

Abstract
ZINC is a free public resource for ligand discovery. The database contains over twenty million commercially available molecules in biologically relevant representations that may be downloaded in popular ready-to-dock formats and subsets. The Web site also enables searches by structure, biological activity, physical property, vendor, catalog number, name, and CAS number. Small custom subsets may be created, edited, shared, docked, downloaded, and conveyed to a vendor for purchase. The database is maintained and curated for a high purchasing success rate and is freely available at zinc.docking.org.
Introduction
ZINC is a research tool for investigators seeking chemical matter for their biological targets. It incorporates purchasable compounds from over one hundred vendors and annotated compounds from over twenty databases. Since it first appeared eight years ago,1 ZINC has grown over 10-fold in size, the quality of its 3D molecular representations have been improved, and new features have been added to its interface. Whereas it retains its original focus on biologically relevant forms for molecular docking, ZINC now supports other chemoinformatic techniques.
To be useful for research, a database for ligand discovery should be big, the compounds purchasable, the molecules relevant, and in biologically applicable forms, i.e. representations that actually bind to proteins. It should be available in useful subsets, easy to search and download, and ready to use without additional processing. To maximize its coverage of chemical space, the database should include as many catalogs of biologically relevant molecules as possible. Hypothesis testing of computationally predicted ligands for proteins is fastest if the compounds are purchasable, and thus current information about expected delivery is crucial. The user should have some say in how long he is willing to wait for delivery. To minimize screening artifacts, it is common practice to filter out compounds containing problematic functional groups,2−4 but reasonable people disagree about exactly which rules should be applied. Ideally, the user should have some choice about whether to be strict or permissive about including molecules that are only sometimes problematic.
Using the relevant protonated and tautomeric molecular form for molecular docking is important, as exemplified by recent studies.5−7 A suitable database should include all relevant protonated and tautomeric forms and as few irrelevant ones as possible. Some forms such as deprotonated sulfonamides, thiols, and aromatic alcohols are only relevant at high pH, for instance in the binding site of a zinc metalloenzyme. On the other hand, some protein sites bind protonated anilines,8 but these same forms would be irrelevant decoys for most binding sites. Therefore pH dependent representations of the screening library are required.
Current opinion favors general purpose screening libraries that are filtered by physicochemical properties, particularly for molecules that have low complexity.9 Two are particularly popular: “lead-like”10 for assays and techniques where binding is not observed directly and requires higher affinities and therefore more mass; and “fragment-like”,11 for techniques such as xray crystallography, nuclear magnetic resonance, and surface plasmon resonance where high concentrations and weak affinities can be detected. For compatibility with historical studies in the field and to be generally useful, the library should also be available as a “drug-like”12 subset.
The database should allow known compounds to be looked up by the target they bind. Experimentally known compounds can be used as experimental controls, chemical probes, as starting points for hit-to-lead optimization or to calibrate docking calculations. Whereas compound bioactivity data are available in the literature, actually assembling sets of purchasable bioactives has remained labor intensive. Database searching should be fast and easy for nonexperts while offering flexibility and power for experts. It should be possible to also search by molecular similarity, substructure, physical properties, delivery time, and even name and CAS number. Libraries of purchasable natural products, metabolites, drugs and experimental compounds would be useful for research, because it would allow known bioactives to be docked and then acquired for testing. For instance, docking metabolites can be used for protein function prediction,13 and docking drugs might be useful for predicting off-target effects or repurposing.
ZINC has a unique focus, but inevitably overlaps to some extent with other databases. ChEMBL,14 PubChem,15 DrugBank,16 BindingDB,17 and TCM@Taiwan,18 for instance, all contain biological activity data but lack ZINC’s focus on docking and purchasability. ChemSpider (www.chemspider.com) combines both biological activity data and purchasing information but does not have ZINC’s focus on biologically relevant representation for docking, or its organization into discovery-oriented subsets. Procurement agents and compound vendors offer purchasability and, increasingly, e-commerce sites and downloadable screening libraries, including target-focused libraries for compounds in their collection, but they do not share ZINC’s focus on relevant 3D forms and subsets for docking.
In an effort to improve ligand discovery and virtual screening, we describe here an improved public resource of purchasable molecules that are relevant for medicinal chemistry and chemical biology. The salient criteria for ZINC are as follows. Compounds should be purchasable for rapid testing of docking hypotheses. Subsets of molecules should be easy to create. The database should contain biologically relevant molecules represented in biologically relevant protonated and tautomeric forms and be organized into subsets that are ready for screening. The database should be quick to search and download, and it should be straightforward to obtain regular updates. In many cases, it should be possible to simply look up the biological activities of molecules, or to look up compounds that are active against a particular target.
Methods
Compound Sources and Filters
ZINC was loaded from 134 commercial supplier catalogs and 36 annotated catalogs (Table 1). If a salt, the largest organic component is taken, and molecules containing an atom other than H, C, N, O, F, S, P, Si, Cl, Br, or I are removed, a limitation due to our use of the Merck Forcefield MMFF94. Only molecules passing the primary filtering rules are loaded. Filtering rules are implemented in OpenEye’s OEChem19 and are listed in text and graphical form20 at filtering.docking.org (see the Supporting Information).
Table 1. Summary of ZINC Contents by (A) Catalogs and (B) Moleculesa.
| (A) Catalogs in ZINC | Count |
|---|---|
| commercial screening compounds | 114 |
| commercial building blocks | 29 |
| commercial make-on-demand | 8 |
| procurement agents | 9 |
| boutique (expensive for screening) | 9 |
| total purchasable catalogs | 169 |
| annotated for bioactivity | 20 |
| lab use | 25 |
| total number of catalogs | 214 |
| (B) molecules in ZINC | Approximate countb |
|---|---|
| commercially available | 20 000 000 |
| annotated for bioactivity | 1 500 000 |
| “lead-like”, rule of 3.5 | 4 500 000 |
| “fragment-like”, rule of 2.5 | 550 000 |
| “Drug-like”, rule of 5 | 14 000 000 |
| total unique molecules | 34 000 000 |
Additional detailed information is provided in the Supporting Information and online at zinc.docking.org/catalogs/.
NB numbers are approximate because ZINC contents change frequently.
