

Abstract
Human mesenchymal stem cells (hMSCs) are bone marrow-derived stromal cells, which play a role in tumor progression. We have shown earlier that breast cancer cells secrete higher levels of interleukin-6 (IL-6) under hypoxia, leading to the recruitment of hMSCs towards hypoxic tumor cells. We found that (i) MDA-MB-231 cells secrete significantly higher levels of lactate (3-fold more) under hypoxia (1% O2) than under 20% O2 and (ii) lactate recruits hMSCs towards tumor cells by activating signaling pathways to enhance migration. The mRNA and protein expression of functional MCT1 in hMSCs is increased in response to lactate exposure. Thus, we hypothesized that hMSCs and stromal carcinoma associated fibroblasts (CAFs) in the tumor microenvironment have the capacity to take up lactate expelled from tumor cells and use it as a source of energy. Our 13C NMR spectroscopic measurements indicate that 13C-lactate is converted to 13C-alpha ketoglutarate in hMSCs and CAFs supporting this hypothesis. To our knowledge this is the first in vitro model system demonstrating that hMSCs and CAFs can utilize lactate produced by tumor cells.
Keywords: Lactate metabolism, Glycolytic tumor cells, Stromal carcinoma-associated, fibroblasts (CAFs)
Introduction
The use of chemotherapeutic agents to treat cancer is limited due to the incomplete understanding of tumor metabolism. Metabolic reprogramming in the tumor microenvironment is very dynamic and the molecular mechanisms that underlie metabolic changes are not well understood [1]. However, it is clear that defects in oxidative phosphorylation (OxPhos), lactate production/extrusion, overexpression of glucose transporters and enhanced glycolysis all play a role in the development and progression of cancer [1].
Glycolysis, the conversion of glucose to pyruvate, is an essential metabolic pathway for the generation of energy in the form of ATP. Otto Warburg, over 80 years ago, first observed that cancer cells take up glucose and produce lactic acid at higher rates than normal cells [2,3]. Interestingly, Warburg noted that cancer cells prefer to utilize glucose for glycolysis even when oxygen tension is elevated. Subsequently, this phenomenon of aerobic glycolysis became known as the “Warburg effect” [4]. Although less efficient than OxPhos, aerobic glycolysis is indeed more advantageous to the cancer cell than OxPhos because of its rapid generation of ATP and production of other biomolecules supporting tumor growth and proliferation [2-5].
A major reason for tumor cells undergoing the continuous metabolism of glucose to lactic acid is due to their adaptation to fluctuating gradients of oxygen [6]. Notably, lactate which is produced as a consequence of hypoxia within the tumor cell is expelled to the tumor microenvironment in order for tumors to continue their glycolytic metabolism [7,8]. In addition, studies have shown that extruded lactate favors cancer cell invasion and metastasis, due to the acidification of the tumor milieu [9]. Inhibition of lactate dehydrogenase A (LDH-A) leads to inhibition of cell proliferation [10-12], suggesting that the generation of therapeutic inhibitors of glycolysis may be useful in halting cancer progression by suppressing tumor growth and metastases. Fate of lactate in the milieu exterior is less well characterized, although studies suggest that aerobic (oxidative) tumor cells take up the lactate and convert it to pyruvate for further metabolic processing [13,14].
Immunohistochemical analyses have shown that stromal fibroblasts within the tumor microenvironment express monocarboxylate transporters MCT1 and MCT2 and may recycle lactate produced by tumor cells [15]. MCTs function primarily as symporters, facilitating the efflux of lactate, pyruvate and butyrate across the plasma membrane [16,17]. Inhibition of MCT1 has been shown to decrease by 85% the lactic acid transported across glioblastoma cells in vitro [18]. Thus, metabolic cooperation between the anaerobic tumor cells and the aerobic stromal cells via MCT1 for efficient ATP production and intracellular homeostasis may play a role in maintaining metastatic potency and proliferative capacity of cancer cells.
Human mesenchymal stem cells (hMSCs) are bone marrow-derived stromal cells, which play a role in tumor progression. We have shown that human breast cancer cells produce increased amounts of interleukin-6 (IL-6) under hypoxia, leading to the recruitment of hMSCs towards hypoxic tumor cells [19]. These hMSCs have been shown to be a source of carcinoma-associated fibroblasts (CAFs), which undergo myo-fibroblast-like differentiation [20]. Exercising muscle produces and secretes lactate in a similar fashion as that observed in cancer cells which is then taken up by a variety of cell types and used as energy source [21,22]. We hypothesized that in the tumor microenvironment, the stromal CAFs have a similar capacity to take up lactate expelled from tumor cells and use it as a source of energy. Results of the present study support this hypothesis.
