

Abstract
Rationale
Little is known about the contribution of bone marrow-derived progenitor cells (BMPCs) in the regulation endothelial barrier function as defined by microvascular permeability alterations at the level of adherens junctions (AJs).
Objective
We investigated the role of BMPCs in annealing AJs and thereby in preventing lung edema formation induced by endotoxin (LPS).
Methods & Results
We observed that BMPCs enhanced basal endothelial barrier function and prevented the increase in pulmonary microvascular permeability and edema formation in mice following LPS challenge. Co-culture of BMPCs with endothelial cells induced Rac1 and Cdc42 activation and AJ assembly in endothelial cells. However, transplantation of BMPCs isolated from sphingosine kinase-1-null mice (SPHK1-/-), having impaired S1P production, failed to activate Rac1 and Cdc42 or protect the endothelial barrier.
Conclusion
These results demonstrate that BMPCs have the ability to re-anneal endothelial AJs by paracrine S1P release in the inflammatory milieu, and the consequent activation of Rac-1 and Cdc42 in endothelial cells.
Keywords: progenitor cells, endothelial permeability, S1P signaling, RhoGTPases
Introduction
The endothelial cell monolayer lining blood vessels forms a semi-permeable barrier that performs the vital tasks of regulating tissue fluid homeostasis and transmigration of blood cells. Bone marrow-derived endothelial progenitor cells induce angiogenesis, and have been touted for therapeutic angiogenesis in the treatment of myocardial ischemia and other diseases requiring re-vascularization(1). Recent studies showed that transplantation of bone marrow-derived mesenchymal cells can also limit lung inflammatory in experimental models (2, 3). As disruption of the endothelial barrier in vascular inflammation leads to tissue edema and injury and there are no current means to mitigate vascular leakage, we addressed the possible role of bone marrow-derived progenitor cells (BMPCs) in regulating endothelial barrier function.
The phospholipid sphingosine-1-phosphatase (S1P), generated by platelets, RBCs and endothelial cells, has an endothelial barrier protective function (4, 5). Sphingosine kinases phosphorylate sphingosine leading to formation of S1P (6). Platelets lack the S1P degradation enzyme sphingosine-1-lyase, thus release S1P in the circulation (7). In addition, RBCs and endothelial cells produce abundant amounts of S1P (8-10). The endothelial barrier promoting effect of S1P is concentration-dependent and characterized by a rapid increase in transendothelial electrical resistance (4, 11). S1P signaling on the basis of its effects on immune and endothelial cells is also a modulator of immune and inflammatory responses (12). In the murine model of LPS-mediated acute lung injury, S1P was shown to attenuate pulmonary vascular leakage and inflammation (12, 13). The S1P effects depend on ligation of Gi coupled Edg-1 and Edg-3 receptors (14). In the present study, we surmised that BMPCs are an important source of S1P, and thereby contribute to BMPC-mediated effects on the endothelium.
We previously showed that re-establishing endothelial contact and junction integrity following injury requires the activation of the RhoGTPases Cdc42 and Rac1 in endothelial cells (15). Thus, we also investigated their involvement in mediating the underlying effects of BMPCs on endothelial junctions. We demonstrate here that homing of BMPCs to lungs profoundly enhanced endothelial barrier function at the level of adherens junctions (AJs) and prevented the LPS-induced increase in endothelial permeability by the generation of S1P and consequent activation of Rac1 and Cdc42 signaling.
Materials & Methods
Mice
All mice were bred and maintained according to NIH guidelines and experiments were approved by the Animal Care and Use Committee.
Isolation of mouse BMPCs
Mouse BMPCs were isolated using modifications of methods based on published markers and methods used for human and rat BMPC isolation (see online Material and Methods)(16-18). The phenotype of confluent cell population was assessed by determining expression of protein markers by FACS analysis (17).
Mouse lung vascular endothelial cells
Mouse lung vascular endothelial cells were isolated as described (19).
Pulmonary uptake of BMPCs
BMPCs labeled with rhodamine fluorophore (CMTMR, Molecular Probes) were injected into mice (3×105 cells/mouse) through the external jugular vein. The animals were then sacrificed at different times to determine the location of BMPCs within mouse lung microvasculature by confocal microscopy. In other studies, we labeled 3×105 BMPCs with 111Indium oxine (20) to determine organ uptake of BMPCs following i.v. injection.
Flow cytometry
Cells were detached using 1 mmol/L EDTA in PBS, and incubated with the following antibodies for 30 min prior to FACS analysis: phycoerythrin (PE)-labeled anti-murine Sca1 (BD PharMingen), and anti-murine CD31 (Becton-Dickinson); allophycocyanin (APC)-labeled anti-murine c-Kit and CD34 antibodies (BD PharMingen); mouse anti-human CD133 (BD PharMingen), rabbit anti-human VE-cadherin (Santa Cruz), and rat anti-mouse CD45 (BD PharMingen). Rabbit anti-mouse or goat anti-mouse FITC (Vector) or Alexa Fluor-labeled anti-goat 594 was used as the secondary antibody (Invitrogen).
BMPC injection
Mice in all cases were injected with 3×105 BMPCs (in 200 μl of EBM-2MV medium) through the external jugular vein (17).