Molecule Preparation Protocol
Catalogs are obtained as 2D SDF files and converted to isomeric SMILES using OpenEye’s OEChem software.19 We generate up to four stereoisomers for stereochemically ambiguous molecules. A trial 3D structure is first generated using Molecular Networks’ Corina program21 to generate a single canonical conformation with the best ring puckering if applicable (arguments are -d neu, wh, rc, mc = 1, canon). Molecules are generated in four pH ranges using Schrodinger’s Epik version 2.120922 as follows. At pH of 7.05, a single best configuration is generated using the arguments: “-ph 7.05 -ms 1”. For the range pH of 6–8 (i.e., 7 ± 1), additional protonated and tautomeric forms are generated such that they have a relative population of at least 20% within that pH range using the arguments: “ph 7.0 -pht 1.0 -tp 0.20”. Similarly for high pH of 7–9.5 (i.e., 8.75 ± 0.75) and low pH of 4.5–6 (i.e., 5.25 ± 0.75), the arguments are “-ph 8.75 -pht 0.75 -tp 0.20” and “-ph 5.25 -pht 0.75 -tp 0.20” respectively. For flexibase files used by DOCK 3.6,23,24 conformations are calculated using OpenEye’s Omega library25 with the following settings: Warts(True), FromCT(False), FixMaxMatch(1), EnumNitrogen(false), EnumRing(false), EnergyWindow(12.5), MaxConfGen(100000), MaxConfs(600), RMSThreshold(0.80). Atomic charges and desolvation are calculated using AMSOL26,27using a protocol we have reported previously.28 The ZINC processing pipeline continues to evolve and is described online in more detail at http://wiki.bkslab.org/index.php/ZPP.
Calculations of Physical Properties
We use Molinspiration’s mib software (www.molinspiration.com) to calculate logP, polar surface area (PSA), molecular weight, number of hydrogen-bond donors and acceptors, and number of rotatable bonds. We use AMSOL27,29 to calculate polar and apolar desolvation energies, using the protocol of Wei.28
Graphical User Interface Software
The ZINC user interface has been completely rewritten in PHP and JavaScript/AJAX. This flexible architecture allows for easy implementation of new chemical tools, which will further facilitate development of ZINC. The interface uses jQuery 3.2 and WebME (www.molinspiration.com). This software, zinc12gui, is available for free from our Web site and will be published separately.
Clustering and Library Diversity
We assess the chemical diversity of a subset by clustering the molecules. First, we sort ligands by increasing molecular weight. Then, we use the SUBSET 1.0 algorithm30 to progressively select compounds that differ from those previously selected by at least the Tanimoto cutoff, using ChemAxon (Budapest, Hungary) axonpath fingerprints in JChem. The resulting representatives have two interesting properties. First, each representative differs from all the others by at least the Tanitmoto cutoff. Second, all the molecules in the subset are within the Tanimoto cutoff of at least one representative. Thus the representatives can be said to cover the chemical space of the subset at a given Tanimoto level.
Natural-Product-like Library
We took all natural products known to us via public sources and fragmented them with mib (www.molinspiration.com) using the −r1 flag. We accepted only fragments that had ten or more non-hydrogen atoms. We then compared them to ZINC, accepting all molecules that had a Tanimoto coefficient of 80% or higher to at least one natural product or its fragments using ChemAxon axonpath fingerprints in JChemBase (Budapest, Hungary).
Clustering ChEMBL Annotations
We wanted to combine annotations for highly related species (e.g., DRD2_RAT, DRD2_MOUSE, DRD2_HUMAN) and also to recognize that annotated ligands can be from very different chemical series. To do this, we first used the organism_class table in ChEMBL12 to group annotations of 10uM or better into six organism classes: Archaea, Bacteria, Eukaryotes, Fungi, Viruses, and Other (unclassified). For each annotation with a SwissProt code, we looked up the UniProt code and clustered annotations by UniProt prefix. SwissProt codes for which we could not find a UniProt code were left unchanged. For instance, DRD1_HUMAN and DRD1_RAT were grouped into a single annotation “DRD1-E”, E for eukaryotic whereas DYR_HUMAN and DYR_ECOLI were in two different groupings, DYR-E and DYR-B, respectively. For each grouped annotation, we used single level sphere exclusion clustering (sphex) with a minimum separation between cluster centroids of 0.85 as implemented in the JKlustor program version 5.8.2 (ChemAxon, Budapest, Hungary). This resulted in typically one to three clusters for most grouped targets; a few targets required as many as ten clusters because of the chemical diversity among the ligands.
Results
A new version of ZINC that is substantially enlarged and improved since the previous paper1 is now available. The number of catalogs of purchasable compounds has grown from 9 to over 150 (Table 1). Each molecule is now represented in multiple biologically relevant 3D forms organized into four pH ranges to better model the appropriate molecular species. The database is organized into subsets that better reflect current opinion in the field. The molecules in each subset have physical properties suitable for drug-discovery (Figure 1). There are now more subset choices: by properties, by delivery time, and by whether potentially reactive groups are present. Filtering by functional group (clean subsets) or delivery time (now subsets) results in subsets that, while smaller, retain similar property distributions (Figure 2). Twenty catalogs of compounds annotated for biological activity such as ChEMBL14 have been added, and new special subsets of natural products, metabolites, drugs, and building blocks have been created, enabling new research. The user interface and the software behind ZINC have been completely rewritten to enable new queries, such as searches by biological target and delivery time. An authentication and shopping cart system allow the user to organize research in progress. The ZINC database is updated continuously, property subsets quarterly or better. The last update of each catalog is shown on its catalog detail page. ZINC aims to follow the 90/90 rule: 90% of the molecules in ZINC are verified as being for sale within 90 days. We begin with new and improved features, followed by examples of how ZINC may be used for research.
Figure 1.

Physical properties of popular standard subsets. Color scheme: lead-like (blue); fragment-like (red); drug-like (green), all-purchasable (purple), everything (cyan). Nine properties: (A) molecular weight (Daltons); (B) calculated LogP; (C) heavy atom count; (D) rotatable bonds; (E) polar surface area (A2); (F) hydrogen-bond donors; (G) hydrogen-bond acceptors; (H) polar desolvation energy (kcal/mol); (I) apolar desolvation energy (kcal/mol).
Figure 2.

Physical properties of lead-like subsets. Color scheme: lead-like (blue); clean-leads, containing only benign chemical functionality (red); leads-now, in stock for two week delivery (green). Nine properties: (A) molecular weight (Daltons); (B) calculated LogP; (C) heavy atom count; (D) rotatable bonds; (E) polar surface area (A2); (F) hydrogen-bond donors; (G) hydrogen-bond acceptors; (H) polar desolvation energy (kcal/mol); (I) apolar desolvation energy (kcal/mol).
New Catalog Types
Purchasability is critical to rapid hypothesis testing and thus is a central focus of ZINC. In the previous version, there was only one kind of catalog: purchasable screening compounds. Now, there are eight catalog types corresponding to variation in cost, delivery time, and quantity for sale, allowing subsets and search results to better match user requirements. Building blocks are available in gram quantities for synthesis and are often also available in milligram quantities for screening. Screening compounds are available in milligram quantities at an average price of up to $100 per sample. Procurement agents incorporate many examples of these two catalog types and can simplify the logistics of compound acquisition. These three classes are all treated as in stock for delivery within two weeks, with a purchase success rate, in our experience around 85%. Make-on-demand catalogs of both building blocks and screening compounds have delivery times of 6–10 weeks and in our experience a 65% purchase success rate or better. Natural product catalogs contain compounds that the vendor attests are from natural sources. Collabocules, a portmanteau of collaborate and molecules, denotes molecules that are not for sale but may be available via collaboration, such as Brazilian natural products or compounds made by investigators in the Enzyme Function Initiative.31Annotated catalogs are databases, not vendors, in which many compounds have a biological measurement or annotation, often available via a URL. Boutique catalogs contain compounds that do not fit into any of these categories, often because they are building blocks that are not sold in small pack sizes and therefore are thought to be too expensive for screening. We assess vendors by their responsiveness and ability to supply compounds in a timely fashion to our own requests and requests of our colleagues. Unresponsive vendors are rare, but when we have evidence they are removed from ZINC.
