

Abstract
The identification of susceptibility genes for human disease is a major goal of current biomedical research. Both sequence and structural variation have emerged as major genetic sources of phenotypic variability and growing evidence points to copy number variation as a particularly important source of susceptibility for disease. Here we propose and validate a strategy to identify genes in which changes in dosage alter susceptibility to disease-relevant phenotypes in the mouse. Our approach relies on sensitized phenotypic screening of megabase-sized chromosomal deletion and deficiency lines carrying altered copy numbers of ∼30 linked genes. This approach offers several advantages as a method to systematically identify genes involved in disease susceptibility. To examine the feasibility of such a screen, we performed sensitized phenotyping in five therapeutic areas (metabolic syndrome, immune dysfunction, atherosclerosis, cancer and behaviour) of a 0.8 Mb reciprocal chromosomal duplication and deficiency on chromosome 11 containing 27 genes. Gene dosage in the region significantly affected risk for high-fat diet-induced metabolic syndrome, antigen-induced immune hypersensitivity, ApoE-induced atherosclerosis, and home cage activity. Follow up studies on individual gene knockouts for two candidates in the region showed that copy number variation in Stat5 was responsible for the phenotypic variation in antigen-induced immune hypersensitivity and metabolic syndrome. These data demonstrate the power of sensitized phenotypic screening of segmental aneuploidy lines to identify disease susceptibility genes.
Keywords: disease susceptibilty, gene dosage, human 17q21.2 region, phenotypic analysis
INTRODUCTION
Common diseases such as metabolic syndrome, heart disease, and mental illness are complex disorders with both genetic and environmental risk factors. Genome-wide association studies confirm that a significant fraction of genetic risk for common disorders derives from many subtle genetic variants with small effect sizes (Manolio & Collins, 2009). However, a significant portion of the heritability observed for common diseases cannot be explained by additive genetic effects and alternative sources of genetic and epigenetic heritability, including gene–gene and gene–environment interactions, rare large-effect variants, somatic mutations, and epigenetic modifications are being actively investigated (Knight, 2009).
Although the majority of genetic variation in mammals occurs in the form of single nucleotide polymorphisms (SNPs), it is becoming increasingly clear that structural variants, including translocations, inversions and copy number variants (CNVs, nucleotide duplication or deficiency, also called segmental aneuploidy) are a major source of phenotypic variation (Beckmann et al, 2007, 2008). Recent data reveal a high frequency of small and medium sized CNVs (<100 kb) in both the human and mouse genomes and a smaller, but significant number of large CNVs that are likely to change the copy number of one or more entire genes (>100 kb). At least 65–80% of humans harbour a large CNV, with 5–10% of individuals carrying a variant >500 kb and 1% harbouring a CNV >1 Mb (Itsara et al, 2009). Importantly, although large CNVs were originally studied as determinants of rare Mendelian disorders (Stankiewicz & Lupski, 2010), increasingly they are associated with altered risk for common diseases, including HIV infection, lupus, Crohn's disease, chronic pancreatitis, autism spectrum disorder, Alzheimer's, and Parkinson's (Beckmann et al, 2007). Unlike most non-synonymous genetic variation, large CNVs are likely to incur disease risk via predictable changes in gene dosage that are relatively straightforward to model in genetically tractable organisms.
The mouse has emerged as the premier model organism to study the genetics of human disease. Gene targeting in embryonic stem (ES) cells allows virtually unrestricted manipulation of the mouse genome, including the engineering of large segmental aneuploidies (Liu et al, 1998; Ramirez-Solis et al, 1995) and many pathologies relevant to human disease can be induced in the mouse using environmental, pharmacological or genetic sensitization protocols.
Systematic production and phenotypic screening of single gene null mutations in the mouse are currently ongoing and promise to reveal important information about gross gene function and physiology (Austin et al, 2004; Auwerx et al, 2004; Brown et al, 2005; Friedel et al, 2007). However, because phenotyping of individual knockout lines is expensive and time consuming present screens are aimed at uncovering phenotypes with high penetrance and expressivity and are likely to miss susceptibility phenotypes that require sensitized screening. One way to increase the efficiency of screening is to manipulate more than one gene in a single individual, a strategy employed in chemical mutagenesis (∼100–1000 genes/individual) and deficiency screens (∼10–100 genes/individual) (Michaud et al, 2005; Rinchik et al, 2002). In non-mammalian species deficiency screening has been a routine method to rapidly identify genetic modifiers in sensitized backgrounds (Deutschbauer et al, 2002; Lindsley et al, 1972; Steinmetz et al, 2002). Although such an approach has been proposed for the mouse (Liu et al, 1998) and the necessary chromosome engineering technology to produce deficiencies and duplications in vitro (Adams et al, 2004; Ramirez-Solis et al, 1995) and in vivo (Herault et al, 1998; Spitz et al, 2005; Wu et al, 2007) exist and has been successfully used to identify genes affected by the gene dosage causing rare genomic disorders (Bi et al, 2007; Carmona-Mora et al, 2009; Merscher et al, 2001; Molina et al, 2008), the systematic screening of mouse lines with the aim to identify genes modulating susceptibility to common diseases has not been reported. Here we demonstrate the power of sensitized phenotypic screening of segmental aneuploidy lines to uncover genes where dosage (1:2:3 copies) moderates susceptibility to environmentally and genetically induced disease-related phenotypes. In the first tier screen, we developed and applied an unbiased phenotyping screen focusing on five therapeutic areas (metabolic syndrome, immune dysfunction, atherosclerosis, cancer, and behaviour) to identify gene dose-dependent phenotypic changes in reciprocal 0.8 Mb deficiency (Df11[1]/+) and duplication (Dp11[1]/+) lines on chromosome 11 containing 27 genes (Liu et al, 1998). This region was chosen because it shows near-perfect synteny with human chromosome 17q21 and contains a cluster of genes with known roles in human disease (e.g. HAP1, JUP1, NAGLY, HCRT, STAT3, STAT5) (Hwa et al, 2005; Kofoed et al, 2003; McKoy et al, 2000; Metzger et al, 2008; Minegishi et al, 2007; Thannickal et al, 2000). In addition, the syntenic 17q21 locus has been associated with susceptibility to several human diseases, such as Crohn's disease (Barrett et al, 2008), non-alchoholic liver disease (Sookoian et al, 2008), and tuberculosis (Jamieson et al, 2004). Gene dose-dependent phenotypes were identified in antigen-induced contact hypersensitivity (CHS), white blood cell and CD8+ T cell counts, glucose tolerance, high-fat diet-induced cholesterol and body fat, ApoE-induced atherosclerosis, anxiety, and home cage activity. In the second tier screen we tested single and compound heterozygous null alleles in two candidate genes residing in the segmental aneuploidy region and found Stat5ab to be responsible for the antigen-induced CHS, white blood cells and CD8+ T cell count, and glucose homeostasis phenotypes. These data demonstrate the potential of unbiased sensitized screening of segmental aneuploidy lines to identify susceptibility genes for common disease phenotypes in the mouse.
RESULTS
Sensitized phenotyping screen
Our screen was aimed at capturing the effect of copy number variation on both baseline and challenge-evoked phenotypes in five therapeutic areas: behaviour (novelty exposure and spatial learning), immune function (antigen-induced CHS), metabolic function (high-fat diet), cardiovascular function (ApoeKO-induced atherosclerosis), and cancer (ApcMin-induced intestinal neoplasia; Fig 1). Sensitized phenotyping assays were a critical feature of our screen and served to expose disease-relevant gene function not revealed under baseline conditions. The two genetic sensitizing mutations (ApoeKO and ApcMin) increased the incidence of atherosclerosis and cancer, respectively, phenotypes that do not spontaneously develop in the wild-type (WT) mouse strains used in our study.
Figure 1. Sensitized phenotyping screen.
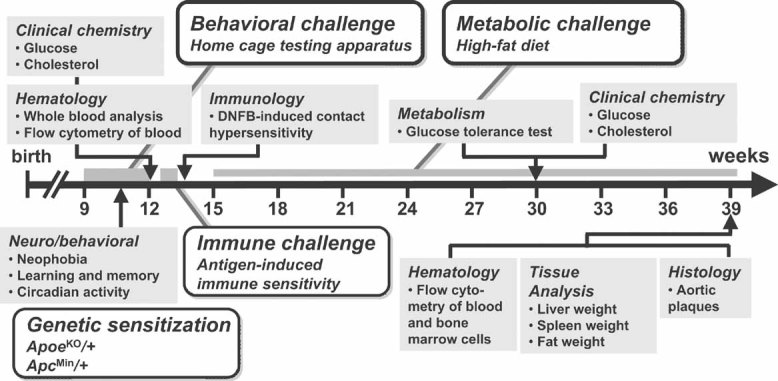
Animals were subjected to a battery of tests designed to reveal effects of gene dosage on physiological parameters (grey boxes) relevant to common human diseases in five therapeutic areas: behaviour, metabolic syndrome, immune dysfunction, atherosclerosis, and cancer. Each therapeutic area was accompanied by a sensitizing environmental, pharmacological or genetic challenge (open boxes). The first cohort of mice were ApoeKO/+ and were subjected to the full phenotyping battery, while a second cohort were ApcMin/+ and were only tested for intestinal neoplasia-induced death. At 9 weeks of age, animals were tested for neophobia, gross activity and spatial learning in a home cage behavioural testing apparatus. At 12 weeks, blood parameters (cell counts and flow cytometry, cholesterol, glucose) were measured and at 14 weeks animals were tested for DNFB-induced CHS. At 15 weeks, animals were placed on a high-fat diet, and after 15 weeks of high-fat diet, blood parameters for clinical chemistry were collected and glucose tolerance measured. After 24 weeks of high fat diet, mice were sacrificed and organs collected. Aortic samples were examined for atherosclerotic plaques and bone marrow cells were extracted and analysed by flow cytometry (all dates are ±2 weeks).