Materials and methods
Tumor cell lines
Tumor cells were cultured in DMEM with 10% fetal bovine serum and 1% penicillin–streptomycin. Cells used in all experimental conditions were maintained under normoxia (20% O2), and hypoxia (1.5% O2) for 6 h and/or 24 h.
MDA-MB-231 and MCF7 human breast cancer cells were obtained from ATCC, Manassas, VA and were determined to be free of pathogens.
Primary cells
Human bone marrow-derived mesenchymal stem cell (hMSC) pooled donor cell line was obtained from Lonza Walkersville, Inc. (Walkersville, MD). Cells were cultured in α-MEM with 10% fetal bovine serum and 1% penicillin–streptomycin and maintained below passage 8. Flow cytometry was utilized to determine cell surface markers expressed on hMSCs, including Stro-1, CD105, CD90, HLA-ABC and CD44 using FITC-labeled antibodies (BD Biosciences, Franklin, NJ.). These cells were determined to be negative for CD45, HLA-DR and CD11b.
Cells were counted using trypan blue exclusion via the Beckman Coulter™ Counter (Beckman, Brea, CA) and viability was quantified using its Vi-CELL™ (v 1.01) software.
Hypoxia
Hypoxia (1.5% O2) was achieved by placing cells in a modular chamber (Billups-Rothenberg, Inc., Del Mar, CA) according to manufacturer’s instructions.
Lactate measurements
Tumor cell conditioned media (TCM) was collected from MDA-MB-231 and MCF7 cells following 24 h conditioning under normoxia and hypoxia. Briefly, 1×105 human breast cancer cells were plated in 6-well plates and grown in DMEM with 10% fetal bovine serum and 1% penicillin–streptomycin for 24 h. The plates were then conditioned under normoxic or hypoxic conditions for a further 24 h. TCM was centrifuged at 1000 rpm for 5 min, 30 μL of TCM was collected from each sample, applied to a lactate test strip and analyzed using a lactate analyzer (Roche), in accordance with manufacturer’s instructions.
Immunoblotting
Cell lines were homogenized in 1× RIPA buffer with protease inhibitors. Protein concentration was determined with a Bradford assay and lysates were separated on an 8–12% SDS-polyacrylamide gel. Proteins were then transferred to nitrocellulose membranes for 1 h. Membranes were then blocked in PBST buffer plus 5% nonfat dry milk for 1 h, probed with primary and secondary antibodies, and developed using ECL chemiluminescence reagent (Pierce, Thermo-Fisher Scientific, Rockford, IL).
Real-time reverse-transcription PCR (qRT-PCR) analysis
Total RNA was extracted from MDA-MB-231 cells or hMSCs or CAFs with TRIzol Reagent or an RNeasyPlus Kit (QIAGEN, Valencia, CA). RNA was reverse transcribed using SYBR Green® One-Step RT-PCR Master Mix Reagents Kit (Applied Biosystems, Minneapolis, MN) followed by qRT-PCR with primer sequences for human MCT-1 and GAPDH mRNA (Applied Biosystems). Gene expression was normalized to GAPDH and gene expression changes were quantified as relative fold changes using the comparative delta Ct method.
For semi-quantitative RT-PCR, primer-probe sequences for MCTs, LDH-A and LDH-B, PDH, and β-actin were all pre-designed and purchased commercially (Table 1). Gene expression was normalized to β-actin.
Table 1.