Improved Functional Group Filtering
Filtering out compounds with potentially problematic functional groups is common practice,2−4 but reasonable people disagree about exactly which rules should be applied. In the previous version, a single set of filtering rules prevented the most reactive compounds such as triflates, alkyl halides, and percholorates from being loaded. This filtering remains in effect in the current version. But what about compounds with more nuanced reactivity, such as Michael acceptors, aldehydes, and thiols? Our approach is to load these compounds, and then flag them as not having benign functionality. Now ZINC users have a choice: standard subsets, which exclude the most reactive and problematic compounds, and clean subsets, which contain only molecules having benign functional groups (Table 2). We ourselves often choose a standard subset, because they contain a broader diversity of chemistry and nonbenign compounds are not necessarily a problem. For instance, in a recent experimental screen of 70 000 compounds against AmpC beta lactamase, none of the nonbenign compounds were hits.32 In a recent study against Dopamine D3, several hits were classified nonbenign and would have thus been missed if the clean subset had been screened.33 ZINC gives the user a choice.
Table 2. Property subsets of ZINCa.
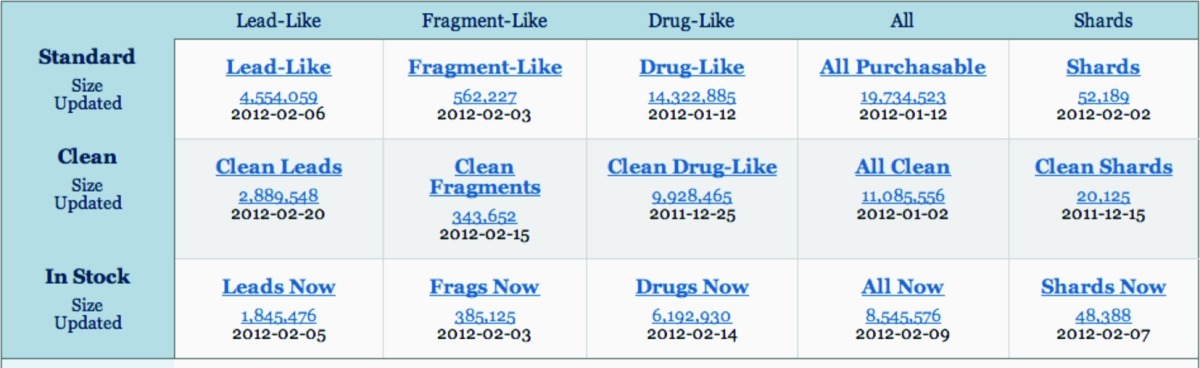
These are general purpose screening libraries representing current opinions in the field. Standard subsets are the biggest and have the most chemical diversity with 0–10 week delivery times. Clean subsets have only molecules with benign functionality; all molecules with potentially problematic functionality such as thiols, aldehydes, and Michael acceptors have been removed. Now subsets are in stock only for rapid 2 week delivery times (see Figure 2).
Improved Biologically Relevant 3D Molecular Models
Recent studies continue to demonstrate the importance of using the relevant protonated5 and tautomeric6,7 molecular form for docking. In the previous version, we calculated the most relevant forms for docking without regard to pH. As a consequence, deprotonated sulfonamides and thiols necessary for docking to metalloenzymes were also included in the docking library when no metal was present. In this context, they were, at best, biologically irrelevant distractions and, at worst, decoys that could be the cause of much wasted effort. Now, molecular representations are enumerated and classified into four pH ranges (see Methods) allowing pH-dependent subsets of ZINC. Most proteins are screened near physiological pH, requiring representations between pH 6 and 8. Some metalloenzymes require higher pH representations, between 8 and 9.5, where most thiols and sulfonamides and some aromatic alcohols can deprotonate. At the other end of physiological pH, targets such as the W191G mutant of cytochrome C peroxidase binds protonated aniline, requiring that representations between pH 4.5 and 6 be included in a docking screen8 (Figure 3). Every ZINC subset is now available in pH dependent subsets, and every search result and shopping cart may be downloaded in representations filtered by pH range using the representation selector in the results control bar (Figures 4 and 5).
Figure 3.

pH dependent representation of molecules in ZINC illustrated by examples. (A) Protonation of histidine, (B) tautomerization of folate, (C) deprotontation of sulfonamide for metals, and (D) protonation of aniline at low pH.
Figure 4.

ZINC combination search page . The drawing panel is to the left, and the SMILES field displays and allows editing of the SMILES of the current molecule. The Redraw button updates the current SMILES in the drawing panel. The similarity level selector may be used to select the Tanimoto cutoff to be used, or substructure. The (+) button adds a new SMILES. The report control bar controls how the results will be filtered and formatted. It contains page size specification, format selector, representations selector, purchasability selector, and a Run Query button. To the right is shown the property selection control bar, including the preset selector (top right) allowing easy selection of popular choices.
Figure 5.

ZINC search results as 2D tiles, the default report format. Molecules may be added to the active cart by clicking on them, making them blue. Hovering the mouse over a molecule reveals a popup with additional detail about purchasability, physical properties, and other similar molecules. The report control bar allows additional report formats and changes to purchasability and pH. It contains page number, next page button, page size specification, format selector, representations selector, purchasability selector, Add all to add all molecules to the active cart, and Refresh to apply any changes that are made.
New Bioactive Compounds
Annotated compounds can be important controls for docking calculations and thus have an important role to play in ZINC. These include catalogs of metabolites such the Human Metabolome Database35 and Metacyc,36 natural products such as TCM@Taiwan,18 approved drugs and clinical compounds such as Drugbank16 and Drugstore14 as well as actives from the medicinal chemistry literature such as ChEMBL,14 BindingDB,17 and PubChem.15 For each annotated catalog, ZINC offers both the full library and a subset containing only those that may be purchased, which we call purchasable bioactive compounds (PBCs). PBCs are of particular interest because they are both biologically active and commercially available and thus may be immediately useful as controls, probes, or chemical starting points for optimization.