All animals in the main cohort were heterozygous for a null allele of Apoe (ApoeKO/+) and the order of multiple testing was chosen to reduce potential interactions between procedures. First, a group housed home cage monitoring system was used to track behavioural responses to novelty, home cage activity and spatial learning. Second, contact dermatitis responses to the antigen 2,4-dinitro-1-fluorobenzene (DNFB) were assessed. Third, mice were exposed for 15 weeks to a high-fat diet before and after which multiple peripheral metabolic measures were assessed. Finally, histological analysis was performed to examine atherosclerotic plaque development, bone marrow cell content and organ weights. A separate group of mice heterozygous for the multiple intestinal neoplasia allele of Apc (ApcMin/+) was assessed solely for latency to cancer-related death.
Production and validation of Df11(1) and Dp11(1) mice
Mouse ES cells (129/SvEvBrd background) carrying a Cre-loxP engineered reciprocal duplication and deficiency of a 0.8 Mb region on chromosome 11 (Df11[1]/Dp11[1]; Liu et al, 1998) were injected into blastocysts to obtain chimeric founders that were subsequently used to establish independent breeding colonies of Df11(1)/+ and Dp11(1)/+ mice and their WT littermates (Fig S1 of Supporting Information). The rearrangement encompasses 27 known and one novel gene and is syntenic to human chromosome 17q21 containing a cluster of human disease susceptibility loci (Fig 2A and Table S1 of Supporting Information). Comparative genome hybridization (CGH) confirmed 1 and 3 copies of the region in Df11(1)/+ and Dp11(1)/+ mice, respectively (Fig 2B), and failed to reveal any other significant aneuploid region in either line (data not shown). To evaluate changes in the expression of genes within the rearrangement we performed microarray-based transcriptome analysis on mRNA samples extracted from cultured T cells of Df11(1)/+, WT and Dp11(1)/+ mice. Among 46 probesets interrogating the 27 known genes in the region, 26 probsets (14 genes) reached threshold for expression detection in WT samples. Statistical analysis performed on detected probesets revealed a significant gene dosage effect on gene expression for all but two genes (Fig 3). Expression changes reached significance for 12 genes in the Df11(1)/+ versus WT comparison, but only 6 in the Dp11(1)/+ versus WT comparison, consistent with the smaller fold change in the latter case (1.5:1 for Dp:WT vs. 2:1 for Df:WT). Because the magnitude of expected expression change was modest we set the threshold for significance at p < 0.25. These data confirm that for the majority of genes in the rearrangement gene expression scaled with copy number, a finding consistent with previous studies in engineered (Kahlem et al, 2004; Laffaire et al, 2009; Li et al, 2009; Prescott et al, 2005) and endogenously occurring CNV in mice (Henrichsen et al, 2009). Cohorts of mice for phenotyping were established by breeding male Df11(1)/+ and Dp11(1)/+ mice with female ApoeKO/KO or ApcMin/+ mice (Fig S1A of Supporting Information). The resulting offspring carried one, two or three copies of the rearrangement region (Df11[1]/+, WT, Dp11[1]/+). Initially Df/11(1)+ and Dp11(1)/+ mice were compared to their respective WT littermates, but because the independent WT groups did not differ statistically all mice were grouped and overall effects of genotype were assessed by analysis of variance (ANOVA) and Kruskall–Wallis tests for parametric and non-parametric distributions, respectively.
Figure 2. Mouse lines carrying reciprocal Df and Dp on chromosome 11.
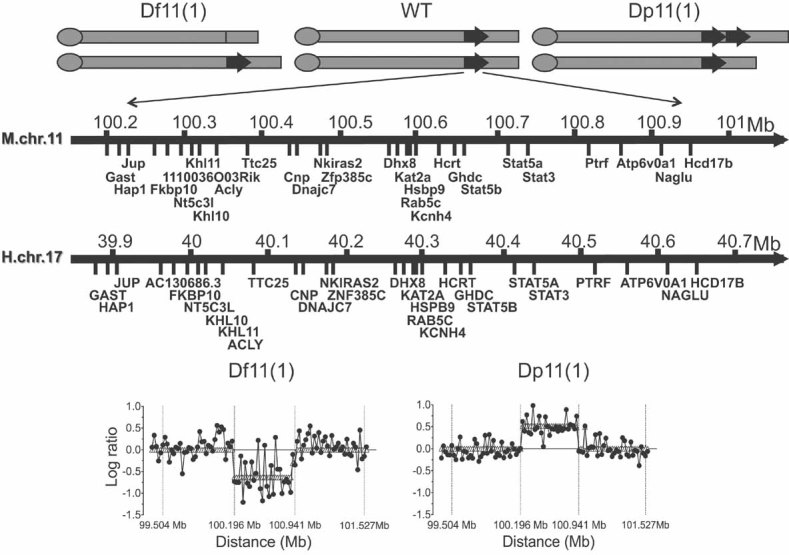
- Schematic representation of the segmental aneuploidy region on chromosome 11 containing 26 annotated and one unknown gene (mouse NCBI m37 assembly, April 2007, C57BL/6J reference strain) and syntenic region on human chromosome 17 (Human GRCh37 assembly February 2009). Df11(1)/+, WT and Dp11(1)/+ mice carried one, two and three copies, respectively, of genes in the interval.
- CGH analysis of Df11(1)/+ versus WT and Dp11(1)/+ versus WT littermates confirmed expected gene dose changes across the rearrangement. Negative and positive log ratios indicate, respectively, loss and gain of genetic material.
Figure 3. Gene expression changes.

Microarray hybridization analysis of expression patterns of genes within the rearrangement demonstrated changes in expression consistent with gene dosage. A heat map shows the normalized expression of genes in the Df11(1)/+ versus WT and Dp11(1)/+ versus WT samples. Average of two microarray experiments per sample (log2 ratio for each gene was scaled with mean = 0 and SD = 1; log fold change and adjusted p values are indicated). Genes with a significant change in expression level are indicated (*p < 0.25).
Altered anxiety in Dp11(1) and Df11(1) mice
To assess a wide repertoire of behavioural measures under both baseline and challenge conditions animals were placed into an automated home cage monitoring system starting at 9 weeks of age. In this system, up to eight animals were housed together and visits of each mouse to four corner chambers with access to water were recorded by a telemetric sensor. Initially, free access was allowed to all four corners to examine exploratory behaviour and neophobia. A significant effect of genotype (F[2,1199] = 3.95, p = 0.020) was observed for total visits during the first 2 h in the novel cage with Dp11(1)/+ mice making significantly less visits compared to Df11(1)/+ mice (Fig 4A). There was a trend for this effect to diminish over the habituation period suggesting decreased neophobia in the Df11(1)/+ animals. However, analysis of corner visits during the ensuing free exploration period showed that significant differences in activity persisted also under more familiarized conditions (F[2,1739] = 3.03, p = 0.020; Fig 4B). Again, Dp11(1)/+ mice made significantly fewer corner visits compared to Df11(1)/+ mice with the differences being most apparent during the dark/active period. These data suggest that there is a gene dose-dependent effect on locomotor drive that is revealed under conditions of behavioural arousal.
Figure 4. Behavioural testing.

- A significant gene dose-dependent effect was observed for number of corner visits during the first 2 h after being placed in the monitoring apparatus (10 min intervals; mean ± SEM; Df11(1)/+, N = 22; WT, N = 45; Dp11(1)/+, N = 33).
- No significant genotype effect on general day/night activity (number of corner visits) of animals was observed (3 h intervals; mean ± SEM; Df11(1)/+, N = 22; WT, N = 41; Dp11(1)/+, N = 24).
- Significant gene dosage effect was observed on percentage of incorrect visits (2 h intervals). Dark and light phases are indicated by the dark and light bars, respectively (mean ± SEM; Df11(1)/+, N = 22; WT, N = 41; Dp11(1)/+, N = 24).
- Significant gene dosage-dependent effect is observed on total numbers of nose pokes to incorrect corners (2 h intervals; mean ± SEM; Df11(1)/+, N = 22; WT, N = 41; Dp11(1)/+, N = 24; *Dp vs. Df, # Dp vs. WT and Df vs. WT; *p < 0.05, **p < 0.01, ***p < 0.001).
Next, mice were subjected to a spatial learning task where animals had free access to all four corners but water was provided in only one of them. In this situation mice rapidly learn to suppress visits to corners without water (errors) and learning can be quantified by a decrease in percentage of incorrect visits (Fig 4C) and number of nose pokes in incorrect corners (Fig 4D). Although no effect of genotype was seen on the learning curves, a significant negative gene dose-dependent effect was seen for total visits (F[2,1565] = 16.2, p < 0.001) and nosepokes (F[2,1565] = 46.2, p < 0.001). Although a significant interaction between genotype and time was seen for nosepokes (F[2,1565] = 1.67, p = 0.010), the reduced activity seen in Dp11(1)/+ mice was more likely related to their decreased locomotor drive rather than altered learning. To determine whether the differences in locomotion reflected an underlying alteration in anxiety, we tested an independent cohort of mice in two tests of innate anxiety, the elevated plus maze (EPM) and open field (OF, Table S2 of Supporting Information). In the EPM, a significant negative gene dose effect was seen for % time spent in the open arms (F[2,49] = 3.32, p = 0.045) and a trend for a negative gene dose effect was seen for number of visits and distance travelled in the open arms (Table S2 of Supporting Information). Similar results were found in the OF, where trends for a negative gene dose effect were seen for time in centre, centre entries and total locomotion (Table S2 of Supporting Information). These findings support a contribution of altered anxiety to the locomotor phenotype.