Primers
| Gene | Primer sequence |
|---|---|
| MCT1 | 5’-ACC AGC AGT TGG AGG TCC AGT TGG ATA-3’ |
| 5’-AAC TGA TTA ATT GTT TGG AAG ACT GAT-3’ | |
| MCT2 | 5’-ACT GAG CTC ATG CCA CCA ATG CCA AGT GCC-3’ |
| 5’-AGA GGT ACC TTA AAT GTT AGT TTC TCT TTC-3’ | |
| MCT4 | 5’-AGG GGC CGT GGT GGA CGA GGG CCC CAC-3’ |
| 5’-GAC GAA GAG CCC CAG CAC CAT GAC CGA-3’ | |
| LDH-A | 5’-GGC CTG TGC CAT CAG TAT CT-3’ |
| 5’-GCC GTG ATA ATG ACC AGC TT-3’ | |
| LDH-B | 5’-AGG ATT CAT CCC GTG TCA AC-3’ |
| 5’-CCC ACA GGG TAT CTG CAC TT-3’ | |
| PDH | 5’-AGC TGC CAA GAC CTA CTA CAT-3’ |
| 5’-ATC CCG AAT GGC TGA TTT-3’ | |
| β-Actin | 5’-CCT CGC CTT TGC CGA TCC-3’ |
| 5’-CGG GCC ACC TGC CTC A-3’ |
Transwell migration assay
The migration assay was carried out as described previously [19]. Blank media (α-MEM, 2% FBS, Invitrogen, Carlsbad, CA), 5 mM lactate- or 15 mM lactate-containing media was plated into the bottom chamber of 24-well, notched tissue culture plates (BD Falcon™, BD Biosciences, Franklin Lakes, NJ) and cell culture inserts containing 2.5×104 hMSCs were placed in the upper chamber. Migration of hMSCs toward lactate-containing media was allowed to continue across the 8 μm uncoated polythene membrane for 18 h following which migrated cells were stained with crystal violet and counted manually using an inverted microscope. For migration inhibition studies hMSCs were treated with blank media or media containing 2 mM CHC (alpha-cyano-4-hydroxy-cinnamic acid, dissolved in DMSO, Sigma, St.Louis, MO), an inhibitor of MCT1, for 1 h. Subsequently, cells were trypsinized and 2.5×104 untreated hMSCs or CHC-treated hMSCs were plated for the migration assay, as indicated above, using 750 μL of 15 mM lactate-containing media or tumor cell-conditioned media (TCM; from an 80–100% confluent T-150 flask of MDA-MB-231 cells) for the bottom chamber.
Lactate uptake assay
CAFs were induced by exposure of hMSCs to tumor cell-conditioned medium produced by MDA-MB-231 cells as described before [20]. CAFs or hMSCs were plated into 24-well plates at 5×104 cells/well. Eighteen hours later media were aspirated and cells were treated with 200 μL of uptake buffer (10 mM HEPES, 5 mM KCl, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.5) containing 0.05 μCi14C-U-lactate (uniformly labeled; Perkin Elmer, Waltham, MA) for 1 h or 2 h at 37 °C, 5% CO2. Cells were washed thrice using 200 μL uptake buffer followed by 15 min incubation with 200 μL uptake buffer containing 1% SDS. Cell lysates were collected into scintillation vials for quantification of 14C-lactate uptake. Labeled lactate uptake in presence of 10 mM CHC (alpha-cyano-4-hydroxy-cinnamic acid, dissolved in DMSO, Sigma, St. Louis, MO) was studied similarly.
Lactate-3-13C exposure, perchloric acid extraction, and NMR spectroscopy
CAFs were induced as previously described using MDA-MB-231 cell-conditioned medium [20]. CAFs or hMSCs were allowed to expand to 90% confluence in T150 flasks. A single flask of hMSCs was treated with cell culture media for 4 h while two flasks each of hMSCs or CAFs were incubated with cell culture media supplemented with 10 mM sodium l-lactate-3-13C (Isotec, Sigma-Aldrich, St. Louis, MO) for 4 h. Cells were harvested and extracted using perchloric acid (PCA), based on previously published methods [23,24]. Briefly, cells were harvested by trypsinization, collected, washed twice with ice-cold 1× PBS, and counted. All subsequent steps were performed on ice in a 4 °C cold room. Ice-cold 8% PCA (4 mL) was added to the cell pellet and the cells homogenized for 10 s using a homogenizer. The homogenate was transferred to a 35 mL glass centrifuge tube, the extraction tube rinsed with 1 mL ice-cold 8% PCA and the content transferred as well. The PCA/cell mix was homogenized in the glass tube for an additional 2 min on ice, and then spun down at 15,000g for 15 min at 4 °C. The supernatant was then transferred to a clean 35 mL glass centrifuge tube and neutralized to pH 7 using 3 M K2CO3/1 M KOH while gently vortexing. The neutralized solution was spun at 15,000g for 15 min at 4 °C. The supernatant was transferred to a clean 35 mL glass centrifuge tube. A 10 mL syringe containing ~1 mL glass wool was prepared prior to the next step. A spatula tip’s worth of Chelex 100 (Sigma C-7901, St. Louis, MO) was added to the supernatant followed by vortexing. The mixture was allowed to incubate on ice for 5 min followed by another round of brief vortexing. The mixture was immediately transferred to the syringe prepared in previous step and collected into a 50 mL plastic conical tube. The sample was flash frozen on dry ice, lyophilized for 24 h, and stored at below −20 °C until NMR spectroscopy. For NMR spectroscopy, each sample was resuspended in 650 μL 10 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) in D2O and 600 μL of resolved sample transferred to a 5 mm NMR tube. The NMR measurements were performed in the NMR Analytical Core Facility at MSKCC on a 600 MHz Bruker-Avance III NMR spectrometer with a 13C/1H cryprobe. One-dimensional 1H-decoupled 13C NMR spectra were acquired using a 30° flip angle, 1536 averages, 39,063 Hz spectral width, 1.7 s acquisition time, 134,144 number of points, and a relaxation delay of 2 s. After applying a 1 Hz exponential line broadening filter, the free induction decays (FIDs) were Fourier transformed, phased, and the reference standard sodium 2,2-dimethyl 2-silapentane-5-sulfonate (DSS) set to 0 ppm.