LPS challenge
All mice (except those used for mortality studies) received a single sub-lethal dose of 10 mg/kg bw LPS (E. coli 0111:B4, Sigma L-2630) i.p. Animals were sacrificed at 6 hr after LPS administration for Kf,c determinations (see below) and at 12 hr for final lung extravascular lung water measurements (21).
Rho GTPase activity assays
Rac1, Cdc42, and RhoA activities were measured using the GSTrhotekin-Rho binding domain that precipitates activated Rac1 and Cdc42 (22, 23). Mouse endothelial cell monolayers were co-cultured with BMPCs for different times. Cell lysates were clarified by centrifugation at 4 °C and 1000 × g for 5 min and equal volumes of lysates were incubated with PAK-GST protein beads (Cytoskeleton Inc.) at 4 °C for 2 h. Beads were washed 3× and bound Rac1 or Cdc42 was eluted by boiling each sample in Laemmli sample buffer to elute samples. Total cell lysates were electrophoresed on 12.5% SDS-polyacrylamide gels and immunoblotted with either mouse anti-Rac1 or rabbit anti-Cdc42 antibody.
siRNA transfection
siRNA constructs were transduced in cells by electroporation. Mouse endothelial cells, 60% confluent, were trypsinized, mixed with 2.5μg siRNA for Rac1, 10 μg for Cdc42, or 3.0 μg for non-silencing siRNA along with 100μl of transfection solution (Thermo Scientific Dharmacon® siGENOME®, USA). Cells were electroporated using a nucleofector device with manufacturer's protocol (VPI-1001, Amaxa, MD). Cells were removed, mixed in EBM-MV2, and plated on either 100-mm dishes, or Transwell chamber filters, or gold-plated 10-well electrodes for functional analyses.
Statistical analysis
Data are presented as mean ± SEM. Differences were tested using analysis of variance (ANOVA with post-hoc comparisons using unpaired t-test or Mann-Whitney tests as appropriate). Survival data were analyzed using Peto-Peto-Wilcoxon test. p< 0.05 denotes significant difference.
Results
FACS analysis
Cultured wt BMPCs were positive for various stem/progenitor cell markers CD133 (92%), Sca1 (83%), CD34 (75%) (Figure 1A). BMPCs based on their high expression of these markers are therefore referred to as progenitor cells. Only a small number of BPMCs expressed the mature endothelial markers CD31 (3%) and VE-cadherin (12%), and generic myeloid cell marker CD45 (0.3%) (Figure 1A). Quantitative real-time RT-PCR analysis also showed minimal expression of CD45 in BMPCs (online Figure 1C).
Figure 1. FACS analysis and lung uptake of mouse bone marrow-derived progenitor cells (BMPCs).
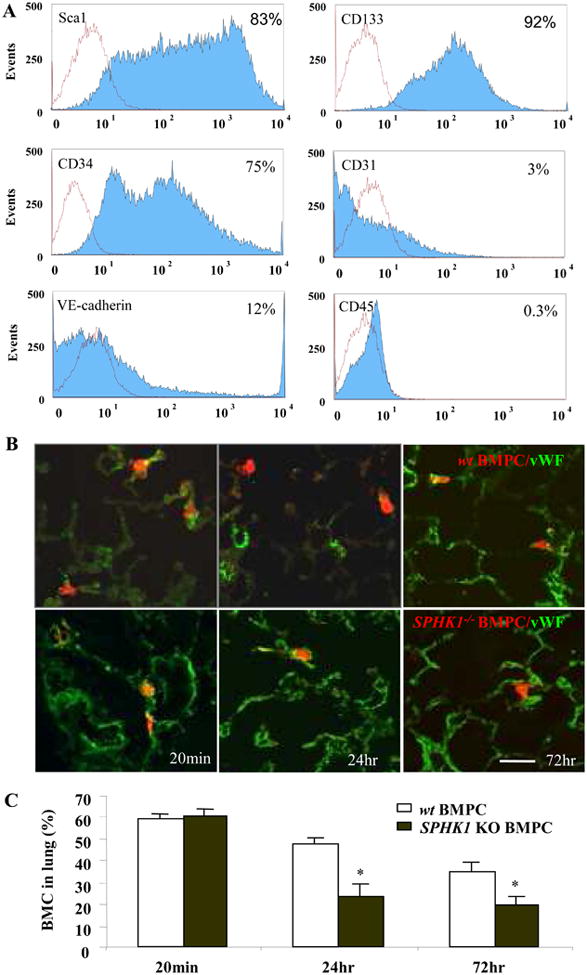
A) FACS analysis was performed using cultured BMPCs on day#21 for hematopoietic progenitor/stem cell markers (Sca-1, CD133, and CD34), endothelial cell markers (CD31, and VE-cadherin), and myeloid markers (CD45). Histogram plots for these markers are shown in blue and negative control antibody results are shown in red. Results are representative for 3 experiments. B) Morphological assessment of BMPC sequestration in lungs. Rhodamine-labeled BMPCs were localized within lung microvessels as evident by BMPCs surrounded by lung vascular endothelial cells stained with FITC-conjugated von Willebrand Factor (vWF) at 20 min post-injection (left panel) and in lung parenchyma distinct from vessel lumen at 24 hr and 72 hr after injection (right panel, bar=30μm) C) Quantification of total number of CMTMR-labeled wt BMPCs and SPHK1-/-BMPCs at 20min, 24hr and 72hr following cell injection.