Haematological deficits in Df11(1) and Dp11(1) mice
Peripheral blood cell counts were assessed at 12 weeks of age to examine gene dosage effects on haematological parameters. While the number of erythrocytes was not affected by gene dosage (data not shown), a significant positive gene dose-dependent effect was seen for white blood cells (F[2,83] = 14.785, p < 0.0001; Fig 5A) and platelets (F[2,87] = 14.695, p < 0.0001; Fig 5B). Flow cytometry of peripheral blood nucleated cells revealed a normal percentage of B220+, MAC1+ and CD4+ cells (Fig 5C, D and E), but a significant positive gene dose-dependent change in percentage of CD8+ cells (F[2,99] = 8.695, p = 0.003; Fig 5F). These data suggest that change in dosage of one or more genes within the rearrangement affects the production and/or maintenance of CD8+ T cells.
Figure 5. Peripheral blood cytometry.
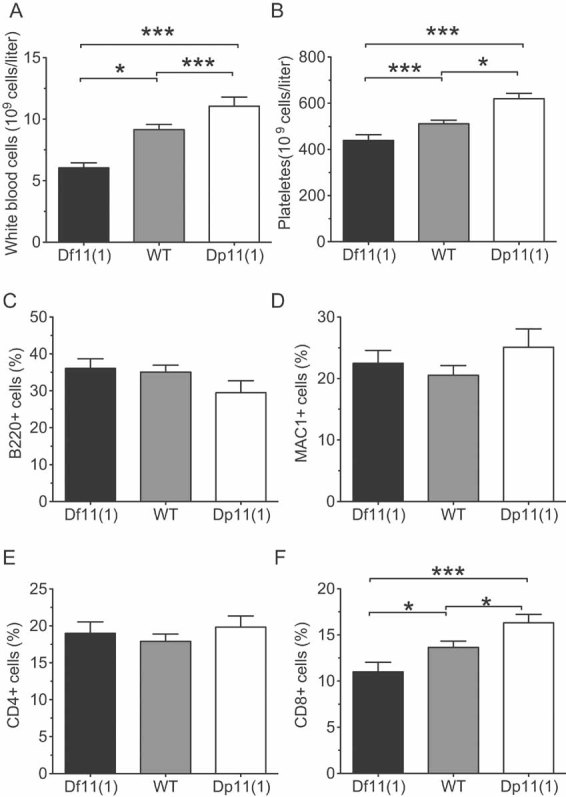
- A,B. A significant positive gene dose-dependent effect was observed for (A) white blood cells, and (B) platelet counts at 12 weeks of age (mean ± SEM; Df11(1)/+, N = 21; WT, N = 41; Dp11(1)/+, N = 20; *P < 0.05, **P < 0.01, ***P < 0.001).
- C–F. Flow cytometry revealed a significant positive gene dose-dependent effect for percentage (F) CD8+, but not (C) B220+, (D) Mac1+, (E) CD4+ positive cells in peripheral blood (mean ± SEM; Df11(1)/+, N = 19; WT, N = 58; Dp11(1)/+, N = 25; *P < 0.05, **P < 0.01).
Impaired contact hypersensitivity in Df11(1) and Dp11(1) mice
To evaluate gene dose-dependent effects on immune system function, we performed DNFB-induced CHS, a model for allergic contact dermatitis that recruits diverse components of the innate immune system. CHS measures the T cell-mediated skin inflammatory reaction elicited following repeated prior contact of the skin with allergenic haptens. Following application of DNFB to the abdominal skin, the compound is processed and presented by cutaneous antigen presenting cells that subsequently migrate to local lymph tissue. In the lymph nodes, antigen presenting cells activate naive CD8+ T cells via the help of antigen-specific memory T cells, that subsequently migrate to the skin and mediate inflammatory responses to future applications of the same hapten. Reapplication of DNFB to the ear elicits an enhanced inflammatory response that can be measured by tissue swelling. When Df11(1)/+, WT and Dp11(1)/+ mice were tested for ear swelling responses following re-exposure to DNFB, a significant positive gene dose dependent effect was observed (repeated measured ANOVA: F[2,81] = 15.432, p < 0.0001; Fig 6A). Notably, while the ear swelling responses peaked in Df11(1)/+ mice at 24 h post-exposure, they peaked only at 72 h in Dp11(1)/+ mice. These findings suggest that the level of expression of one or more genes within the rearrangement is critical to determine both the magnitude and kinetics of the CHS response.
Figure 6. Contact hypersensitivity and T cell function.
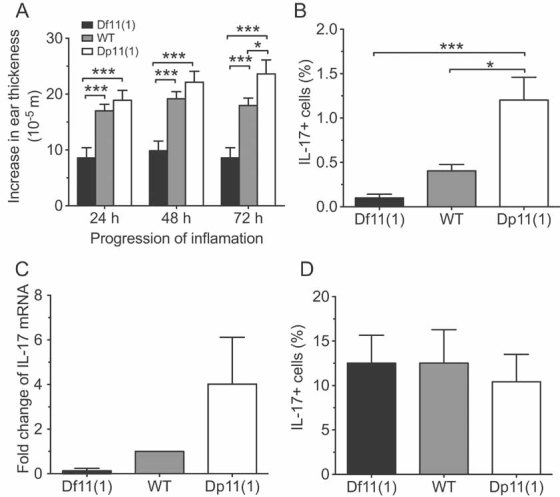
- A significant positive gene dose-dependent effect was observed for ear swelling following local application of DNFB. Ear thickness was measured at 24, 48 and 72 h following DNFB treatment in mice that had been sensitized by DNFB pre-treatment (mean ± SEM; Df11(1)/+, N = 22; WT, N = 42; Dp11(1)/+, N = 20; *p < 0.05, ***p < 0.001).
- A significant positive gene dose-dependent effect was observed for the fraction of cultured naïve splenic T cells (Th0 cells) immunostaining for intracellular IL-17 (N = 4).
- Quantitative real-time PCR revealed a positive gene dose-dependent effect on IL-17 mRNA expression in Th0 cells cultured for 5 days and stimulated acutely with cytokines. Data represents fold change compared to WT animals. N = 2 for each genotype.
- The gene dose-dependent effect on fraction of IL-17+ T cells was occluded in cytokine-induced polarized T cells (Th17 cells, mean ± SEM, N = 4).
CD8+ T cells are a critical component of the CHS response and the number of these cells in peripheral blood showed a positive gene dose-dependent effect (Fig 5F). To determine whether tissue resident CD8+ T cells also showed gene dose-dependent changes, we performed flow cytometry on spleen cells. Similar to what we found in the periphery, CD8+ (but not CD4 +) cell counts showed a significant positive gene dose-dependent effect (F[2,9] = 4.63 p = 0.04; Fig S2 of Supporting Information). These findings open the possibility that variation in the number of resident CD8+ T cells may play a role in the altered CHS response observed in Df11(1)/+ and Dp11(1)/+ mice.
To further investigate potential cellular deficits underlying the CHS phenotype, we examined the functionality of cultured splenic T cells. Secretion of interleukin-17 (IL-17) by regulatory T helper (Th-17) cells is a critical paracrine signalling step in the inflammatory response to sensitizing antigens and mice lacking IL-17 show a blunted CHS response similar to that seen in Df11(1)/+ mice (Nakae et al, 2002). To determine whether Th-17 cell maturation and/or IL-17 secretion was affected in Df11(1)/+ and Dp11(1)/+ mice, we stimulated naïve splenic T cells (Th0 cells), in culture with IL-6 and TGF-β to become Th-17 cells and used flow cytometry coupled to intracellular immunostaining and real-time PCR to determine production of IL-17. In non-polarized T cells (Th0 cells), IL-17 expression showed a positive gene dose-dependent effect as determined by quantitative RT-PCR (Fig 6C). Moreover, the fraction of Th0 cells positive for IL-17 showed a significant genotype effect, demonstrating that one or more genes within the rearrangement is critical for the function and/or differentiation of IL-17 expressing cells (F[2,9] = 13.496, p = 0.002; Fig 6B). However, following IL-6/TGF-β polarization the fraction of IL-17 positive cells and expression of IL-17 protein appeared similar in all genotypes (Fig 6D) suggesting that under polarizing conditions, such as those induced following allergen treatment, Th-17 cell function did not differ between genotypes. These findings suggest that, although IL-17 cell expression is altered in Th0 cells of Df11(1)/+ and Dp11(1)/+ mice under baseline conditions, these differences may not underlie the CHS phenotype.