Improved User Interface
The prior interface to ZINC had numerous weaknesses: slow, unreliable queries, and a single inflexible result format, for instance. Some basic queries were either arcane or impossible to formulate, and some functions were simply unreliable. To address these problems and enable new research, the web interface was redesigned. The new modular interface makes interesting questions easy to construct for both beginners and experts (Figure 4). Queries may be composed using the search menu, the quick search bar, or by composing a URL directly (see the Supporting Information). Results may be formatted in over ten ways to suit a range of needs and tastes. Premade subsets by physical properties and catalog are available for immediate download. Small subsets of up to 5000 molecules by biological target, combinations of searches, or arbitrary criteria may be created, edited, shared, prioritized, and downloaded quickly and easily. The user’s compounds may be uploaded and processed using the standard ZINC molecule processing pipeline, even for compounds that are not already in ZINC, provided they pass the basic ZINC filters (see Methods). Physically matched decoys for docking benchmarks may be generated.38 New tools to simplify compound acquisition are available. A growing number of vendors now support direct transfer of the ZINC shopping cart to their e-commerce sites. In all, seven new and four improved search types are available, which we now take up in turn.
New and Improved Search
In the first version, substructure and similarity searches were unreliable and could take minutes. It was not difficult to create searches that never finished. Now, ZINC uses ChemAxon’s JChemBase for similarity and substructure search. As a result, users can now expect to search twenty seven million molecules in a few seconds in most cases. The software that translates what is entered on the web page into an executable query, runs it, and presents the results has been completely redesigned.
New and Improved Structure Query Composition
To support more users and more devices, we now use the WebME drawing tool, a Java-free AJAX technology that runs in all browsers and on all devices we have tried. SMILES are now presented in a text window as the structure is drawn, and both the SMILES and its depiction are alternately editable. Multiple SMILES may be searched at once, each at its own similarity level. Each SMILES may be viewed and edited separately, and up to one hundred SMILES may be pasted from the clipboard and searched in a single transaction.
Improved Physical Property Specification
In the prior version, limiting values were typed as text into clumsy text fields. Now, the physical property range specification tool allows maximum and minimum values to be specified either by typing or by sliding graphical controls. Presets such as lead-like, fragment-like, and drug-like simplify the most popular choices.
Improved Combination Search
In the original version, all searches were always combined on a single page. Now, each search type has its own page, and a combination page allows complex queries when required.
New Text Queries
In the previous version, it was not possible to search ZINC by just typing text. Now, a quick search bar enables many queries with Google-like simplicity. In the prior version, ZINC did not keep track of CAS numbers and drug names. In this version names, synonyms and CAS numbers as supplied by vendors and annotated catalogs have been loaded and may be searched using the quick search bar. Text search can also search by target, by catalog, by original catalog code, by SMILES, and by ZINC ID. Pattern matching is supported using * and ? wildcards (see the Supporting Information).
New and Improved Catalog Search
In the prior version, only one catalog could be specified per search. The new version contains a catalog selection tool, allowing fine control over which catalogs will be searched. Catalogs may be selected individually or the purchasability may be specified by catalog purchasability type. The vendor page also allows searching by original catalog code, providing a list of partial matches on the fly using AJAX. Pattern matching is supported. Up to 100 catalog codes may be uploaded by pasting from the clipboard and searched in a single transaction.
New Search by Target
In the prior version, there was no biological activity data in ZINC. Now using ChEMBL, compounds annotated for biological activity may be searched using the target name or UniProt or SwissProt code. Three ChEMBL annotation types are supported: binding affinity, functional assay, and ADME/T assay data. Only compounds reported active at 10uM or better are incorporated in ZINC. In addition to original ChEMBL organism-specific annotations labeled by UniProt codes, we have grouped annotations by ChEMBL organism class and clustered them by chemotype to provide a complementary way to organize compound annotations (see Methods).
Improved Report Formats
In the prior version there was a single report format, listing all the information in ZINC for each molecule, in rows. Pagination was unreliable and downloading search results was limited. Now ZINC offers many report formats with several customization options to support many needs. Report formatting and filtering may be selected using the report control bar (Figure 4). The format selector offers six on-screen reports, seven downloadable reports, and five links to third party applications such as Chimera39 (UCSF), PyMol (Schrodinger, Inc.), and Instant JChem (ChemAxon, Budapest, Hungary). The representations selector allows the pH range to be specified. The purchasability selector is used to choose which catalog types to include and thus the purchasability of the compounds. The report control bar is available in search results reports, on the search page, and in the shopping carts.
New Scriptable Interface
In the prior version nearly all ZINC queries required the web interface. Now, all queries available via the graphical user interface may also be scripted. A comprehensive query construction syntax is supported that covers all aspects of selection, filtering, and formatting. As a result, other databases may now link to ZINC directly using queries by target, molecule, name, properties, and other criteria (see the Supporting Information). The query details tab on the results page includes embeddable JavaScript, a reusable URL and ZINC command language for each query.
New Authentication and Shopping Cart
In the previous version, there was no way to combine the results of several searches, or to retain results for future use. In the current version, a shopping cart allows the user to collect molecules from multiple searches, review them, and share them with colleagues. Compounds in a shopping cart may be prioritized by opinion using collaborative tools and an academic grading scheme of A+ through C–. The shopping cart can generate a purchasability report ranking vendors in descending order of the number of molecules in the cart they sell. The contents of the cart for each vendor can be sent to that vendor by e-mail, or directly to the vendor’s e-commerce site, if supported. Users who request a free login and authenticate can access additional features, such as multiple and persistent shopping carts and collaborative tools for prioritizing compounds.
New Decoy Maker
A new feature allows authenticated ZINC users to create physically matched decoys for the contents of a shopping cart. The decoys are drawn from ZINC using the DUD approach.38 For each ligand, 33 compounds with similar physical properties (molecular weight, logP, charge, H-bond donors, H-bond acceptors) but chemically distinct are selected from ZINC. The shopping cart is currently limited to 1500 compounds, and therefore, a maximum of 45 actives are currently used for finding decoys (45 × 33 = 1485).
Improved Upload of Molecules to Shopping Cart
In the previous version, a facility was available to upload molecules to the ZINC Web site to prepare molecules for docking using the ZINC processing pipeline, regardless of whether the molecules were already in ZINC. In the new version, the upload facility has been updated and incorporated into the shopping cart facility and is only available to authenticated users.
Having described the new and improved features of ZINC, we now turn to examples of how ZINC can be used to discover chemical reagents for biology. The first five focus on acquisition of libraries for docking screens or chemical informatics research. Three address topics that were not easy before ZINC but are now straightforward. We conclude with some not-so-simple questions that can now be answered easily using ZINC.