In comparing wt and SPHK1 KO BMPCs, we found similar CD133 expression (92% for wt and 90% for SPHK1 KO BMPCs). Although Sca1 and CD34 expression was elevated in both cells, their expression was greater in wt BMPCs (for Sca1, 83% in wt BMPCs and 40% in SPHK1 KO BMPCs and for CD34, 75% in wt BMPCs and 40% for SPHK1 KO BMPCs). There were also differences in CD31, VE-cadherin and CD45 expression in wt and SPHK1 KO BMPCs (for CD31, 2.6 % in wt BMPCs and 66% in SPHK1 KO BMPCs, for VE-cadherin, 12% in wt BMPCs and 40% for SPHK1 KO BMPCs, and for CD45, 0.3% in wt BMPCs and 15% for SPHK1 KO BMPCs) (online Figure 1A).
Localization of BMPCs in lungs
We next determined the uptake of transplanted BMPCs in lungs. At day#3 following i.v. injection of 111Indium oxine-labeled BMPCs, lungs contained 80% of the injected BMPCs compared to other organs (online Figure 1B). To visualize BMPCs , they were incubated with the fluorophore CMTMR (25 μM) for 40 min, then washed, and injected into the external jugular vein. We found similar 60% of the wt and SPHK1 -/- BMPCs were retained at 20 min (with most in lung microvessels) after injection (Figure 1B & 1C). By 24 hr, total number of fluorescent cells was decreased in both groups with a greater decrease in SPHK1-/- BMPCs (24 % in SPHK1-/- BMPCs and 47% in wt BMPCs (p<0.05). The location of cells in both groups was the same with most cells detected in the pulmonary parenchyma or lodged extramurally in vessels (Figure 1B). At 3d after injection, there was a further decrease in accumulation of both wt and SPHK1-/- BMPCs (34% and 20%, respectively, p<0.05 between two groups), with most cells localized in the parenchyma (Figure 1B).
BMPCs prevent increase in lung vascular permeability
We quantified alterations in pulmonary microvascular permeability by determining the microvessel filtration coefficient (Kf,c) in mice receiving either BMPCs or mouse lung microvessel endothelial cells, ECs, (in each case 3×105 cells were injected i.v.). The mice were challenged at day#3 after BMPC injection with a sub-lethal LPS dose (10 mg/kg i.p.). BMPC transplantation 75% of the increase in Kf,c induced by LPS whereas injection of ECs afforded no barrier protection (Figure 2A). Lung extravascular water content increased in control mice (7.0±1.8g/g) and EC-injected group (5.9±1.1) in response to LPS whereas it was normal in LPS-challenged mice transplanted with BMPCs (4.3±1.0) (Figure 2B).
Figure 2. Transplantation of BMPCs in mice prevents increased lung vascular permeability and edema and improves survival after LPS challenge.

A) Kf,c was measured to determine changes in pulmonary microvascular permeability. In PBS-treated lungs, Kf,c increased 3-fold compared to basal (PBS alone) at 12 hr following i.p. LPS injection (10 mg/kg BW). By contrast, transplantation of BMPCs (3×105 cells, i.v.) prevented the LPS-induced increase in Kf,c whereas injection of same number of ECs provided no protection. Data are shown as mean ± SEM (n=5 mice/group). *, P < 0.05 versus either LPS or EC+LPS. B) BMPC transplantation prevents LPS-induced pulmonary edema formation. Extravascular lung water content was determined 12 hr post-LPS challenge (10 mg/kg BW, i.p.). Data are shown as mean ± SEM (n= 8 mice/group). *, P < 0.05 versus LPS alone or EC+LPS. Extravascular lung water content was significantly reduced in BMPC-transplanted group compared to transplantation of ECs or injection of PBS. C) BMPCs transplantation promotes survival in mice challenged with LPS. At 3 d post-transplantation of either BMPCs or mouse lung microvascular ECs (3×105 cells/each, i.v.), or PBS control, mice were challenged with LPS (22 mg/kg BW, i.p.). In PBS-treated mice, only 11% of mice survived 3 d post-LPS challenge. In comparison, BMPC transplantation significantly reduced LPS-induced mortality with 83% survival seen at 3d post-LPS. Differences in survival between BMPC-treated group vs either EC-treat or PBS-treated group were significant (n = 15-18 mice per group. *, P < 0.001 BMPC treated vs. either EC or PBS treated group).
We also determined the effects of BMPC transplantation on survival post-LPS insult (Fig 2C). Mice in this case were challenged with a lethal dose of LPS (22 mg/kg, i.p.) at day#3 after BMPC injection or either EC injection or PBS injection as controls. In PBS-injected mice, only 11% of mice survived 3 days post-LPS challenge. However, mice receiving 3×105 BMPCs i.v. exhibited 83% survival. In contrast, LPS-challenged mice receiving 3×105 ECs (Fig 2C) or 3×105 blood monocytes (not shown) showed no reductions in mortality.
BMPCs induce Rac1 and Cdc42 activation in endothelial cells
To address mechanisms of BMPC-induced endothelial barrier protection, we first studied alterations in endothelial junctions that are crucial for maintenance of barrier integrity. Transendothelial electrical resistance (TER) was measured in confluent endothelial cell monolayers to provide assessment of junction alterations (19). BMPCs added to cultured EC monolayers (∼15 × 103 ECs in the confluent monolayer) in increasing numbers (from 0 to 5×103 cells) increased TER (Figure 3A). The response peaked at 30 min, then decreased but remained elevated above baseline during the experiment period (Figure 3A). In contrast, ECs added in same numbers had no effect on TER (Figure 3B). The maximum increase in TER induced by BMPCs was 4-fold greater than that by ECs (Figure 3C).