Altered cholesterol and glucose tolerance in Df11(1) and Dp11(1) mice
To assess metabolic function under baseline and challenge conditions several measures of energy homeostasis were taken after 15 weeks high-fat diet treatment and in age-matched controls maintained on a normal diet. No significant genotype effect on body weight was observed before or during the high-fat diet treatment, indicating no gross metabolic abnormality (Fig 7A). However, while mice on a normal diet showed a trend for a negative gene dose-dependent effect on fasting blood glucose, glucose tolerance and cholesterol (with Dp11(1)/+ mice showing a significant reduction in fasting blood glucose, when compared to WT animals; Fig 7B), high-fat diet treated animals showed a significant negative gene dose-dependent effect on glucose tolerance and cholesterol (glucose tolerance: F[2,65] = 3.196, p = 0.04; cholesterol: F[2,79] = 3.9, p = 0.02; Fig 7C and D and Fig S3 of Supporting Information). Moreover, under high-fat diet conditions fasting blood glucose levels became normalized in Dp11(1)/+ mice (Fig 7B) demonstrating that these animals were partially able to compensate for dietary challenge. Finally, normalized gonadal fat, but not liver weight was significantly reduced in Df11(1)/+ mice compared to either WT or Dp11(1)/+ mice (F[2,66] = 3.782, p = 0.02; Fig 7E and F). These findings suggest a dissociation between gene dose-dependent effects on homeostasis of blood metabolites and fat deposition.
Figure 7. Metabolic parameters.

- A. Normal body weight in Df11(1)/+ and Dp11(1)/+ mice 4, 7, 10 and 13 weeks after initiation of high-fat diet (mean ± SEM; Df11(1)/+, N = 14; WT, N = 18; Dp11(1)/+ N = 11).
- B. Significant negative gene dose-dependent effect on fasted blood glucose concentration in mice on normal (week 15), but not after 15 weeks of high-fat diet treatment (week 30).
- C. Significant negative gene dose-dependent effect on glucose clearance following intra-peritoneal dextrose challenge after high-fat diet treatment for 15 weeks (AUC = area under the curve; Df11(1)/+, N = 20; WT, N = 36; Dp11(1)/+, N = 15 Controls were age-matched mice on a normal diet: Df11(1)/+, N = 7; WT, N = 19; Dp11(1)/+, N = 11 *p < 0.05).
- D. Significant negative gene dose-dependent effect on plasma cholesterol in mice treated for 15 weeks with high-fat (week 30), but not normal diet (week 14 mean ± SEM; Df11(1)/+, N = 23; WT, N = 49; Dp11(1)/+, N = 22).
- E,F. Significant positive gene dose-dependent effect on normalized (E) gonadal fat, but not (F) liver weight (mean ± SEM; Df11(1)/+, N = 20; WT, N = 35; Dp11(1)/+, N = 15). Weight of Gonadal fat and liver for each animal were normalized to total body weight.
Altered incidence of atherosclerosis in Df11(1) and Dp11(1) mice
Next, we examined the risk for ApoE-induced atherosclerosis following continuation of high-fat diet treatment for a total of 25 weeks (Fig 1). It has been shown previously that ApoeKO/+ animals develop frequent atherosclerotic lesions in proximal aorta in the presence of an atherogenic diet (Bobkova et al, 2004; Zhang et al, 1994). Consistent with these observations, ApoeKO/+ mice on our genetic background showed aortic atherosclerotic plaques, while no or only very small plaques were detected in non-ApoeKO animals (data not shown). Haematoxylin and Texas Red double staining was carried out on serial sections of the proximal aorta and unbiased image processing was used to quantify the percentage of lumen area with plaque material (Fig 8A). Consistent with previous studies (Purcell-Huynh et al, 1995), a significant effect of sex (F[1,36] = 19.05, p = 0.0001) was observed on plaque area occupied by the lesions, with females showing larger lesion than males (Fig 8B). In addition, there was an opposite gene dose-dependent effect on plaque area in males and females, with Df11(1)/+ females showing significantly smaller fractional plaque area than WT females (Fig 8B) and a trend for larger fractional plaque area in Df11(1)/+ males when compared to Dp11(1)/+ males. These findings suggest that changes in dosage of one or more genes in the rearrangement affects the penetrance of aortic plaque formation or maintenance in ApoeKO/+ mice in a sex-dependent manner.
Figure 8. Atherosclerotic aortic plaques.

- A. Representative histological sections of proximal aorta showing significant staining of plaque material (red, indicated by arrows) in all genotypes. Opposite gene dose-dependent effects on relative area of aortic plaques were seen in females (Df11(1)/+, N = 6; WT, N = 10; Dp11(1)/+, N = 3) and males (Df11(1)/+, N = 6; WT, N = 10; Dp11(1)/+, N = 5). In females, Df11(1)/+ mice showed significantly smaller plaques that WT mice, whereas in males, Df11(1)/+ mice showed a trend for increased plaque size compared to Dp11(1)/+ mice (mean ± SEM, *p < 0.05).
- C. Kaplan–Meier survival plot of ApcMin/+ animals with Df11(1) or Dp11(1) alleles and their WT littermates. No gene dose-dependent moderation of survival in ApcMin/+ mice was observed. (Df11(1)/+; ApcMin/+, N = 7; +/+; ApcMin/+, N = 9; Dp11(1)/+; ApcMin/+, N = 5).
Normal progression to cancer in Df11(1) and Dp11(1) mice
To investigate potential gene dose-dependent effects on cancer incidence, we examined survival rates of a separate cohort of Df11(1)/+, WT and Dp11(1)/+ mice that had been crossed to the ApcMin/+ mutation. ApcMin/+ mice develop multiple intestinal adenomas and rarely survive beyond 4 months of age on the C57BL/6 background (Moser et al, 1992). Starting at day 120, mice were examined weekly for signs of illness or weakness and moribund animals were sacrificed. No mice survived beyond 400 days of age and no significant gene dose-dependent effect on survival was detected (Fig 8C). These data demonstrate that gene dosage in this region does not affect progression to death due to intestinal neoplasia in ApcMin/+ mice, although more subtle effects on tumor development or quality could not be ruled out.
Stat5 is responsible for contact hypersensitivity phenotype and glucose homeostasis
The discovery of multiple gene dose-dependent phenotypes associated with Df11(1) and Dp11(1) suggested that one or more genes within the region has a critical dose sensitive effect on behavioural, haematological, immune, metabolic, and atherosclerotic phenotypes. Moreover, these genes may be acting independently or together to influence these phenotypes. An analysis of genes in the interval revealed three genes previously implicated in immune function, Stat5a, Stat5b and Stat3 (O'Shea & Murray, 2008). All three genes encode transcriptional regulators that act as major mediators of cytokine/growth factor signalling and are critical for the initiation and progression of inflammatory responses (O'Shea & Murray, 2008). To examine whether changes in gene dose in one or more of these candidate genes could be responsible for the immune phenotypes we observed, we obtained and tested single and double heterozygous null mutations in Stat5ab (a deletion of both Stat5a and Stat5b genes, Cui et al, 2004) and Stat3 (Alonzi et al, 2001) and their WT littermates for changes in haematological parameters and DNFB-induced CHS.
Analysis of changes in ear thickness following re-exposure to DNFB revealed significantly reduced swelling in heterozygous Stat5ab knockout, but not heterozygous Stat3 knockout mice when compared to WT littermates (repeated measured ANOVA: F[3,49] = 6.995, p = 0.0005; Fig 9A). Double heterozygous Stat5abKO/Stat3KO mice were indistinguishable from Stat5abKO/+ littermates arguing for a lack of interaction between the two mutations. Importantly, the magnitude of the reduced sensitivity to DNFB in heterozygous Stat5ab knockout mice was similar to that seen in Df11(1)/+ mice (Fig 6A) suggesting that heterozygosity in this gene pair alone was responsible for the deficiency phenotype.
Figure 9. Contact hypersensitivity in heterozygous Stat5ab and Stat3 knockout mice.
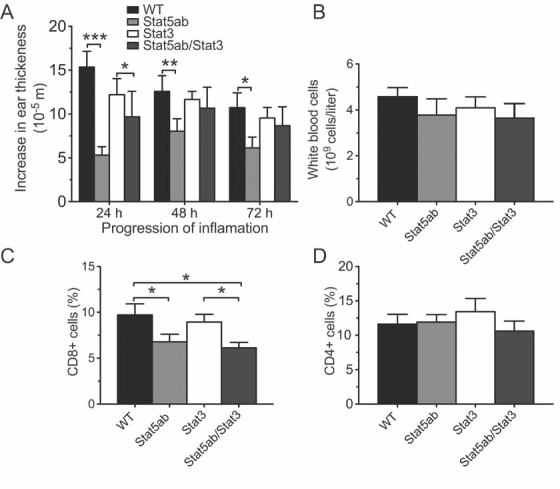
- A. A significant reduction of ear swelling was observed in heterozygous Stat5abKO, but not Stat3KO mice following local application of DNFB. Ear thickness was measured at 24, 48 and 72 h following DNFB treatment in mice that had been sensitized by DNFB pre-treated on the abdomen 5 days earlier (mean ± SEM; WT, N = 15; Stat5abKO/+, N = 13; Stat3KO/+, N = 10; Stat5abKO/Stat3KO, N = 6; *p < 0.05, **p < 0.01, ***p < 0.001).
- B–D. Significant reduction in peripheral blood (C) CD8+, but not (D) CD4+ T cells or (B) total white blood cells (mean ± SEM; WT, N = 9; Stat5abKO/+, N = 9; Stat3KO/+, N = 7; Stat5abKO/Stat3KO, N = 6; *p < 0.05, **p < 0.01, ***p < 0.001).