Results
Hypoxia enhances lactate production and secretion in breast cancer cells
Several observations have indicated that hypoxic cells produce excess lactate [7,14]. Lactic acid produced by tumor cells has been shown to suppress anticancer immune effectors [25]. Consequently, altered metabolic flux in the tumor microenvironment may enhance the metastatic potential of cancer cells [26]. To investigate whether hypoxia affects the production of lactate in breast cancer cells, we measured lactate secretion in these cells following hypoxic conditioning.
We utilized a lactate analyzer to measure the production of lactate in MDA-MB-231 and MCF-7 breast cancer cells following culture under normoxia (20% O2) or hypoxia (1.5% O2) for 24 h. We found that MDA-MB-231 cells secrete significantly higher levels of lactate (2.5-fold more) under hypoxia than under normoxia (Fig. 1A). Similarly, MCF-7 cells also exhibited ~2-fold enhanced production and secretion of lactate upon hypoxic conditioning as compared to the normoxic control (Fig. 1B).
Fig. 1.
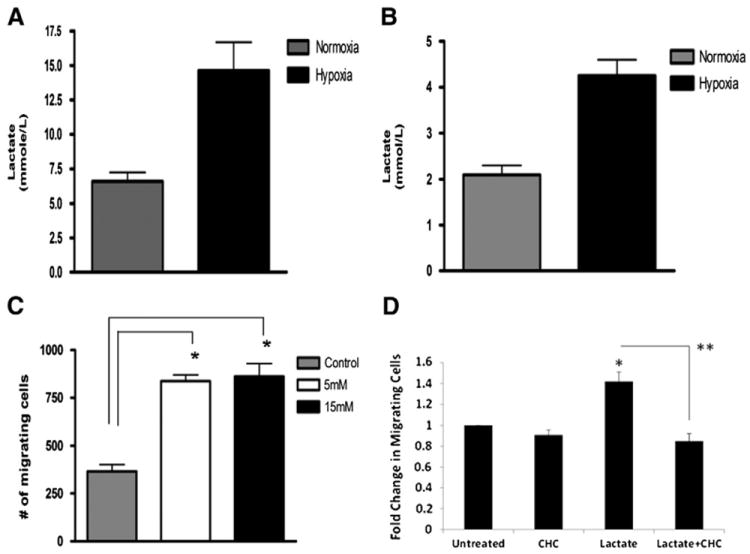
A, B: The breast cancer cell lines MDA-MB-231(A) and MCF-7 (B) secrete higher levels of lactate under hypoxia (1.5% O2) than normoxia (20% O2) (*p <0.05, unpaired t-test, n=3). Tumor cell-conditioned media (TCM) were collected after 24 h conditioning and levels of secreted lactate quantified using a lactate analyzer (Roche Diagnostics, Mannheim, Germany). C: Lactate stimulates hMSC migration. The migration of hMSCs towards culture media containing different doses of lactate, or control medium in 18 h was measured using a transwell migration assay. The number of migrating hMSCs towards lactate-containing culture medium was significantly higher than towards control medium (*p<0.05, unpaired t-test, n=4). D. CHC inhibits lactate-mediated increase in hMSC migration. The migration of untreated or CHC-treated hMSCs towards culture media containing 15 mM lactate, or control medium in 18 h was measured using a transwell migration assay. The number of migrating hMSCs for each condition was normalized to untreated controls. (*p<0.05, unpaired t-test, n=3, **p<0.005, paired, unequal variance t-test, n=3.)