Figure 3. BMPC-mediated endothelial barrier protection requires activation of Rac1 and Cdc42 in endothelial cells.
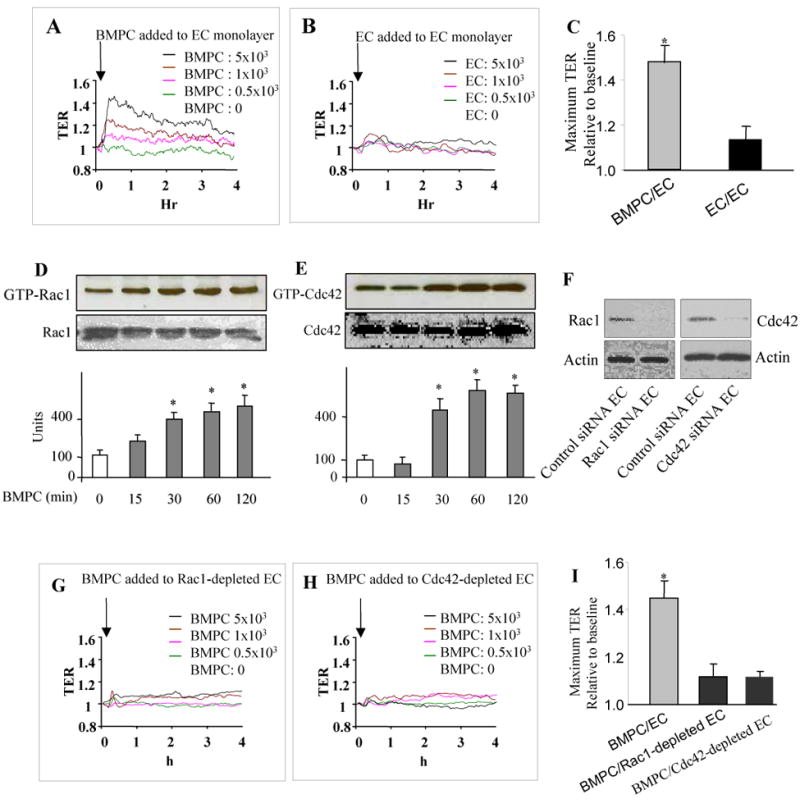
A-C) Real-time TER was measured after addition of increasing numbers of BMPCs (A) or ECs (B). BMPCs were added to EC monolayers (∼15 × 103 ECs in the confluent monolayer) in increasing numbers (from 0 to 5×103 BMPCs). Data are representative of 3 experiments and show that BMPCs enhance endothelial junction barrier in a cell number-dependent manner (A). Addition of ECs had no significant effect on TER (B). Maximum TER values seen with addition of either BMPCs or ECs are shown as mean± SEM in (C) (n=3 per group). D, E) BMPCs differentially activate Rac1 and Cdc42 in mouse lung microvessel ECs. ECs were grown to confluence on gelatin-coated 100mm dishes for 3 days and then co-incubated with BMPCs for up to 120 min (ratio of ECs to BMPCs 3:1). Active GTP-bound Rac1 or Cdc42 in ECs was determined using anti-Rac1 or anti-Cdc42 antibodies. BMPCs addition activated Rac1 and Cdc42 in ECs within 30 min, which remained elevated after BMPC addition. Total amount of Rho GTPases did not change in any group. Data are representative of 3 experiments. Densitometric analysis is shown in bar graphs as mean ± SEM (n=3). *, P < 0.05 versus basal (0 min). F-I) Depletion of Cdc42 and Rac1 in ECs by siRNA prevent BMPC-mediated stabilization of endothelial junction barrier. siRNA-mediated suppression of Rac1 and Cdc42 in ECs 96 hr post-transfection (F). ECs treated with scrambled siRNA did not reduce expression of Rac1 and Cdc42 (F). TER was measured in confluent Rac1 depleted (G) or Cdc42 depleted (H) ECs. BMPCs did not alter TER in either Rac1 or Cdc42 depleted ECs. Data are representative of 3 experiments. Maximum TER values following addition of BMPCs to either control ECs or ECs depleted of either Rac1 or Cdc42 (in each case 5×103 cells BMPCs were added) are shown as mean ± SEM (n=3) (I). *, P < 0.001 versus either Rac1- or Cdc42-depleted ECs.
Since monomeric RhoGTPases Rac1 and Cdc42 regulate AJ assembly (11, 15), we next investigated their role in signaling junction barrier enhancement observed with BMPCs. BMPC addition to EC monolayers induced activation of Rac1 and Cdc42 in ECs within 30 min of BMPC addition and activities of both GTPases remained elevated for the study duration (Figure 3D,E). Using siRNA to suppress the expression of Rac1 or Cdc42 in ECs (Figure 3F), we addressed the role Rac1 and Cdc42 in signaling the barrier enhancement. Reduction in the expression of either Rac1 or Cdc42 in ECs prevented the BMPC-induced increases in TER (Figures 3G, 3H, and 3I), suggesting that BMPC induce endothelial barrier enhancement by the activation of Rac1 and Cdc42. Specificity of siRNA-mediated silencing was verified by Western blot using Rac1 siRNA treated cells. Rac1 siRNA was reduced Rac1 activity without affecting Cdc42 (not shown).