Next, we examined haematological parameters of peripheral blood of single and double heterozygous Stat5ab and Stat3 knockout littermates. While total numbers of red blood cells and platelets were unaltered (data not shown), we observed a trend towards decrease in white blood cells counts (Fig 9B). Flow cytometry revealed a significant and selective reduction in CD8+ cells in heterozygous Stat5ab knockouts F[3,27] = 3.178, p = 0.04 (Fig 9C). No significant effect of genotype was seen for CD4+, Mac1+ or B220+ cells (Fig 9D and data not shown). The magnitude of reduction in peripheral CD8+ cells in heterozygous Stat5ab knockout mice was similar to that seen in Df11(1)/+ animals (Fig 5F) demonstrating that changes in Stat5ab gene dose were sufficient to explain both altered CD8+ cell number and reduced CHS in the deficiency mice. To examine whether increases in Stat5ab gene dose were necessary for the immune deficits observed in Dp11(1)/+ mice, we crossed the two alleles together. Increased peripheral white blood cell counts in Dp11(1)/+ mice were normalized in Dp11(1)/+;Stat5abKO/+ double mutant mice (F[3,30] = 4.987, p = 0.0063; Fig 10A). Likewise, the trend for increased CHS in Dp11(1)/+ mice was also reversed in Dp11(1)/+;Stat5abKO/+ double mutants (Fig S7 of Supporting Information).
Figure 10. Contact hypersensitivity and metabolic parameters in Dp11(1)/+; Stat5ab KO /+ double mutant mice.

- A,B. The significant (A) increase in total white blood cells and (B) decrease in baseline glucose seen in Dp11(1)/+ mice compared to WT mice was reversed in Dp11(1)/+;Stat5abKO/+ double mutant littermates (Dp11(1), N = 7; Dp11(1)/Stat5ab, N = 9; WT, N = 10; Stat5ab, N = 8).
- C. The trend for increased glucose tolerance seen in Dp11(1)/+ mice compared to WT mice was reversed in Dp11(1)/+;Stat5abKO/+ double mutant littermates (Dp11(1), N = 7; Dp11(1)/Stat5ab, N = 9; WT, N = 14; Stat5ab, N = 7; mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001).
Finally, we investigated whether changes in Stat5ab gene dosage might also underlie the metabolic deficits observed in Df11(1)/+ and Dp11(1)/+ mice. Reduced baseline glucose seen in Dp11(1)/+ mice was normalized in Dp11(1)/+;Stat5abKO/+ double mutant littermates (F[3,33] = 3.081, p = 0.04; Fig 10B), and the trend for increased glucose tolerance was similarly reversed (Fig 10C). Notably, glucose tolerance was significantly greater in Dp11(1)/+ mice (three copies of Stat5ab) when compared to Stat5abKO/+ littermates (one copy of Stat5ab; Fig 10C). These findings argue that Stat5ab gene dosage is critically involved in both the immune and metabolic susceptibility observed in Dp11(1)/+ and Df11(1)/+ mice.
DISCUSSION
We have demonstrated that deletion or duplication of a 0.8 Mb chromosomal region on mouse chromosome 11 alters susceptibility to multiple disease-relevant phenotypes. A previous study with the same rearrangement revealed a dose-dependent increase in corneal hyperplasia and thymic neoplasia in Dp11(1)/+ and Dp11(1)/Dp11(1) mice at >10 months of age (Liu et al, 1998). No phenotype in Df11(1)/+ mice has been previously reported, although these animals demonstrated higher susceptibility to tumours when located in trans to a null p53 allele, presumably due to an increased frequency of deletions encompassing p53 on the deficiency chromosome (Biggs et al, 2003). Here we used a broad, unbiased and sensitized screening approach to uncover susceptibility to immune, metabolic, cardiac, cancer, and behavioural phenotypes. The phenotypes observed could be divided into two groups, those showing positive (Df < WT < Dp) or negative (Df > WT > Dp) gene dose-dependent effects across all three genotypes and those showing phenotypes in only one rearrangement line (Df or Dp ≠ WT). The first group included alterations in anxiety and home cage activity (Fig 4 and Table S2 of Supporting Information), number of peripheral white blood cells and platelets (Fig 5A and B), peripheral and splenic CD8+ T cells (Fig 5F and Fig S2 of Supporting Information), DNFB-induced CHS (Fig 6A), number of cultured splenic IL-17+ T cells (Fig 6B), and dextrose-induced glucose clearance (Fig 7C). Phenotypes in the second group included fasting blood glucose (Fig 7B), high-fat diet induced blood cholesterol (Fig 7D), gonadal fat (Fig 7E), and ApoE induced aortic atherosclerotic plaques (Fig 8A and B). Several annotated genes within the rearrangement were candidates for the observed phenotypes and follow-up studies with single and double heterozygous knockout mutations in Stat5ab and Stat3 showed that reduced Stat5ab gene dosage was sufficient to cause the deficits in CHS response and CD8+ T cell number seen in Df11(1)/+ mice (Fig 9A and C) and necessary for the increased peripheral white blood cell counts see in Dp11(1)/+ mice (Fig 10A). Altered Stat5ab gene dosage was also necessary for the reduced baseline and dextrose-induced glucose levels in Dp11(1)/+ mice (Fig 10B and C).
CGH demonstrated the expected changes in DNA copy number across the rearrangement, confirmed its end points and ruled out changes in gene copy number at other genomic locations in the Dp11(1)/+ and Df11(1)/+ lines (Fig 2B). Expression profiling of mRNA extracted from cultured splenic T cells revealed changes in gene expression that matched gene copy number for 12 of 14 genes in the region (Fig 3). Changes detected by microarray hybridization were confirmed by real time PCR for Acly, Kat2a, Dhx28, and Stat5a (data not shown). In addition, immunoblotting confirmed gene dose-dependent effects on Stat3 and Stat5b protein expression in liver tissue (Fig S4 of Supporting Information). The lack of a copy number-mRNA correlation for two genes in the rearrangement (Klhl11 and Dhx58, Fig 3) presumably reflected the engagement of compensatory transcriptional mechanisms for these genes, although a selective discrepancy in the amplification and/or quantification of mRNA for these genes could not be ruled out. Furthermore, we could not draw conclusions about copy number driven changes in expression for the remaining genes in the rearrangement (13/27 annotated genes, Fig 2A) as these were not detected in T cells. Our gene expression findings are consistent with other studies in mouse deficiency and duplication lines where the expression of the large majority of genes reflected DNA copy number (Kahlem et al, 2004; Li et al, 2009; Pereira et al, 2009; Prescott et al, 2005). Importantly, only 1 of 39 genes closely linked to the rearrangement (Psmc3ip) showed expression differences (data not shown) demonstrating that cis-acting effects of the rearrangement are essentially limited to the rearrangement boundaries.
An important feature of our screen was its use of genetic and environmental sensitizers, including heterozygous ApoeKO and ApcMin mutations, exposure to environmental novelty and spatial learning, hapten challenge, and high-fat diet treatment (Fig 1). In the mouse, focused sensitized screens have been successful at identifying susceptibility mutations using quantitative trait loci (QTL, e.g. modifiers of ApcMin; Dietrich et al, 1993) and chemical mutagenesis (ENU, e.g. Matera et al, 2008; Rubio-Aliaga et al, 2007) approaches. However, for expediency most unbiased screens have been restricted primarily to baseline phenotypes (Brown et al, 2005; McGuinness et al, 2009; Svenson et al, 2007). Arguably, several of our observed phenotypes would not have been detected without challenges and this feature underlines the critical importance of the use of multiple sensitizers in our screen.
The diversity of phenotypic changes seen in the Df11(1)/+ and Dp11(1)/+ lines suggested a contribution of multiple genes. However, it was also possible that one or a small number of genes contributed to multiple phenotypes. Support for the first hypothesis came from our studies with single gene knockouts. Mutations in Stat5ab and Stat3 have been associated with deficient T cell function in mice and humans and these genes were strong candidates for the haematological and CHS phenotypes (Hoelbl et al, 2006; Holland et al, 2007; Minegishi et al, 2007; Moriggl et al, 1999; Yao et al, 2006). For example, T cell-specific Stat3 null mutations lead to loss of Th17+ T cells and compromise their ability to secrete IL-17, a critical mediator of the CHS response (Harris et al, 2007; Nakae et al, 2002). Likewise, heterozygous Stat5ab null mice show reduced peripheral CD8+ T cells and this class of T cells plays an important facilitating role in CHS (Vocanson et al, 2006). However, our single gene knockout studies clearly showed that copy number variation in Stat5ab, but not Stat3 modulated the CHS response (Fig 9A). Flow cytometry also revealed a clear dissociation between the effects of Stat5ab and Stat3 heterozygosity, with the former showing significantly reduced CD8+, but not CD4+ T cells in peripheral blood (Fig 9C and D and Fig S2 of Supporting Information). These findings corroborate previous reports of reduced thymic CD8+ T cells in heterozygous Stat5ab mice (Hoelbl et al, 2006) and support the hypothesis that altered numbers of CD8+ T cells in skin and lymph tissue might be responsible for the altered CHS seen in Df11(1)/+ and Dp11(1)/+ mice (Fig 6A). It remains to be determined whether Stat5ab-dependent modulation of T cell number determines the degree of CHS response. Interestingly, while we were unable to detect a correlation between the percentage of CD8+ T cells in peripheral blood and the intensity of CHS response, we did observe a significant correlation between total white blood cells and CHS response (R2 = 0.641, p = 0.001; Fig S7 of Supporting Information).
Furthermore, despite a known role for Stat3 signal transduction in IL-17 secretion and the related gene dose-dependent change in the expression of this cytokine in cultures of quiescent T cells (Fig 6B and C), alterations in IL-17 function under stimulated conditions (Fig 6D) were not correlated with the CHS phenotype. Moreover, there was no indication of a genetic interaction between Stat3 and Stat5ab alleles (Fig 9) as might have been expected given their proposed synergistic role in immunological responses (Zhu & Paul, 2008). These data suggest that Stat3-dependent changes in T cell function were not sufficient to moderate CHS and support a novel, purely Stat5-dependent gene dosage effect. Our findings of a role for Stat5ab in glucose homeostasis is consistent with earlier work (Jackerott, 2006; Lee et al, 2007). In one study, partial inhibition of Stat5ab function in pancreas was associated with higher basal glucose levels and glucose intolerance, while expression of constitutively active Stat5b was associated with increased glucose tolerance (Jackerott et al, 2006). These findings are in agreement with our data and suggest that Stat5ab gene dosage in pancreatic beta cells may underlie the metabolic phenotypes we observed.
