess or a deficiency in the gene dosage of one particular gene within the affected genomic interval with a certain phenotype was already identified (Zhang et al, 2009). However, at present, we do not know what proportion of genetic disease is caused by CNVs.
Generally, CNVs can either be inherited or caused by de novo mutations and can encompass from one kilobase up to several megabases in size. Several molecular mechanisms are responsible for the occurrence of CNV within the genome such as non-allelic homologous recombination, non-homologous end-joining, retrotransposition (mostly of L1 elements) as well as fork stalling and template switching. This can lead to either too many or too few dosage sensitive genes, which might result in phenotypic variability, complex behavioural traits and disease susceptibility.
Interestingly, CNVs have not only been associated with disease, but also with genome evolution and adaptive traits. The AMY1 gene, which encodes a protein that catalyzes the first step in digestion of dietary starch and glycogen, constitutes an interesting example. It has been found that the copy number of this gene is three times higher in humans compared to chimpanzees, suggesting that humans were favoured in the gene dosage due to a concomitant increase of starch consumption. Moreover, in this study, the authors found a positive correlation between the increase in AMY1 copy number and expression levels of salivary amylase protein (Perry et al, 2007). Thus, in terms of evolution, changes in the gene dosage of certain genes or regions, especially duplications, may act as a reservoir for accumulating changes, which in the end, may produce adaptive phenotypes.
Copy number variations are found in the apparently healthy population. Today, thanks to the advances in genome analysis platforms, including comparative genomic hybridization arrays, single nucleotide polymorphism genotyping platforms and next-generation sequencing techniques, the number of identified CNVs is rapidly increasing. So far, over 66,000 CNVs of approximately 16,000 loci have been identified in the apparently healthy population (Database of Genomics Variants, http://projects.tcag.ca/variation). Moreover, it was found that any individual in average carries ∼1000 CNV ranging from 443 bp to 1.28 Mb, with a median size of 2.9 kb (Conrad et al, 2010). The amount of CNVs mentioned sounds impressive. But what are the consequences of having those CNVs? In other words, does CNV mean disease?
Despite the extreme complexity of CNV and related outcomes, we can now, also with the help of mouse models, partially answer these questions. One important aspect herein is which gene within a genomic rearrangement is responsible for the final phenotype. An example of how this can be answered was reported for the Potocki–Lupski syndrome (PTLS) using the Retinoic acid inducible (Rai1) knock-out mouse. An analysis of compound heterozygous mice carrying a duplication in one chromosome 11 along with a null allele of Rai1 in the other chromosome 11 homologue showed that normal disomic Rai1 gene dosage was sufficient to rescue the complex physical and behavioural phenotypes observed in the PTLS mouse model, despite altered trisomic copy number of the other 18 genes present in the rearranged genomic interval. These data provided a model for variation in copy number of single genes that could account for the syndromic clinical presentation and also influence common traits such as obesity and behaviour (Walz et al, 2006). Another important question is the relative contribution of structural changes and gene dosage alterations on phenotypic outcomes. To answer this, phenotypes of wild type mice were compared with balanced 2n compound heterozygous mice carrying deletion/duplication copies of the Smith–Megenis syndrome (SMS)/PTLS genomic region. Interestingly, the presence of a genomic structural change, as well as gene dosage imbalance, contributes to the ultimate phenotype (Ricard et al, 2010).
Importantly, today we can assert that many CNVs, which affect specific genes and chromosomal regions, can lead to susceptibility and predisposition to certain diseases such as HIV, lupus, nephritis, pancreatitis and psoriasis among many other phenotypes. However, often the simple gene dosage difference cannot explain a certain difference in phenotype. It has been shown, for example that individuals who carry the same dosage for a particular gene or region, for instance within an affected family, show differences in the manifestation of the investigated phenotype. This has been demonstrated for a 520 kb microdeletion in 16p12.1 which was identified to be non-syndromic, associated with variable phenotypes and inherited from a parent in 95% of the cases (Girirajan et al, 2010a). Within the affected family, the carrier parents showed subclinical manifestations of mild neurosychiatric illness, while the proband was affected to a higher degree. Further studies demonstrated that the differences in the phenotypic variability were due to the presence of an additional large deletion or duplication (second hit) in the probands that resulted in a sensitized genetic background and consequently a more pronounced phenotype. These findings were later summarized in the Two Hit Model for phenotypic variability (Girirajan et al, 2010a,b), which suggests that one hit is sufficient to induce some features of a given phenotype while the second hit pushes towards a more severe manifestation. In this case, the first hit is represented by the 520 kb microdeletion and the second hit is the additional large deletion or duplications observed in the probands' genomes. Thus, the overall number of CNVs in the genome can determine different sensitized genomic backgrounds, which result in different disease outcomes.