Example 1. Acquire a General Screening Library for Docking
The original design goal of ZINC was to be a source of general purpose screening libraries for docking. Among these, lead-like and fragment-like most closely reflect current thinking in the field. We address two common concerns by offering two variants of standard subsets. Clean subsets sacrifice chemical diversity for the expectation of fewer screening artifacts due to reactivity by filtering out even marginally reactive compounds. Now subsets trade rapid sourcing from vendor stock for decreased coverage of chemical space (Table 2). We also offer ready-to-download-and-dock versions of every catalog in ZINC. For annotated catalogs, there are two forms: the full catalog, and a subset of those that can be purchased, termed purchasable bioactive compounds (PBCs). Unique to ZINC, we also offer special subsets of purchasable bioactive compounds drawn from many catalogs: drugs, metabolites, natural products, and natural product-like compounds (see Methods). Problem: The user wishes to discover ligands with new chemistry for a protein target using docking and, thus, requires a library of purchasable lead-like compounds. Approach: Six steps are required. (A) From the Subsets menu in ZINC, choose Properties (Figure 4). (B) From among the kinds of subsets on the horizontal axis, choose lead-like. (C) From among the types of subsets on the vertical axis, choose standard. (D) Click on the word lead-like in the top left corner of the table to go to the lead-like subset detail page. (E) To download the subset in SDF format on a Unix or Mac computer in the usual physiologically relevant forms, click on the SDF link next to Unix download under Quick Links at the bottom of the subset detail page. (F) For finer control, click on the Downloads tab just below the main menu, where fine control over pH-dependent subsets is available. Variations. (i) The user may also download the database in MOL2, flexibase, or SMILES formats. (ii) The user may also select clean subsets when the tolerance for screening artifacts is very low or now subsets when compounds are needed immediately. (iii) The user may also select fragment-like, drug-like, all purchasable, and everything subsets.
Example 2. Acquire a Target-Focused Library
Target-focused subsets using data from ChEMBL can be useful in silico, to test whether a docking model can recapitulate known actives, and in vitro as commercially available experimental controls. ZINC provides target-focused purchasable docking libraries for over 2400 protein targets, 700 functional assays, and 140 ADME/T assays, all active at 10 μM or better, as per ChEMBL. Each bioactive refers back to the original literature via ChEMBL. Problem. The user wants a set of purchasable actives as controls. For instance, if the user is docking against a homology model of Schistosoma mansoni HMG CoA Reductase, he might wish to investigate how well approved drugs for the human form (statins) dock to the model. Approach. Four steps are required. (A) From the Search menu in ZINC select Targets. (B) In the text field under ChEMBL/SwissProt, type HMG. A popup menu appears after a second or two allowing the user to select HMDH_HUMAN, the first choice. (C) The user clicks on Run Query, and in a few seconds, approximately 50 compounds appear as tiles. (D) The user downloads these in the usual biologically relevant forms in mol2 format by selecting mol2 from the format selector in the results control bar and then clicking on Refresh. Variations. (i) The user may also choose to download in SDF or SMILES format. (ii) The user may request only a single form at pH 7 by selecting single in the representations selector and clicking on Refresh. (iii) The user may specify all compounds, not just the purchasable ones, by choosing everything from the purchasability selector and clicking on Refresh. (iv) The user may type the word all into the page size field or click the All button to specify that all available compounds should be downloaded. Authenticated users may download more molecules than anonymous users. (v) A complete listing of all available purchasable annotated libraries using ChEMBL annotations is available by choosing Targets from the Subsets menu. (vi) Annotations that have been clustered by organism type (Archaea, Eukaryotes, Bacteria, Fungi, Viruses, and Other) as per ChEMBL organism class and UniProt prefixes are also available by using the selector under Clustered Target instead of ChEMBL/SwissProt.
Example 3. Find Purchasable Analogs by Catalog
The user might want to explore around an initial hit by testing analogs. Analogs may also be used to expand a set of known actives from ChEMBL to include possible actives for docking or testing. In cases where no ChEMBL actives are for sale for an annotation, searching for purchasable analogs may yield commercially accessible controls or probes. There are two ways to look for analogs: overall similarity illustrated here and substructure, illustrated in example 4 below. Both are supported in ZINC using ChemAxon’s JChemBase. Problem. Find purchasable analogs of an experimental hit. For instance, in a phenotypic screen, the drug cetirizine was a hit. The user thus wants to dock and purchase analogs of cetirizine, an H1 receptor antagonist, to investigate structure–activity relationships. Approach. Eight steps are required. (A) In the quick search bar, enter cetirizineand click Go. Both enantiomers are displayed. (B) To see full details about the compound on the right, click on its ZINC ID number, 19364230, in the top left corner of the tile to display its molecule detail page. (C) To find close analogs, click on the 80% link under the word Analogs, on the right side of the page. At time of writing, 26 purchasable compounds, including stereoisomers, are available. (D) Download them as in the previous examples. (E) Add them all to the shopping cart by clicking on Add all in the top right corner of the page. (F) Switch to the shopping cart by clicking on Active cart in the top right corner of the page. (G) Click on purchasability report for an analysis of how to buy them. As of the time of writing, Toronto Research Chemicals (TRC) sold more of these compounds than any other vendor. (H) To enquire about price and availability, click on email quote request. If the user is logged in, the e-mail may be sent directly. If anonymous, the text may be cut and pasted to an e-mail client. Variations. (i) To find more analogs, select a more permissive similarity level, e.g. 70%. (ii) Instead of using Add all, put compounds in the cart individually by clicking on them. Compounds in the active cart are colored light blue. (iii) Authentication allows persistent carts and other features not available to anonymous users.
Example 4. Acquire Fragment- and Scaffold-Based Libraries
Problem. Obtain a purchasable chemical library for docking. For instance, the user has read that molecules containing quinuclidine (SMILES: C1CN2CCC1CC2) are often active against muscarinic targets. To investigate this, the user wishes to download a library of fragment-like compounds containing this functional group for docking. Approach. Seven steps are required. (A) From the Search menu in ZINC, select Combination. (B) Clear any previous search by clicking on the “blank sheet” icon to the right of the smiling face in the WebME drawing tool. (C) In the text field above the drawing panel, enter C1CN2CCC1CC2, click the update drawing icon, and select substructure from the similarity level selector or draw it using the drawing tool. (D) In the properties panel to the right, select the predefined set fragment-like from the preset selector in the top right corner. (E) Run the query by clicking Run query. (F) Page through five pages to see the results using the next button in the results control bar. At the time of writing, there were 220 purchasable compounds. (G) To download these in SMILES text format, select SMILES from the format selector, type ALL in the Page Size field, and click Refresh. Variations. (i) To download in mol2 or SDF formats, make the appropriate choice from the format selector and click Refresh. (ii) To choose only compounds available for 2 week delivery, select in-stock only from the purchasability selector and click Refresh.