As VE-cadherin localization at junctions is required for AJ barrier integrity (15), we next determined the effects of BMPCs on VE-cadherin assembly. VE-cadherin immunostaining showed increased membrane localization of VE-cadherin in ECs in the presence of BMPCs (online Figure 2A). However, knockdown of Rac1 or Cdc42 in ECs inhibited BMPC-induced AJ annealing (online Figure 2A).
We quantified transendothelial 125I-albumin permeability using Transwell microporous filters coated with a confluent endothelial monolayer to determine the functional effects of BMPCs on barrier function (Figure 4A). Permeability of 125I-albumin increased following LPS exposure (Figure 4B), and addition of 50 × 103 BMPCs in the lower chamber not only prevented the increase in endothelial permeability but also BMPCs reduced basal endothelial permeability to a value below baseline. This experiment also demonstrated that endothelial barrier protection induced by BMPCs did not require direct contact with the EC monolayer, indicating the involvement of a paracrine mechanism. Depletion of either Rac1 or Cdc42 in EC monolayers prevented the BMPC-induced reduction in transendothelial 125I-albumin permeability that was seen in control cells (Figure 4B). Neither Rac1 nor Cdc42 silencing in ECs significantly altered the basal endothelial permeability values relative to that observed in control EC monolayers (Figure 4B).
Figure 4. BMPCs prevent increase in transendothelial 125I-albumin permeability by Rac1 and Cdc42 activation in endothelial cells.

A) Schematic of Transwell chamber used to separate BMPCs from ECs and measure transendothelial 125I-albumin permeability. ECs were grown to a confluent monolayer on top of the microporous filter for 48 hr and either ECs or BMPCs were grown at bottom of the lower chamber to 60-70% confluence for 48 hr. We used 50×103 of each cell type on the filter and the bottom chamber. In some experiments, endothelial monolayers were depleted of either Rac1 or Cdc42 using siRNAs. Endothelial monolayers were challenged with LPS (2 μg/ml) for 2 h and transendothelial 125I-albumin permeability was measured. B) LPS addition shows an increase in transendothelial 125I-albumin permeability whereas the increase is prevented by addition of BMPCs. Knockdown of Cdc42 or Rac1 in endothelial monolayer prevented the BMPC-mediated protective effect on transendothelial 125I-albumin permeability. Values are mean ± SEM (n = 4-6 per group). *, P < 0.01 versus other 3 LPS treated groups. #, P < 0.05 LPS challenged versus Basal without LPS challenge. C) BMPCs but not bone marrow-derived CD45+ cells blocked LPS-indcued increase in transendothelial 125I-albumin permeability. Values are mean ± SEM (n = 4-6 per group). *, P < 0.01 versus either ECs or CD45+ cells treated group.
As paracrine mediators of progenitor-like cells from a monocyte/myeloid lineage could also mediate endothelial repair (24), we addressed the possibility that barrier protection induced by BMPCs was the result of a CD45+ myeloid cell subpopulation. However, addition of an enriched population of CD45+ cells (50 × 103) did not increase endothelial barrier function (Figure 4C).
BMPC expression of SPHK1 is required for endothelial barrier annealing
We next addressed the possibility that endothelial barrier protection was the result of production of S1P by BMPCs because of the importance of S1P in regulating endothelial AJ barrier integrity (11). We thus isolated BMPCs from sphingosine kinase-1-null mice (SPHK1-/-) in which S1P production is severely impaired, and measured TER in the presence of SPHK1-/- BMPCs added in the lower chamber. Addition of SPHK1-/- BMPCs failed to increase TER in contrast to wild-type BMPCs (Figures 5A, B, C). The SPHK inhibitor, SKI-II, significantly decreased the BMPC-induced enhancement of endothelial junction barrier (online Figure 1D). We also determined Kf,c in mice injected with either wild-type BMPCs or SPHK1-/- BMPCs. Administration of LPS to SPHK1-/- mice resulted in similar increase in Kf,c as in wild-type mice challenged with LPS (Figure 5D). Transplantation of BMPCs from SPHK1-/- mice into either wild-type mice or SPHK1-/- mice failed to prevent the LPS-induced increase in lung vascular permeability. However, transplantation of wild-type BMPCs into SPHK1-/- mice prevented the increase in lung vascular permeability induced by LPS (Figure 5D). Pulmonary edema formation followed the same pattern as alterations in lung vascular permeability; pulmonary edema did not develop following transplantation of wild-type BMPCs into lungs of SPHK1-/- mice (Figure 5E).
Figure 5. BMPC generated S1P prevents increased lung vascular permeability and edema in mice following LPS challenge.