Our findings demonstrate the potential of unbiased sensitized screening of segmental aneuploidy lines to identify susceptibility genes for common disease phenotypes in the mouse. The large-scale characterization of engineered CNV mouse lines may offer an efficient approach to identify susceptibility genes not uncovered via other systematic screening methods currently being pursued. In addition, this approach taps into a rich source of natural copy number variation that is increasingly recognized to play a key role in disease susceptibility. Identifying susceptibility mutations is a key strategy to uncover novel pathological mechanisms amenable to therapeutic intervention.
MATERIALS AND METHODS
Ethics statement
All mice were handled according to protocols approved by the Italian Ministry of Health and commensurate with NIH guidelines for the ethical treatment of animals.
Animals
AB2.2 ES cells (129/SvEvBrd) carrying the Cre-loxP engineered reciprocal Dp11(1)/Df11(1) rearrangement (Liu et al, 1998) were kindly provided by Allan Bradley and were injected in C57BL/6J blastocysts to derive chimeric mice. Male chimerae were crossed to both WT 129/SvEvBrd (kind gift of Allan Bradley) and C57BL/6J mice. Only C57BL/6J crosses gave rise to offspring transmitting the Df11(1)/+ and Dp11(1)/+ alleles and these were then backcrossed to 129/SvEvBrd to establish the male parents of the phenotyping cohort, Df11(1)/+ and Dp11(1)/+, respectively. Mothers were C57BL/6J mice either homozygous for the ApoeKO allele (B6.129P2-ApoEtm1Unc/Crl, Charles River Laboratories, Calco, Italy) or heterozygous for the Apcmin allele (stock #002020, The Jackson Laboratory, Bar Harbor, ME). Thus, offspring used for phenotyping were a mixed B6x[129xB6] background. WT mice were littermates. All mice in the ApcMin/+ cohort were homozygous for the modifier of Min (Mom-1) allele of Pla2g2a (Santos et al, 1998). Stat3 conditional knockout (Alonzi et al, 2001) and Stat5ab constitutive knockout mice (Cui et al, 2004) were imported from the Ludwig Bolzmann Institute and maintained on a C57BL/6J background. Stat3 constitutive knockout mice were derived by crossing Stat3 conditional knockouts (Alonzi et al, 2001) with a general Cre-deleter line and deletion was verified by PCR of tail DNA. Mice for the single knockout experiments were produced by breeding Stat5abKO/+ mice with Stat3KO/+ mice. Mice of all four genotypes were littermates. Animals were weaned at 3 weeks and group housed. The colony was maintained on a 12 h light/dark cycle (lights on at 7:00) with free access to food and water. Animals were kept on a standard diet (2018 Teklad Global 18% Protein Rodent Diet, Harlan, San Pietro al Natisone, Italy). High fat diet (‘Paigen’ diet, catalog #4021.37, Hope Farm, Woerden, Netherlands) was initiated at 15 weeks of age. All mice were handled according to protocols approved by the Italian Ministry of Health and commensurate with NIH guidelines for the ethical treatment of animals.
Home cage behavioural testing
Testing was carried out in a home cage continuous monitoring apparatus (IntelliCage, New Behavior, Zürich, Switzerland). This system allowed for automatic monitoring of spontaneous and learned behaviour in a home cage environment (Galsworthy et al, 2005; Knapska et al, 2006; Onishchenko et al, 2007). The apparatus consisted of a large cage with food, bedding and shelter and an operant chamber located in each corner that had a small opening leading to a chamber where two nosepoke detectors linked to two liquid delivery stations were located. Only a single mouse could occupy the chamber at any time. A sensor detected animal entries via subcutaneous implanted transponders. Access to each drinking station could be blocked by closing an automatic door and licking was recorded by electrical contact with the drinking spout.
Animals were transferred to the testing room and transponders (T-IS 8010 FDX-B, Datamars, Switzerland) were injected subcutaneously under light anaesthesia using isoflourane 4 days before being placed in the apparatus. During the initial phase (5 h) mice were allowed access to all corners and water was freely available in each chamber. During the second phase (72 h) mice had free access to all corners but had to nosepoke to access water. After gaining access to water on any given visit to a corner, the door stayed open for 7 s and then remained closed for the rest of the visit. During the third phase (48 h), mice only had access to water in their previously least visited corner and the percentage of incorrect visits and total nosepokes in incorrect corners were measured.
Elevated plus maze and open field
The EPM consisted of two open arms (67 cm × 7 cm) and two enclosed arms (67 cm × 7 cm × 50 cm) extending from a central platform and raised 50 cm above the ground. Mice were placed in the central platform and behavioural measures (number of entries, time spent in open and closed arms, total distance) scored using a videotracking system (TSE Systems, Bad Homburg, Germany) for 5 min. For the OF, mice were placed against one side of a grey, plastic box (50 cm × 50 cm × 30 cm) and allowed to explore for 30 min. Behavioural measures (total distance, time spent, entries and total distance in centre) were scored automatically by a videotracking system (TSE Systems).
Haematology
Haematocrit and red cell, platelet and white cell counts were obtained using a haematology analyser (Hemavet 950, Drew Scientific, Waterbury, CT). Blood was collected by tail incision and collected into 10 µl of 500 mM EDTA and analysed within 1 h of collection. For flow cytometry blood was collected and lysed using Gay solution (10 mM KHCO3 and 75 mM NH4Cl, pH 7.4) and immunodetection was performed using (unless otherwise indicated purchased from eBioscience, San Diego, CA) anti-B220-phycoerthyrin (12-5892), anti-B220-allophycocyanin (17-0452), anti-CD4-PE (553052, Pharmigen, San Diego, CA), anti-CD8a-PE (12-0081) and anti-CD11b-APC (17-0112-83). Dead cells were stained with 7-amino-actinomycin D (00-6993) and excluded from the analysis.
Contact hypersensitivity
DNFB induced CHS was performed at 14 weeks of age. In brief, 35 µl of 0.5% DNFB in acetone diluted in olive oil (1:4) was applied to the shaved abdominal skin and five days later animals were challenged by applying 10 µl of 0.2% DNFB on both sides of one ear. The thickness of the ear was measured before and 24, 48 and 72 h following challenge using a micrometer (Mitutoyo, Kawasaki, Japan).
Glucose tolerance
Blood glucose concentration was measured following overnight fasting (14–16 h) using glucometer strips (OneTouch, AccuCheck Active, Roche Diagnostics, Monza, Italy) at 0, 15, 30 and 120 min after injection of a single dose of dextrose (2 mg/g, i.p. in water). Area under the curve (AUC) for plasma glucose was calculated using trapezoidal analysis.
Atherosclerotic plaques
Analysis of aortic lesions was performed as previously described (Rubin et al, 1991). In brief, mice were fasted for 16 h, given a lethal dose of avertin, and perfused with PBS. The thoracic cavities were dissected and fixed for 24 h in paraformaldehyde. The heart with attached aorta was removed, cryoprotected with sucrose overnight, frozen in OCT for serial sectioning at 10 µm, and stained with oil red and counterstained with haematoxylin. Aortic lesion size was calculated as a fraction of total lumen area in four section separated by 50 µm using a custom NIH ImageJ (Bethesda, MD) toolset macro based on colour deconvolution (OilRedQuantifier, Tiago Ferreira).
The paper explained
PROBLEM
Genetic factors play a major role in determining susceptibility to disease in human. Recently it has been shown that both changes in the sequence as well as in the number of copies of genes can influence disease risk. To find association between gene copies number and disease we developed and tested phenotyping screen in mice with duplication or deletion of large chromosomal regions in five therapeutic areas: metabolic syndrome, immune dysfunction, atherosclerosis, cancer and behaviour.
RESULTS
To test the feasibility of such a screen we tested mouse lines carrying a duplication and/or deletion of a 0.8 Mb region on chromosome 11 for physiological changes relevant to human disease. Specific deficits were found in metabolic, immune, heart and behavioural markers, showing that changes in copy number of genes in the tested region modify disease susceptibility. Follow up studies demonstrated that changes in copy number of Stat5ab explained the altered immune and metabolic function.
IMPACT
These data demonstrated that phenotyping analysis of mice with large chromosomal rearrangements is a viable approach to identify novel dosage sensitive genes affecting disease susceptibility and highlight potential role of the gene dosage of Stat5ab in susceptibility to metabolic and immunological illnesses.
Tissue collection
Animals were fasted overnight, given a lethal dose of avertin and perfused with PBS. Hearts were dissected and fixed with 4% PFA for further analysis. Liver, spleen and gonadal fat were weighed and frozen on powdered dry ice. For bone marrow analysis two tibia and two femurs were dissected.
Comparative genome hybridization
CGH was performed using the Agilent 44 K oligonucleotide probe chip (Agilent Technologies, Waldbronn, Germany; mean spacing: 22.3 kb, median spacing: 13.1 kb). Experiments were performed in duplicate with swapped dyes. Analysis was performed using DNA Analytics software (Agilent Technologies).