Example 5. Acquire Screening Libraries of Bioactive Compounds
Problem. The user is interested in discovering the function of proteins by docking and, therefore, wishes to only dock biogenic molecules. Approach. Four steps are required. (A) From the Subsets menu in ZINC select Properties. (B) Click on the Special tab to view the special ZINC subsets. (C) Select Znp–ZINC Natural Products, by clicking on the Znp link. (D) Download as before, either under Quick Links or using the Downloads tab. Variations. (i) Other special subsetsinclude drugs, research compounds, building blocks, and natural-product-like compounds. (ii) Additional libraries of purchasable bioactive compounds are available as purchasable bioactive compounds(PBCs), by choosing Catalogs from the Subsets menu and clicking on the PBCs tab.
Example 6. Identify Specialist Vendors
Problem. The user wishes to discover which vendors sell the most bioactives for a particular target. For instance, the user is interested in exploring the bias between beta-arrestin and G-coupled signaling pathways of beta-2 adrenergic receptor ligands. To study this experimentally, the user wants to buy many compounds at low cost and would thus like to know which vendor sells the most known to bind at 1 μM or better. Approach. Ten steps are required. (A) From the Subsets menu in ZINC, choose Targets. (B) Click on the Binding 1uM tab. (C) Use the web browser search feature to locate “beta-2 adrenergic”. Note that several species are listed. (D) Next to the human annotation, ADBR2_HUMAN, click on the Catalogs link to perform a market analysis. At the time of writing, the company Sigma Aldrich sold 29 compounds, by far the most of any single vendor, not counting Chemonaut and Molport, which are purchasing agents. (E) To view these compounds, click on Overview. (F) To add these to the shopping cart, click on Add all. (G) There is more than one page, so click Next to go to page 2 and click on Add all again. (H) Click on Active cart in the top right of the page to view the shopping cart. (I) Click on Purchasability Report to see vendors sorted in decreasing order of the number of compounds they sell. (J) Click on e-mail quote request to compose a message to the vendor. Authenticated users may modify and send this email directly to the vendor, whereas anonymous users must copy and paste the text into their own e-mail program to prevent spam. Variations. (i) Over 3000 targets are available to choose based on ChEMBL bioactivity data. (ii) The user may combine the ligands from several annotations (e.g., rat, mouse and human) by adding them each to the shopping cart, and individual compounds in the cart may be removed by clicking on them.
Example 7. Decoys for Docking
ZINC can generate DUD-style decoy molecules with similar physical properties chemically dissimilar to the actives as a bias-controlled docking benchmark.38 Decoys are useful for testing that docking enrichment is not artificial and therefore misleadingly good due simply to gross physical differences between the actives and the decoys. Problem. The user wants to benchmark docking using actives and physically matched decoys. At a high level, the approach involves logging in, putting the actives or their representatives into a shopping cart, clicking on Generate Decoys, confirming, waiting a few minutes, and then downloading from a new cart. Approach. Ten steps are required. (A) Log in by clicking on sign in, in the top right of the page. (B) Create a new cart, select it, and put the actives into it. There are many ways to supply actives: using ChEMBL annotations, uploading ZINC IDs or SMILES while creating a cart, or hand picking compounds from several queries. In this example, we will generate decoys for GRIK4, the glutamate receptor KA-1 (SwissProt code Q16099). (C) Click on Manage my carts (must be logged in). (D) In section 2, for Cart Name, type GRIK4 and click Submit. Note that GRIK4 is now the active cart. (E) From the Search menu in ZINC, choose Targets. (F) From the annotation type selector, choose Binding 1uM. In the text field, enter Q16099 and click Run Query. (G) Click on Add all to add these to the GRIK4 cart. (H) Go to the cart by clicking on Active Cart. (I) In the cart, click on Generate Decoys. On the confirmation page that follows, click Generate. Only the first 45 compounds in the cart will be used for creating decoys. A new cart called GRIK4-decoys is created and is initially empty. The decoy procedure runs asynchronously in the background and loads the decoys into the GRIK4-decoys cart when it has finished, which typically takes a few minutes and depends on system load. (J) When ready, the decoys may be downloaded in SMILES, mol2, or SDF. All other features work in the usual way.
Example 8. Prioritize Hits for Purchase
Problem. The user has identified 100 molecules she likes by docking or chemical informatics. Before purchasing, she wants to enlist the help of her colleagues to help prioritize them for purchase. We call this a hit picking party, a standard procedure in our lab. Buying compounds can be expensive, and a hit picking party can help avoid blunders and solicit experience learned from other projects. Hit picking parties can also be didactically useful, because they foster discussion about the biophysics and trade-offs involved in protein–ligand binding. Approach. (A) Log in to ZINC by clicking on sign in, in the top right of the page. (B) Go to cart management by clicking on Manage my carts (top right). (C) Create a new cart named MyGreatProject by using option 2, typing MyGreatProject in the text field, and clicking on Submit. Optionally, the user may populate the cart by uploading a file of ZINC IDs or SMILES, one per line. In the large text area, provide a description of the project. The new cart is now the active cart. (D) In section 4 of the cart management page, add Teammates to the cart by typing part of their user name or part of their email address in the text field and clicking on the plus (+) button. Each teammate requires a docking.org ID, which is free on request. For subsequent projects, the cart can inherit the teammates from a previous project by clicking on the buttons displayed below. There are two kinds of teammates: Peers and PIs. PIs can see all teammates’ scores, whereas Peers can only see their own scores. When finished, click on update users to finalize changes. (E) Add molecules to the cart. (F) Teammates may view the cart by choosing carts from the subsets menu, then rating the molecules using an A+ to C− scoring scheme. (G) The user may see teammates’ ratings by viewing the cart and clicking on hit picking party above the report control bar.
ZINC can answer many other questions that have been challenging in the past. A few brief examples illustrate some possibilities.
What Targets Does a Vendor’s Library Hit?
Before purchasing a library, the user wishes to learn what targets it hits. Approach. From the Subsets menu in ZINC, choose Catalogs. Click on the Targets or Clustered targets tabs to see a target-focused analysis of the vendor’s library. Click on the catalogs link of any target to see a ranked list of vendors selling compounds for that target. Two complementary kinds of annotation are available: native species-specific ChEMBL annotations and annotations grouped by organism class and then clustered by chemotype.
Which Approved Drugs Can Be Purchased As Pure Compounds?
Approach. From the Subsets menu in ZINC, choose properties and, then, choose the special tab. For drugs, click on Zdd for the ZINC drug database. This subset is ready to download, or click on Sample Molecules to browse. All the molecules in all special subsets are purchasable. The user can also see which drugs are not for sale by clicking on Sample Molecules to browse and then choosing not for sale from the purchasability selector in the results control bar. These are drugs that, according to ZINC, are not for sale as pure substances. If it is available, we would appreciate being informed of where it can be purchased so that we can update ZINC. Results can be obtained for natural products and metabolites also.
Which Library Is Most Diverse?