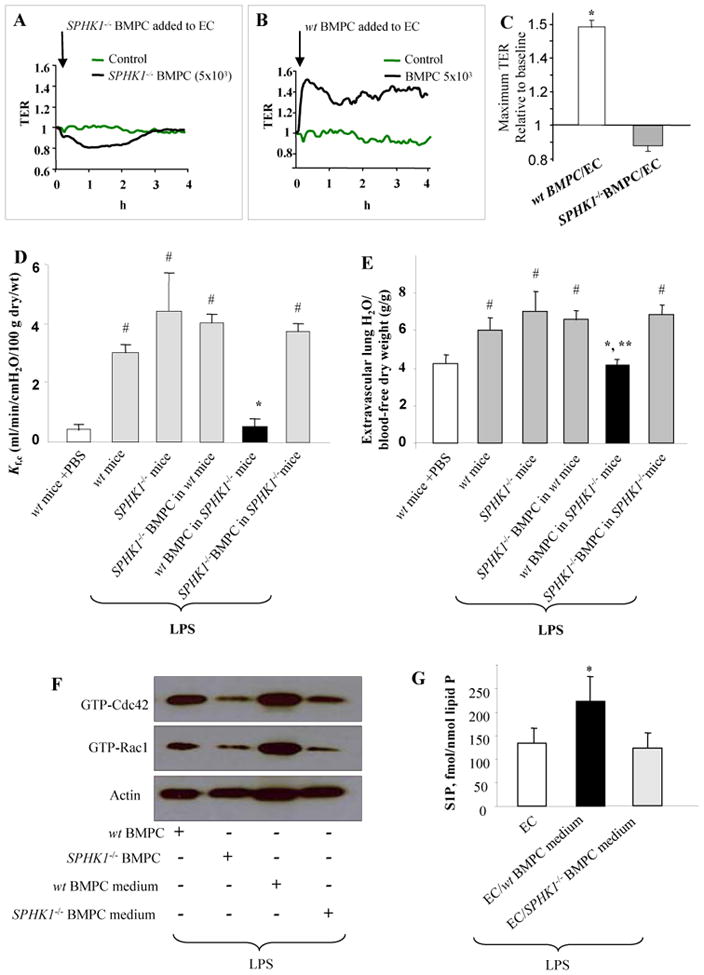
A) SPHK1 in BMPCs is required for endothelial barrier protection. TER was measured in EC monolayers (∼15 × 103 ECs) to which BMPCs (5×103 cells) from either SPHK1-/- (A) or wt (B) mice were added. C) Results are representative of 3 experiments. Bar graph indicates the maximum TER values (mean ± SEM; n=3). *, P < 0.001 versus SPHK1-/- BMPC treated ECs. D) Pulmonary microvascular permeability was measured in wt and SPHK1-/- mice following BMPC transplantation by determining Kf,c 12 hr post LPS challenege (10 mg/kg BW, i.p.). Values are mean ± SEM (n=5-7 mice per group). *, P < 0.001 versus other LPS LPS treated groups. #, P < 0.05 versus wt mice+PBS. Transplantation of BMPCs from wt mice prevented the LPS-induced increase in Kf,c in SPHK1-/- mice. In contrast, transplantation of SPHK1-/- BMPCs was not protective. E) Extravascular lung water content was assessed 12 hr post-LPS challenge (10 mg/kg BW, i.p.). Values are mean ± SEM (n = 5-12 mice per group). *, P > 0.05 versus wt+PBS; **, P < 0.01 versus other LPS treated groups. #, P < 0.05 versus wt mice+PBS. Wt BMPC prevented lung edema formation whereas SPHK1-/- BMPC had no effect. F) Interaction of SPHK1-/- BMPCs to ECs (BMPC: EC ratio of 1:3) or SPHK1-/- BMPC-conditioned medium reduced Cdc42 and Rac1 activation in ECs induced by LPS (2 μg/ml). Studies were made using the Transwell chamber to determine the effects of SPHK1-/- BMPCs or SPHK1-/- BMPC conditioned medium in modulating Rac1 and Cdc42 activities LPS-induced activation in ECs. Both SPHK1-/- BMPCs or SPHK1-/- BMPC conditioned medium reduced Rac1 and Cdc42 activities in ECs compared to wt BMPCs or wt BMPC conditioned medium. Data are representative of 3 experiments. G) LPS induces generation of S1P by wt BMPCs but not SPHK1-/- BMPCs. S1P generation in the media was determined after LPS (2 μg/ml) stimulation of ECs alone and ECs treated with either wt BMPCs or SPHK1-/- BMPCs. Values are mean ± SEM (n=3 per group). *, P < 0.05 versus either EC or EC/SPHK1-/- BMPC medium.
Using the Transwell microporous filter described above, we next determined the activities of Rac1 and Cdc42 in ECs in the presence of wild-type or SPHK1-/- BMPCs cultured on the filter while wild-type ECs were cultured in the lower chamber. Activities of Rac1 and Cdc42 were markedly reduced in LPS-challenged ECs with the addition of either SPHK1-/- BMPCs or with conditioned medium from SPHK1-/- BMPCs compared to ECs with the addition of either wild-type BMPCs or medium from wild-type BMPCs (Figure 5F). We measured generation of S1P in the presence of LPS by either wild-type BMPCs or SPHK1-/- BMPCs cultured with ECs in the Transwell chamber (Figure 5G). S1P production increased with addition of wild-type BMPCs but not with SPHK1-/- BMPCs (Supplement Figure WS-2B), indicating the paracrine role of BMPC-derived S1P in mediating endothelial barrier protection.