T cell culture and analysis
Splenic CD4+ T cells were purified using anti-CD4 MiniMacs beads (Miltenyi Biotech, Bergisch Gladbach, Germany) according to the manufacturer's protocol and were plated at 106 cells/ml in complete RPMI 1640 medium (Sigma) supplemented with 10% fetal bovine serum (FBS), 0.3 mg/ml l-glutamine, 10 µM 2-mercaptoethanol, 100 U/ml penicillin and 0.1 mg/ml streptomycin in six well plates coated overnight with anti-CD3 (5 µg/ml) and anti-CD28 (1 µg/ml) antibodies. After 5 days of culture cells were collected and prepared for intracellular flow cytometry analysis. Cells were suspended at 107 cells/ml in complete medium and cytokine secretion was induced with 10 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma) and 1 µg/ml ionomycin (Sigma). After 2 h protein secretion was inhibited with brefeldin A (1:2000, eBioscience). After 4.5–5 h, cells were washed with PBS and fixed for 20 min at room temperature. Fixed cells were stained with anti-CD4-flourescein (FITC) antibody. For intracellular cytokine detection, cells were permeabilized and stained with anti-IL17-PE and anti-IFN-gamma-APC antibodies for 20 min at room temperature. Non-specific binding was blocked by anti-FcgRII/III mouse antibodies. Cells were washed twice in permeabilization buffer (eBioscience) before flow cytometry. All antibodies were purchased from eBioscience.
Gene expression analysis
RNA from 2 × 106 cultured CD4+ T cells (RNAeasy kit, Qiagen, Hilden, Germany) was isolated following cytokine induction and 2 µg were used to prepare cDNA (cDNA synthesis kit, Amersham, Otelfingen, Switzerland). Quantitative real-time PCR on cDNA was performed using SYBR Green (Invitrogen, Carlsbad, CA) on a Luminex Bioanalyser Light Cycler 480 (Roche Diagnostics, Penzberg, Germany) and primers for IL-17 (5′-CTCCAGAAGGCCCTCAGACTA, 3′-AGCTTTCCCTCCGCATTGACAC) and ubiquitin (5′-GATCCTCTTACCCCCTCGTC, 3′-CCTTTAGGCCACTCCTTCCT), which served as an internal control. Genome-wide expression profiling (mouse 430 2.0 array, Affymetrix, Santa Clara, CA) was performed on RNA extracted from CD4+ T cells as described above. All experiments were performed as biological duplicates. Data were processed using the Bioconductor software suite (Gentleman et al, 2004). Raw .CEL files were preprocessed using RMA (Gautier et al, 2004) and ‘present/absent’ calls were made with the MAS5.0 algorithm. The differential expression analysis was restricted to probesets called ‘present’ in both WT samples, and mapping to genes within the rearranged genomic region (Ensembl v56). Differential expressed genes between WT and Dp or Df samples were identified using a moderated t-test available in the limma software package. A FDR-adjusted (Benjamini & Hochberg, 1995) cut-off of p-value < 0.25 was used as the significance threshold. The microarray dataset has been loaded in ArrayExpress. ArrayExpress accession is E-MTAB-447.
Protein analysis and Western blotting
Individual liver samples were dounce-homogenized in SDS lysis buffer (SDS 1%, glycerol 10% and 50 mM Tris–HCl, pH 6.8, 150 mM NaCl, protein inhibitors cocktail [Sigma]) and centrifuged at 10,000 × g for 15 min. Western blotting was performed as described previously (Towbin et al, 1992). Immunoblots were probed with anti-Stat3 (9132, Cell Signalling, Danvers, MA) and anti-Stat5 (9385, Cell Signalling) and signals visualized with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) and an enhanced chemiluminescent (ECL) system (GE Healthcare). Blots were washed with washing buffer (Tris-buffered saline, 0.1% Tween-20) and reprobed with anti-tubulin antibodies (Cell Signalling) to normalize for protein loading. Signals from ECL films (GE Healthcare) were quantified using NIH ImageJ software.
Statistical analysis
All data were analysed for effects of genotype, sex and treatment (where relevant) by ANOVA. Ear thickness and glucose tolerance data were analysed by repeated measure ANOVA. Post hoc testing in case of significance was performed using Fisher exact test. In cases where data were not normally distributed Kruskall–Wallis was used followed by Duncan post hoc in case of significance. Effects of genotype were assessed using ANOVA. Kaplan–Meier statistics were used to assess differences in survival time. For behavioural analysis we used mixed model of ANOVA (three-, two-, or one-way measures) followed by Holm–Sidak post hoc analysis. In cases where data from male and female mice are not displayed separately, no significant interaction between sex and genotype was identified.
Acknowledgments
We thank A. Bradley and P. Liu for AB2.2 Dp/Df11(1) ES cells and for the 129/SvEvBrd mouse strain, J. Kelly-Barrett for ES cell blastocyst injections, E. Perlas for help with histological analysis, V. Carola for advice with statistical analysis and F. Zonfrillo for animal husbandry. This work was supported by funds from EMBL. OE was supported by the European Commission FP5 EUMORPHIA and FP7 NEUROCYPRSS grant programmes. LL, TF and FC were supported by the EMBL Ph.D. Programme. RM was supported by grant SFB F28 from the Austrian Basic Research Funds (FWF). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Footnotes
→ See accompanying
Closeup by Cesar P. Canales and Katherina Walz, DOI 10.1002/emmm.201000111
Author contributions
CG and OE conceived and designed the experiments. OE, LP, LL, SR and RCP performed the experiments. OE, MA, DF and FC analysed the data. TF, RM, NL and CN contributed reagents/materials/analysis tools. CG and OE wrote the paper.
For more information
EMBL-Monterotondo Mouse Biology Unit:
Ludwig Boltzmann Institute for Cancer Research (LBI-CR):
European Bioinformatics Institute:
Center for Regenerative Medicine:
Ensemble:
http://www.ensembl.org/Mus_musculus/Info/Index
Accompanying Closeup:
http://dx.doi.org/10.1002/emmm.201000111
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
REFERENCES
- Adams DJ, Biggs PJ, Cox T, Davies R, van der Weyden L, Jonkers J, Smith J, Plumb B, Taylor R, Nishijima I, et al. Mutagenic insertion and chromosome engineering resource (MICER) Nat Genet. 2004;36:867–871. doi: 10.1038/ng1388. [DOI] [PubMed] [Google Scholar]
- Alonzi T, Maritano D, Gorgoni B, Rizzuto G, Libert C, Polli V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol Cell Biol. 2001;21:1621–1632. doi: 10.1128/MCB.21.5.1621-1632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin CP, Battey JF, Bradley A, Bucan M, Capecchi M, Collins FS, Dove WF, Duyk G, Dymecki S, Eppig JT, et al. The knockout mouse project. Nat Genet. 2004;36:921–924. doi: 10.1038/ng0904-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auwerx J, Avner P, Baldock R, Ballabio A, Balling R, Barbacid M, Berns A, Bradley A, Brown S, Carmeliet P, et al. The European dimension for the mouse genome mutagenesis program. Nat Genet. 2004;36:925–927. doi: 10.1038/ng0904-925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- Beckmann JS, Sharp AJ, Antonarakis SE. CNVs and genetic medicine (excitement and consequences of a rediscovery) Cytogenet Genome Res. 2008;123:7–16. doi: 10.1159/000184687. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;20:289–300. [Google Scholar]
- Bi W, Yan J, Shi X, Yuva-Paylor LA, Antalffy BA, Goldman A, Yoo JW, Noebels JL, Armstrong DL, Paylor R, et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum Mol Genet. 2007;16:1802–1813. doi: 10.1093/hmg/ddm128. [DOI] [PubMed] [Google Scholar]
- Biggs PJ, Vogel H, Sage M, Martin LA, Donehower LA, Bradley A. Allelic phasing of a mouse chromosome 11 deficiency influences p53 tumorigenicity. Oncogene. 2003;22:3288–3296. doi: 10.1038/sj.onc.1206384. [DOI] [PubMed] [Google Scholar]
- Bobkova D, Honsova E, Kovar J, Poledne R. Effect of diets on lipoprotein concentrations in heterozygous apolipoprotein E-deficient mice. Physiol Res. 2004;53:635–643. [PubMed] [Google Scholar]
- Brown SD, Chambon P, de Angelis MH. EMPReSS: standardized phenotype screens for functional annotation of the mouse genome. Nat Genet. 2005;37:1155. doi: 10.1038/ng1105-1155. [DOI] [PubMed] [Google Scholar]
- Carmona-Mora P, Molina J, Encina CA, Walz K. Mouse models of genomic syndromes as tools for understanding the basis of complex traits: an example with the smith-magenis and the potocki-lupski syndromes. Curr Genom. 2009;10:259–268. doi: 10.2174/138920209788488508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:15530–15535. doi: 10.1073/pnas.202604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- Friedel RH, Seisenberger C, Kaloff C, Wurst W. EUCOMM—the European conditional mouse mutagenesis program. Brief Funct Genomic Proteomic. 2007;6:180–185. doi: 10.1093/bfgp/elm022. [DOI] [PubMed] [Google Scholar]
- Galsworthy MJ, Amrein I, Kuptsov PA, Poletaeva II, Zinn P, Rau A, Vyssotski A, Lipp HP. A comparison of wild-caught wood mice and bank voles in the Intellicage: assessing exploration, daily activity patterns and place learning paradigms. Behav Brain Res. 2005;157:211–217. doi: 10.1016/j.bbr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry G, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, et al. Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, Ruedi M, Kaessmann H, Reymond A, et al. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- Herault Y, Rassoulzadegan M, Cuzin F, Duboule D. Engineering chromosomes in mice through targeted meiotic recombination (TAMERE) Nat Genet. 1998;20:381–384. doi: 10.1038/3861. [DOI] [PubMed] [Google Scholar]
- Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, Beug H, Hennighausen L, Moriggl R, Sexl V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006;107:4898–4906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- Hwa V, Little B, Adiyaman P, Kofoed EM, Pratt KL, Ocal G, Berberoglu M, Rosenfeld RG. Severe growth hormone insensitivity resulting from total absence of signal transducer and activator of transcription 5b. J Clin Endocrinol Metab. 2005;90:4260–4266. doi: 10.1210/jc.2005-0515. [DOI] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackerott M, Moldrup A, Thams P, Galsgaard ED, Knudsen J, Lee YC, Nielsen JH. STAT5 activity in pancreatic beta-cells influences the severity of diabetes in animal models of type 1 and 2 diabetes. Diabetes. 2006;55:2705–2712. doi: 10.2337/db06-0244. [DOI] [PubMed] [Google Scholar]
- Jamieson SE, Miller EN, Black GF, Peacock CS, Cordell HJ, Howson JM, Shaw MA, Burgner D, Xu W, Lins-Lainson Z, et al. Evidence for a cluster of genes on chromosome 17q11-q21 controlling susceptibility to tuberculosis and leprosy in Brazilians. Genes Immun. 2004;5:46–57. doi: 10.1038/sj.gene.6364029. [DOI] [PubMed] [Google Scholar]
- Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, et al. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome Res. 2004;14:1258–1267. doi: 10.1101/gr.1951304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Walasek G, Nikolaev E, Neuhausser-Wespy F, Lipp HP, Kaczmarek L, Werka T. Differential involvement of the central amygdala in appetitive versus aversive learning. Learn Mem. 2006;13:192–200. doi: 10.1101/lm.54706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JC. Genetics and the general physician: insights, applications and future challenges. QJM. 2009;102:757–772. doi: 10.1093/qjmed/hcp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L, Jasper H, Tepper A, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–1147. doi: 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- Laffaire J, Rivals I, Dauphinot L, Pasteau F, Wehrle R, Larrat B, Vitalis T, Moldrich RX, Rossier J, Sinkus R, et al. Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genomics. 2009;10:138. doi: 10.1186/1471-2164-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Gavrilova O, Davani B, Na R, Robinson GW, Hennighausen L. The transcription factors Stat5a/b are not required for islet development but modulate pancreatic beta-cell physiology upon aging. Biochim Biophys Acta. 2007;1773:1455–1461. doi: 10.1016/j.bbamcr.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Roy M, Kuscuoglu U, Spencer CM, Halm B, Harrison KC, Bayle JH, Splendore A, Ding F, Meltzer LA, et al. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med. 2009;1:50–65. doi: 10.1002/emmm.200900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC, Jacobs PA, Miklos GL, Davis BK, Gethmann RC, et al. Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics. 1972;71:157–184. doi: 10.1093/genetics/71.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Zhang H, McLellan A, Vogel H, Bradley A. Embryonic lethality and tumorigenesis caused by segmental aneuploidy on mouse chromosome 11. Genetics. 1998;150:1155–1168. doi: 10.1093/genetics/150.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS. The HapMap and genome-wide association studies in diagnosis and therapy. Annu Rev Med. 2009;60:443–456. doi: 10.1146/annurev.med.60.061907.093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera I, Watkins-Chow DE, Loftus SK, Hou L, Incao A, Silver DL, Rivas C, Elliott EC, Baxter LL, Pavan WJ, et al. A sensitized mutagenesis screen identifies Gli3 as a modifier of Sox10 neurocristopathy. Hum Mol Genet. 2008;17:2118–2131. doi: 10.1093/hmg/ddn110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness OP, Ayala JE, Laughlin MR, Wasserman DH. NIH experiment in centralized mouse phenotyping: the vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am J Physiol Endocrinol Metab. 2009;297:E849–55. doi: 10.1152/ajpendo.90996.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Metzger S, Rong J, Nguyen HP, Cape A, Tomiuk J, et al. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum Mol Genet. 2008;17:1137–1146. doi: 10.1093/hmg/ddn003. [DOI] [PubMed] [Google Scholar]
- Michaud EJ, Culiat CT, Klebig ML, Barker PE, Cain KT, Carpenter DJ, Easter LL, Foster CM, Gardner AW, Guo ZY, et al. Efficient gene-driven germ-line point mutagenesis of C57BL/6J mice. BMC Genomics. 2005;6:164. doi: 10.1186/1471-2164-6-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- Molina J, Carmona-Mora P, Chrast J, Krall PM, Canales CP, Lupski JR, Reymond A, Walz K. Abnormal social behaviors and altered gene expression rates in a mouse model for Potocki-Lupski syndrome. Hum Mol Genet. 2008;17:2486–2495. doi: 10.1093/hmg/ddn148. [DOI] [PubMed] [Google Scholar]
- Moriggl R, Sexl V, Piekorz R, Topham D, Ihle JN. Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity. 1999;11:225–230. doi: 10.1016/s1074-7613(00)80097-7. [DOI] [PubMed] [Google Scholar]
- Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Ywakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Onishchenko N, Tamm C, Vahter M, Hokfelt T, Johnson JA, Johnson DA, Ceccatelli S. Developmental exposure to methylmercury alters learning and induces depression-like behavior in male mice. Toxicol Sci. 2007;97:428–437. doi: 10.1093/toxsci/kfl199. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira PL, Magnol L, Sahun I, Brault V, Duchon A, Prandini P, Gruart A, Bizot JC, Chadefaux-Vekemans B, Deutsch S, et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet. 2009;18:4756–4769. doi: 10.1093/hmg/ddp438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott K, Ivins S, Hubank M, Lindsay E, Baldini A, Scambler P. Microarray analysis of the Df1 mouse model of the 22q11 deletion syndrome. Hum Genet. 2005;116:486–496. doi: 10.1007/s00439-005-1274-3. [DOI] [PubMed] [Google Scholar]
- Purcell-Huynh DA, Farese RV, Jr, Johnson DF, Flynn LM, Pierotti V, Newland DL, Linton MF, Sanan DA, Young SG. Transgenic mice expressing high levels of human apolipoprotein B develop severe atherosclerotic lesions in response to a high-fat diet. J Clin Invest. 1995;95:2246–2257. doi: 10.1172/JCI117915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378:720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- Rinchik EM, Carpenter DA, Johnson DK. Functional annotation of mammalian genomic DNA sequence by chemical mutagenesis: a fine-structure genetic mutation map of a 1- to 2-cM segment of mouse chromosome 7 corresponding to human chromosome 11p14-p15. Proc Natl Acad Sci USA. 2002;99:844–849. doi: 10.1073/pnas.022628199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265–267. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- Rubio-Aliaga I, Soewarto D, Wagner S, Klaften M, Fuchs H, Kalaydjiev S, Busch DH, Klempt M, Rathkolb B, Wolf E. A genetic screen for modifiers of the delta1-dependent notch signaling function in the mouse. Genetics. 2007;175:1451–1463. doi: 10.1534/genetics.106.067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos J, Herranz M, Perez de Castro I, Pellicer A, Fernandez-Piqueras J. A new candidate site for a tumor suppressor gene involved in mouse thymic lymphomagenesis is located on the distal part of chromosome 4. Oncogene. 1998;17:925–929. doi: 10.1038/sj.onc.1202009. [DOI] [PubMed] [Google Scholar]
- Sookoian S, Castano G, Gianotti TF, Gemma C, Rosselli MS, Pirola CJ. Genetic variants in STAT3 are associated with nonalcoholic fatty liver disease. Cytokine. 2008;44:201–206. doi: 10.1016/j.cyto.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Spitz F, Herkenne C, Morris MA, Duboule D. Inversion-induced disruption of the Hoxd cluster leads to the partition of regulatory landscapes. Nat Genet. 2005;37:889–893. doi: 10.1038/ng1597. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Steinmetz LM, Scharfe C, Deutschbauer AM, Mokranjac D, Herman ZS, Jones T, Chu AM, Giaever G, Prokisch H, Oefner PJ, Davis RW. Systematic screen for human disease genes in yeast. Nat Genet. 2002;31:400–404. doi: 10.1038/ng929. [DOI] [PubMed] [Google Scholar]
- Svenson KL, Von Smith R, Magnani PA, Suetin HR, Paigen B, Naggert JK, Li R, Churchill GA, Peters LL. Multiple trait measurements in 43 inbred mouse strains capture the phenotypic diversity characteristic of human populations. J Appl Physiol. 2007;102:2369–2378. doi: 10.1152/japplphysiol.01077.2006. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotechnology. 1992;24:145–149. [PubMed] [Google Scholar]
- Vocanson M, Hennino A, Cluzel-Tailhardat M, Saint-Mezard P, Benetiere J, Chavagnac C, Berard F, Kaiserlian D, Nicolas JF. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815–820. doi: 10.1038/sj.jid.5700174. [DOI] [PubMed] [Google Scholar]
- Wu S, Ying G, Wu Q, Capecchi MR. Toward simpler and faster genome-wide mutagenesis in mice. Nat Genet. 2007;39:922–930. doi: 10.1038/ng2060. [DOI] [PubMed] [Google Scholar]
- Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, Li D, Durum SK, Jiang Q, Bhandoola A, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Reddick RL, Burkey B, Maeda N. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J Clin Invest. 1994;94:937–945. doi: 10.1172/JCI117460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.