Library diversity is a huge topic with many nuances, but for many users, a simplistic analysis from ZINC may be good enough for some applications. Approach. In the subsets menu in ZINC, choose catalogs and, then, click the diversity tab. This table displays the clustered diversity of every subset. The user can sort by the diversity at calculated at four levels. For instance, clicking on 60% sorts by the number of clustered representatives at the Tanimoto 60% level (see Methods). The results in ZINC show that there is far more chemical diversity represented in annotated catalogs such as ChEMBL and BindingDB with sixteen and twelve thousand representatives at the 60% Tanimoto level respectively than is present in even the more diverse single vendor libraries such as Vitas-M and ChemBridge, with about eight thousand each.
How Can I Dock and Purchase Vitamin K?
Approach. In the quick search bar, type vitamin K and click Go. The molecule, including purchasing information and ready-to-dock files, is available. Drug names are loaded from vendor and annotation catalogs. Wildcards are supported. Thus c?lexawill match celexa and methotr* will match methotrexate and methotrimeprazine. The pattern *azepam will find purchasable drugs that hit GABA and *pride will find drugs that hit dopamine D2, as well as some other compounds. Some names will not hit. This could be because the compound is not in ZINC or we do not yet know about the name used. CAS numbers may be used, also with wildcards. Our list of CAS numbers is drawn from vendor catalogs, and as such may be incomplete.
The protocols and instructions described here will evolve as ZINC develops. For the current versions of these protocols, please visit our Web site, http://wiki.bkslab.org/index.php/Zinc2012paper.
Discussion
Three main results emerge from this work. First, ZINC represents a much larger database of commercially available compounds for ligand discovery and virtual screening than before. The database is actively maintained and is freely available online (http://zinc.docking.org). Second, ZINC molecules are represented in improved, biologically relevant forms and organized into discovery-relevant subsets such as lead-like and fragment-like that are ready for downloading and docking. Third, ZINC can now be used to discover the biological targets for a compound by simple mouse-click, or to find purchasable compounds for a given target, based on literature annotations. We take up each of these results in turn.
At over 20 million purchasable molecules, ZINC is the largest database of commercially available compounds for virtual screening. The over 150 vendor catalogs used to create ZINC provide a diverse range of chemistry, including drugs such as ketoconazole, metabolites such as thebaine, natural products such as harmaline, and synthetic compounds such as 2-(3,5-dihydroxyphenyl)ethylamine. ZINC catalogs are categorized by type and delivery time to allow the user to select molecules that match research goals. For projects with tight deadlines, subsets consisting only of compounds from stock offer rapid delivery and high acquisition success rates, whereas projects that can afford to wait can benefit from on average twice as many molecules, albeit with lower acquisition success rates. When a target screen cannot tolerate reactive groups, subsets containing only benign chemistry can be used, whereas other projects can benefit from subsets with significantly more compounds, but also a higher risk of artifacts.
Docking is often challenging, frequently because of all the things that can go wrong with a docked molecule. Possible problems include incorrect protonation state, irrelevant tautomeric form, wrong conformation, and commercial unavailability. ZINC improves docking compared to previous versions by providing better representations of biologically relevant molecules organized into refined subsets that are relevant for discovery. A primary focus of ZINC is on the protonation state and tautomeric form of a molecule. In our own docking projects, we are constantly examining molecular forms that are automatically generated by our pipeline of programs. When we find errors, we fix the pipeline, or, if necessary, individual molecules themselves. Another common problem is wrong conformations, often associated with less common aliphatic rings or functional group combinations. When we identify these problems, often during a hit picking party, we compare conformations against the Cambridge Structural Database40 and amend the molecule, our protocol, or both as needed. We have found docking to be an outstanding way to find errors in a library and in our molecule-processing pipeline, because incorrect molecules often rise to the top of the docking hit list, demanding scrutiny. Even if the molecule is ideal from the chemical and physical perspective, if it is unavailable to purchase, too expensive, or takes too long to arrive, it is also irrelevant. ZINC tackles these concerns by updating and reclassifying catalogs continuously. A static database would rapidly become less useful since at least one million new compounds become available every year, and more than half as many become unavailable. Database curation for relevance is an ongoing, unavoidable process if we are to have discovery-relevant docking libraries.
When looking for ligands for a target, the simplest place to start is often the literature. ZINC incorporates a subset of ChEMBL, the medicinal chemistry database, to provide a simple way to find purchasable compounds for over 3000 targets and biological activities for hundreds of thousands of compounds. Compounds with experimentally known affinities can be useful as experimental controls, chemical probes, as starting points for hit-to-lead optimization, as well as controls for a docking calculation. Until recently, assembling sets of purchasable bioactives for a target could take weeks of combing the literature. Now with ZINC and ChEMBL, compounds are available in ready-to-dock formats in seconds, making it easier to test docking models whenever ligands are available in the literature. ZINC also provides the ability to generate physically matched yet chemically distinct decoys, which can help avoid library bias in docking calculations.
Building ZINC is prone to numerous pitfalls, perhaps the biggest of which is irrelevant molecules. Among these, incorrect protonation states and tautomeric forms are decoys that waste an investigator’s time. Compounds that are no longer for sale are irrelevant, wasting time with false hopes of acquisition. Keeping these distracting molecules out of ZINC is an almost Sisyphean task. And there is more to drive the perfectionist to tears: catalogs that are not yet in ZINC, biological activity errors in the literature, data transcription errors, and coding errors in the interface. Whenever a problem is brought to our attention, we aim to investigate it and fix it promptly if we can. ZINC by its very nature is imperfect, as each improvement allows new problems to surface.
Notwithstanding these caveats, ZINC has emerged as a pragmatic and useful tool. It supports molecular docking and chemoinformatics and is a general purpose source of relevant molecules for other techniques. Increasingly, biological activity for compounds can be simply looked up, or purchasable bioactives for a given target can be identified. We hope ZINC will help investigators to find reagents for their biological targets and to discover the mechanism of action of their bioactive compounds.
Acknowledgments
We thank NIGMS for financial support GM71896 (to B. Shoichet and J.J.I.) and the National Research Service Award Kirschstein Fellowship F32GM096544 (to R.G.C.). We are grateful to the commercial software suppliers who support ZINC: ChemAxon (Budapest, Hungary) for the use of JChem, Marvin, Instant JChem and Cartridge; OpenEye Scientific Software (Santa Fe, NM) for the use of OEChem, Omega,25 Ogham and Quacpac; Molecular Networks GmbH for the use of Corina; Molinspiration (Bratislava, Slovakia) for the use of mib and WebME; Peter Ertl for the use of Java Molecular Editor. We thank Brian Shoichet and Henry Lin for reading the manuscript. We thank lab members, the reviewers and many ZINC users worldwide for helpful criticism.