»How can we show that a particular CNV can predispose to specific traits…«
How can we show that a particular CNV can predispose to specific traits when no clear clinical manifestation is visible? How can we prove the second-hit hypothesis? One elegant way to answer these questions is described in the present issue of EMBO Molecular Medicine by Ermakova et al (2011) (DOI 10.1002/emmm.201000112). The authors propose and validate a method to identify genes involved in disease susceptibility through phenotypic analysis of mice carrying a large chromosomal rearrangement that are subjected to different ‘second hits’ that can be genetic or environmental. The analyzed cohort of mice carries a 0.8 Mb duplication [Dp11(1)) or its reciprocal deletion (Df11(1)] together with a ApoEKO/+ allele (for four out of five assayed phenotypes). The authors studied the effect of CNVs on both baseline and challenge-evoked phenotype in five broad therapeutic areas using the following challenges: high fat diet for metabolic syndrome, antigen-induced contact hypersensitivity for immune function, the genomic challenge by ApoEKO for evaluating cardiovascular function, novelty exposure and special learning for evaluating behaviour traits and finally ApcMin knock out for cancer susceptibility. They found that the phenotypes observed would not have been detected without the challenges, which underlines the critical importance of using multiple sensitizers in the screening. The authors demonstrated that the described genomic rearrangements on mouse chromosome 11 Dp11(1) or its reciprocal deletion Df11(1) alter the susceptibility to multiple disease relevant phenotypes, that are actually manifested when challenged by a ‘second hit’ (genetic or environmental) (Fig 1).
Figure 1. Copy number variations (CNV) result in subtle differences in genomic background, which can lead to phenotypic variability.
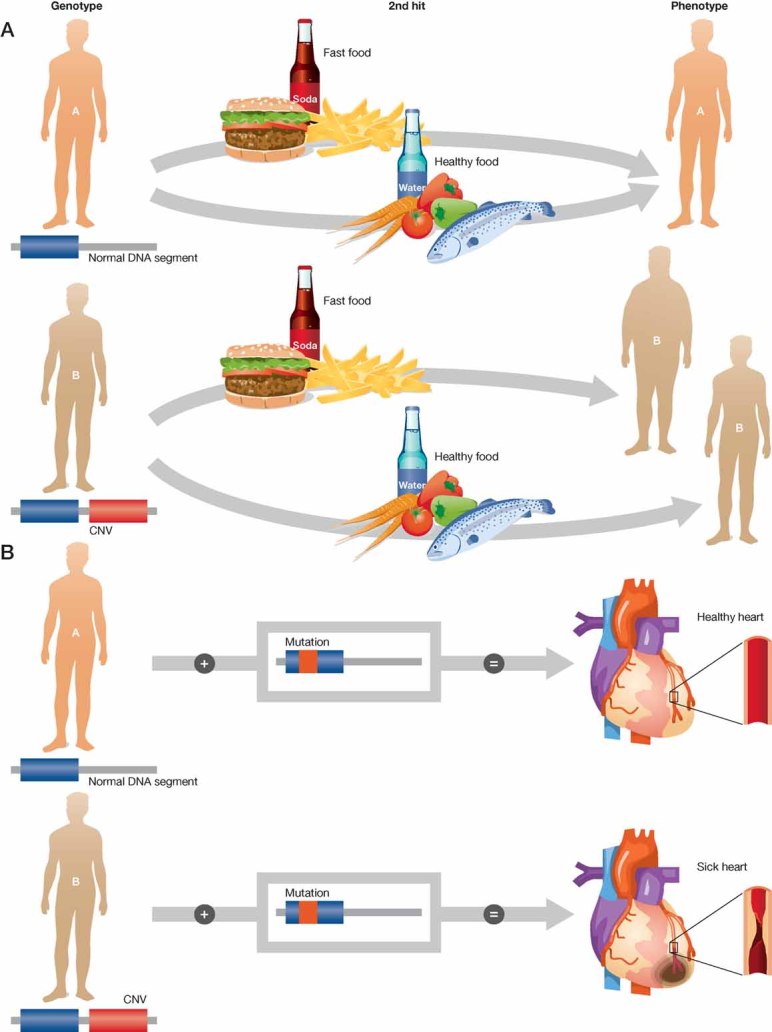
The presence of CNV (first hit) can determine sensitized backgrounds, which set different thresholds for disease development. Not only a genetic second hit can trigger a phenotype, also environmental ‘second hits’ or challenges can activate a set of phenotypes and complex traits.
- Environmental second hit: an individual carries a CNV, which predisposes to obesity under certain environmental conditions. The phenotype is only revealed after challenge (high fat diet).
- Genetic second hit: an individual carries a CNV, which predisposes for the development of a cardiovascular disease. Both individuals have a mutated allele for a gene that predisposes them to a cardiovascular disease, but only the one with the two genetic modifications will develop a cardiovascular phenotype.
»…identify genes involved in disease susceptibility…«
Certainly CNVs, together with many more changes that are present in our genome, make all of us different and susceptible to particular traits. Particular genetic combinations will prevent us from developing an undesired phenotype while others will predispose us to them. The complexity is enormous, and the challenge to understand variability and susceptibility to complex traits is just beginning.
Acknowledgments
The authors declare that they have no conflict of interest.
Footnotes
See related article in EMBO Molecular Medicine by Olga Ermakova et al: http://dx.doi.org/10.1002/emmm.201000112
References
- Conrad D, et al. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakova O, et al. EMBO Mol Med. 2011;3:xx. doi: 10.1002/emmm.201000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, et al. Nat Genet. 2010a;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, et al. Hum Mol Genet. 2010b;19:R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, et al. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Perry GH, et al. Nat Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard G, et al. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000543. e1000543. DOI:10.1371/journal.pbio.1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn LJ, et al. Nat Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- Walz K, et al. J Clin Invest. 2006;116:3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, et al. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]