Discussion
Here we have addressed the role of BMPCs in regulating endothelial barrier function and defined the crucial underlying signaling mechanisms mediating this response. We demonstrate that transplantation of BMPCs prevents the increase in lung vascular permeability and edema formation and significantly reduces mortality in mice in response to LPS challenge, and does so primarily through a S1P-mediated paracrine mechanism. The studies were made using BMPCs expressing the progenitor/stem cell markers Sca-1 and CD133 as well as hematopoietic stem/progenitor cell marker CD34. These cells importantly did not express the pan-leukocytic marker CD45 or a battery of mature endothelial cell markers. The study was designed specifically to test the function of BMPCs in the mechanism of endothelial barrier repair. We carried out the studies in cells, which we defined a BMPCs to avoid the implication that they were a uniform population. In a sense this was fortuitous since a mixed cell population of progenitor cell can act synergistically to induce a high degree of re-vascularization and tissue repair (24).
Analysis of uptake of i.v. injected 111indium oxine-labeled BMPCs showed the sequestration of cells persisting up to 3 days after injection with relatively few cells localizing in other organs. This was also evident by immunostainng of BMPCs in lungs on day#3 after transplantation when BMPCs were localized in the lung parenchyma. The basis of preferential lung uptake is not clear. The uptake is likely the result of BMPCs being trapped in the first vascular bed encountered following their iv injection (17).
Based on both cell culture and lung vascular permeability results, it is evident that BMPC transplantation marked endothelial barrier protection. However, mature mouse endothelial cells, CD45+ leukocytes, or blood monocytes afforded no protection. We also observed that transplantation of BMPCs induced greater than 7-fold increase in survival compared to wild-type mice following a lethal dose of LPS challenge. Such a marked reduction in mortality may be the result of multiple factors besides the re-annealing of endothelial junctions. It is possible that BMPCs also have a direct anti-inflammatory effect that could improve survival (2, 25).
Addition of BMPCs induced the activation of Cdc42 and Rac1 in endothelial cells, whereas siRNA-mediated suppression of Cdc42 or Rac1 expression in endothelial cells abolished BMPC-mediated endothelial barrier protection. These intriguing findings suggest a cross-talk between BMPCs and endothelial cells that resulted in the Rac1 and Cdc42 activation in endothelial cells. Both Cdc42 and Rac1 have been shown to induce the formation of membrane protrusions resulting in the formation of the junction adhesion complexes that thereby enhances barrier integrity (26). We determined alterations in VE-cadherin, the AJ protein regulating endothelial permeability (27), to address whether BMPCs mediated their barrier protective effect by promoting junctional integrity. We observed that BMPCs induced Rac1- and Cdc42-dependent assembly of AJs in endothelial cells indicating that BMPC-induced activation of Rac1 and Cdc42 was required for AJ assembly and thus the repair of endothelial barrier.
To identify mechanisms of BMPC-mediated endothelial barrier protection, we focused on the endothelial barrier annealing mediator S1P, which promotes endothelial junction integrity via Rac1 and Cdc42 signaling (11). Our results here demonstrated that S1P generation by BMPCs and the activation of Rac1 and Cdc42 in endothelial cells plays a crucial role in restoring the integrity of the endothelial barrier. We observed that BMPCs not only released S1P to protect the endothelial barrier but also S1P generation exerted its maximal effects in the presence of LPS. This latter finding is consistent with studies showing that LPS induces the activation of SPHK1 in macrophages and hepatic cells, which can serve to dampen the hyper-immune response induced by gram-negative bacteria (28). Our studies made using BMPCs isolated from SPHK1-/- mice (29), the key enzyme responsible for S1P production in endothelial cells (9), showed that SPHK1-/- BMPCs failed to prevent the increase in lung vascular permeability induced by LPS whereas wild type BMPCs were protective. In addition, an SK inhibitor SKI-II prevented BMPC-induced endothelial barrier protective effect, indicating a causal relationship between BMPC SPHK functional activity and endothelial barrier protection. Thus, these findings demonstrate the fundamental importance of the SPHK1-generated S1P in the mechanism of endothelial barrier protection. Although we have identified a novel mechanism, the present findings do not preclude the possibility that other mediators such as IL-10 released by BMPCs may also be involved in the endothelial barrier protection and dampening the inflammatory response in vivo.
Other factors should also be considered to explain the role of SPHK in the mechanism of endothelial barrier protection. We observed that some antigen markers in SPHK1-/- BMPCs were different from wild-type BMPCs and also that fewer SPHK1-/- BMPCs were sequestered in lungs after LPS challenge than in wild-type BMPCs. While we cannot the rule out contribution of these factors in the reduced endothelial barrier protection in lungs seen with SPHK1-/- BMPCs, we observed in the cell culture experiments that when the same number of wild type and SPHK1-/- BMPCs were added to endothelial monolayer only the SPHK1-/- BMPCs failed to prevent increased endothelial permeability. Thus, while SPHK1-generated S1P is an important paracrine mediator responsible for BMPC-induced endothelial barrier protection, other factors in the whole lung such as the reduced uptake of SPHK1-/- BMPCs could also explain our results.
In conclusion, BMPC sensing of LPS and the consequent activation of SPHK1 and generation of S1P is a critical factor preventing increased vascular permeability. The S1P release function of BMPCs stabilizes endothelial junction barrier by a paracrine mechanism that activates Cdc42 and Rac1 signaling in endothelial cells. The present results provide a rationale for the therapeutic use of BMPCs in inflammatory diseases such as acute lung injury.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the outstanding technical assistance of Qiyan Zhou and Nanjia Yu.