Glossary
Abbreviations and Acronyms
- ADME/T
absorption, distribution, metabolism, excretion and toxicity
- CAS
Chemical Abstracts Service
- FDA
Food and Drug Administration
- ChEMBL
Medicinal Chemistry Database of the European Molecular Biology Laboratory
- PBC
purchasable bioactive compounds
- SMILES
simplified molecular-input line-entry specification
- SwissProt
Swiss Protein Resource
- UniProt
Universal Protein Resource
- URL
uniform resource locator
- ZINC
ZINC is not commercial
Supporting Information Available
Tables summarizing the contents of commercial catalogs and annotated databases in ZINC, filtering rules, a guide to the ZINC query construction syntax including some sample query scripts, a quick search bar guide, how to link to ZINC, and a description of the ZINC processing pipeline. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Irwin J. J.; Shoichet B. K. ZINC: A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charifson P. S.; Walters W. P. Filtering databases and chemical libraries. J. Comput.-Aided Mol. Des. 2002, 16, 311–323. [DOI] [PubMed] [Google Scholar]
- Rishton G. M. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discovery Today. 2003, 8, 86–96. [DOI] [PubMed] [Google Scholar]
- Huth J. R.; Mendoza R.; Olejniczak E. T.; Johnson R. W.; Cothron D. A.; Liu Y.; Lerner C. G.; Chen J.; Hajduk P. J. ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 2005, 127, 217–224. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Raushel F. M.; Shoichet B. K. Virtual Screening against Metalloenzymes for Inhibitors and Substrates. Biochemistry. 2005, 44, 12316–12328. [DOI] [PubMed] [Google Scholar]
- Martin Y. C. Let’s not forget tautomers. J. Comput.-Aided Mol. Des. 2009, 23, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milletti F.; Vulpetti A. Tautomer preference in PDB complexes and its impact on structure-based drug discovery. J. Chem. Inf. Model. 2010, 50, 1062–1074. [DOI] [PubMed] [Google Scholar]
- Brenk R.; Vetter S. W.; Boyce S. E.; Goodin D. B.; Shoichet B. K. Probing molecular docking in a charged model binding site. J. Mol. Biol. 2006, 357, 1449–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann M. M.; Leach A. R.; Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [DOI] [PubMed] [Google Scholar]
- Teague S. J.; Davis A. M.; Leeson P. D.; Oprea T. I. The design of leadlike combinatorial libraries. Angew. Chem., Int. Ed. 1999, 38, 3743–3748. [DOI] [PubMed] [Google Scholar]
- Carr R. A.; Congreve M.; Murray C. W.; Rees D. C. Fragment-based lead discovery: leads by design. Drug Discovery Today 2005, 10, 987–992. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J.. Experimental and computational approaches to estimate solubility and permeability in drug discovery development settings. Adv. Drug Delivery Res. 1997, 23. [DOI] [PubMed] [Google Scholar]
- Hermann J. C.; Marti-Arbona R.; Fedorov A. A.; Fedorov E.; Almo S. C.; Shoichet B. K.; Raushel F. M. Structure-based activity prediction for an enzyme of unknown function. Nature 2007, 448, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Cheng T.; Wang Y.; Bryant S. H. PubChem as a public resource for drug discovery. Drug Discovery Today 2010, 15, 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C.; Law V.; Jewison T.; Liu P.; Ly S.; Frolkis A.; Pon A.; Banco K.; Mak C.; Neveu V.; Djoumbou Y.; Eisner R.; Guo A. C.; Wishart D. S. DrugBank 3.0: a comprehensive resource for ’omics’ research on drugs. Nucleic Acids Res. 2011, 39, D1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Y. TCM Database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in silico. PLoS One 2011, 6, e15939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [DOI] [PubMed] [Google Scholar]
- Schomburg K.; Ehrlich H. C.; Stierand K.; Rarey M. From structure diagrams to visual chemical patterns. J. Chem. Inf. Model. 2010, 50, 1529–1535. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Gasteiger J.; Todeschini R.; Mauri A.; Livingstone D.; Ertl P.; Palyulin V. A.; Radchenko E. V.; Zefirov N. S.; Makarenko A. S.; Tanchuk V. Y.; Prokopenko V. V. Virtual computational chemistry laboratory--design and description. J. Comput.-Aided Mol. Des. 2005, 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [DOI] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 2005, 5, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Flexible ligand docking using conformational ensembles. Protein Sci. 1998, 7, 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer C. J.; Truhlar D. G. An SCF solvation model for the hydrophobic effect and absolute free energies of aqueous solvation. Science 1992, 256, 213–216. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Truhlar D. G. AM2-SM2 and PM3-SM3 parameterized SCF solvation models for free energies in aqueous solution. J. Comput.-Aided Mol. Des. 1992, 6, 629–666. [DOI] [PubMed] [Google Scholar]
- Wei B. Q.; Baase W. A.; Weaver L. H.; Matthews B. W.; Shoichet B. K. A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 2002, 322, 339–355. [DOI] [PubMed] [Google Scholar]
- AMSOL, 6.8; University of Minnesota, Minneapolis, 2002. [Google Scholar]
- Voigt J. H.; Bienfait B.; Wang S.; Nicklaus M. C. Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput. Sci. 2001, 41, 702–712. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Allen K. N.; Almo S. C.; Armstrong R. N.; Babbitt P. C.; Cronan J. E.; Dunaway-Mariano D.; Imker H. J.; Jacobson M. P.; Minor W.; Poulter C. D.; Raushel F. M.; Sali A.; Shoichet B. K.; Sweedler J. V. The Enzyme Function Initiative. Biochemistry 2011, 50, 9950–9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaoglu K.; Simeonov A.; Irwin J. J.; Nelson M. E.; Feng B.; Thomas C. J.; Cancian L.; Costi M. P.; Maltby D. A.; Jadhav A.; Inglese J.; Austin C. P.; Shoichet B. K. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J. Med. Chem. 2008, 51, 2502–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; Coleman R. G.; Setola V.; Irwin J. J.; Fan H.; Schlessinger A.; Sali A.; Roth B. L.; Shoichet B. K. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 2011, 7, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Eisner R.; Young N.; Gautam B.; Hau D. D.; Psychogios N.; Dong E.; Bouatra S.; Mandal R.; Sinelnikov I.; Xia J.; Jia L.; Cruz J. A.; Lim E.; Sobsey C. A.; Shrivastava S.; Huang P.; Liu P.; Fang L.; Peng J.; Fradette R.; Cheng D.; Tzur D.; Clements M.; Lewis A.; De Souza A.; Zuniga A.; Dawe M.; Xiong Y.; Clive D.; Greiner R.; Nazyrova A.; Shaykhutdinov R.; Li L.; Vogel H. J.; Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R.; Altman T.; Dale J. M.; Dreher K.; Fulcher C. A.; Gilham F.; Kaipa P.; Karthikeyan A. S.; Kothari A.; Krummenacker M.; Latendresse M.; Mueller L. A.; Paley S.; Popescu L.; Pujar A.; Shearer A. G.; Zhang P.; Karp P. D. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010, 38, D473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K.; Irwin J. J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Allen F. H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. B. 2002, 58, 380–388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.