Source of founding: The study was support by research grants R01 HL090152 and Illinois Stem Cell Research Program.
Non-standard Abbreviations and Acronyms
- BMPCs
bone marrow-derived progenitor cells
- S1P
sphingosine-1-phosphatase
- AJs
adherens junctions
- CMTMR
rhodamine fluorophore
- PE
phycoerythrin
- APC
allophycocyanin
- Kfc
microvessel filtration coefficient
- TER
Transendothelial electrical resistance
- SKI-II
Sphingosine Kinase inhibitor
Footnotes
Disclosures: None
References
- 1.Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Süselbeck T, Werner W, Haase J, Neuzner J, Germing A, Mark B, Assmus B, Tonn T, Dimmeler S, Zeiher AM. Improved clinical outcome after intracoronary administration of bone-marrow-derived progenitor cells in acute myocardial infarction: final 1-year results of the REPAIR-AMI trial. Eur Heart J. 2006;27:2775–2783. doi: 10.1093/eurheartj/ehl388. [DOI] [PubMed] [Google Scholar]
- 2.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179:1855–1863. doi: 10.4049/jimmunol.179.3.1855. [DOI] [PubMed] [Google Scholar]
- 3.Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007;4:e269. doi: 10.1371/journal.pmed.0040269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garciar JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T. Vascular endothelial cell adherens junction assembly morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- 6.Rosen H, Liao J. Sphingosine 1-phosphate pathway therapeutics: a lipid ligand-receptor paradigm. Curr Opin Chem Biol. 2003;7:461–468. doi: 10.1016/s1367-5931(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 7.Tani M, Sano T, Ito M, Igarashi Y. Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J Lipid Res. 2005;46:2458–2467. doi: 10.1194/jlr.M500268-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Hanel P, Andreani P, Graler MH. Erythrocytes store and release sphingosine 1-phosphate in blood. Faseb J 2007. 2007;21:1202–1209. doi: 10.1096/fj.06-7433com. [DOI] [PubMed] [Google Scholar]
- 9.Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, Hla T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102:669–676. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, Coughlin SR. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316:295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 11.Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem 2005. 2005;280:17320–17328. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- 12.Szczepaniak WS, Zhang Y, Hagerty S, Crow MT, Kesari P, Garcia JG, Choi AM, Simon BA, McVerry BJ. Sphingosine 1-phosphate rescues canine LPS-induced acute lung injury and alters systemic inflammatory cytokine production in vivo. Transl Res. 2008;152:213–224. doi: 10.1016/j.trsl.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 14.Pyne S, Pyne N. Sphingosine 1-phosphate signalling via the endothelial differentiation gene family of G-protein-coupled receptors. Pharmacol Ther. 2000;88:115–131. doi: 10.1016/s0163-7258(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 15.Broman MT, Kouklis P, Gao X, Ramchandran R, Neamu RF, Minshall RD, Malik AB. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res. 2006;98:73–80. doi: 10.1161/01.RES.0000198387.44395.e9. [DOI] [PubMed] [Google Scholar]
- 16.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res 2005. 2005;96:442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 18.Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–2760. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- 19.Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J Biol Chem. 1997;272:25968–25975. doi: 10.1074/jbc.272.41.25968. [DOI] [PubMed] [Google Scholar]
- 20.Cooper JA, Lo SK, Malik AB. Fibrin is a determinant of neutrophil sequestration in the lung. Circ Res. 1988;63:735–741. doi: 10.1161/01.res.63.4.735. [DOI] [PubMed] [Google Scholar]
- 21.Barie PS, Hakim TS, Malik AB. Effect of pulmonary artery occlusion and reperfusion on extravascular fluid accumulation. J Appl Physiol. 1981;50:102–106. doi: 10.1152/jappl.1981.50.1.102. [DOI] [PubMed] [Google Scholar]; chromatography-tandem mass spectrometry. Anal Biochem. 2005;339:129–136. doi: 10.1016/j.ab.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 22.Mehta D, Ahmmed GU, Paria BC, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, Minshall RD, Malik AB. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J Biol Chem. 2003;278:33492–33500. doi: 10.1074/jbc.M302401200. [DOI] [PubMed] [Google Scholar]
- 23.Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem. 2001;276:22614–22620. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- 24.Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophagessecrete angiogenic growth factors. Circulation. 2003;107:1164–1169. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 25.Burchfield JS, Iwasaki M, Koyanagi M, Urbich C, Rosenthal N, Zeiher AM, Dimmeler S. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circ Res. 2008;103:203–211. doi: 10.1161/CIRCRESAHA.108.178475. [DOI] [PubMed] [Google Scholar]
- 26.Kouklis P, Konstantoulaki M, Malik AB. VE-cadherin-induced Cdc42 signaling regulates formation of membrane protrusions in endothelial cells. J Biol Chem. 2003;278:16230–16236. doi: 10.1074/jbc.M212591200. [DOI] [PubMed] [Google Scholar]
- 27.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 28.Wu W, Mosteller RD, Broek D. Sphingosine kinase protects lipopolysaccharide-activated macrophages from apoptosis. Mol Cell Biol. 2004;24:7359–7369. doi: 10.1128/MCB.24.17.7359-7369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, Spiegel S, Proia RL. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.