Lactate stimulates hMSC migration
We have shown previously that hypoxia plays a role in stimulating hMSC migration towards the tumor via enhanced secretion of the chemotactic factor IL-6 [19]. Since lactate secretion is also a consequence of hypoxia in the solid tumor microenvironment, we investigated whether lactate can also serve as a chemoattractant for hMSC migration towards hypoxic tumor cells. We utilized a transwell migration assay in which we placed lactate in the bottom chamber of the assay as a stimulus in a dose-dependent manner, and allowed hMSCs in the top chamber to migrate towards the stimulus for 18 h. We found that lactate significantly stimulates the migration of hMSCs (2.5-fold more) as compared to the control (Fig. 1C). There was no significant difference in the number of migrating cells towards 5 mM or 15 mM lactate. A range of 5–20 mM of lactate falls within the physiological range for the hypoxic tumor microenvironment [26], and we decided to use 15 mM concentration as a stimulus for subsequent experiments. As lactate is taken up via monocarboxylate transporters, particularly MCT1, we next determined if the observed increase in lactate-mediated hMSC migration could be attenuated via CHC, an MCT inhibitor. Results confirmed that lactate increases the total number of migrating hMSCs (Fig. 1D). Further, there was a statistically significant decrease in hMSC migration when they were pretreated with CHC and then exposed to lactate, as compared to lactate alone. CHC alone in complete media did not alter hMSC migration.
Increased expression of MCT1 upon lactate exposure
Previous studies have indicated that tumor-associated fibroblast express functional MCT1 and MCT2 proteins involved in the proton-linked extrusion of lactate and other monocarboxylates [15]. Because MCT1 expression is essential for lactate transport from tumor cells and is necessary for lactate absorption by neighboring cells, we therefore determined whether lactate was able to up-regulate MCT1 expression in hMSCs and CAFs. Utilizing qRT-PCR, we observed that MCT1 expression was induced in both hMSCs (Fig. 2A, left panel) and CAFs (Fig. 2A, right panel) after stimulation with 15 mM lactate as compared to the untreated control. Similarly, by Western blot analysis, we found increased MCT1 protein levels after lactate exposure in hMSCs, mirroring increases observed at the mRNA level (Fig. 2B). In CAFs, the exposure to 15 mM lactate also increased the expression of MCT2, MCT4, as well as LDH-B and PDH mRNA (Fig. 2C, right panel). Interestingly, MCT4 mRNA expression was increased, while MCT1 mRNA expression was decreased in MDA-MB-231 breast cancer cells (Fig. 2C, left panel), as well as in MCF-7 cells (Fig. 2C, bar graph). In CAFs as well as in MDA-MB-231 cells, LDH-A mRNA expression remained unchanged upon exposure to lactate (data not shown). Taken together, these results indicate that hMSCs and CAFs express functional MCT1 transporter and respond to lactate stimulation. The increased expression of LDH-B and PDH in CAFs upon lactate exposure suggests that they may be primed to convert lactate to pyruvate to acetyl-CoA.
Fig. 2.
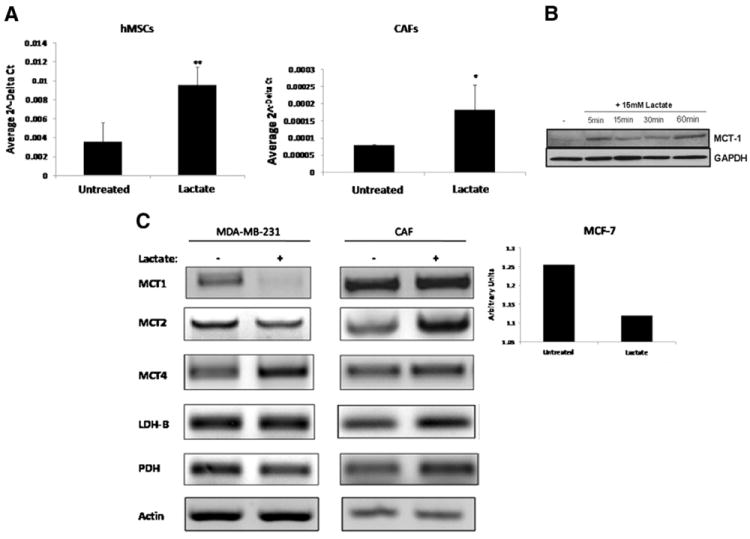
A: MCT expression is induced upon lactate exposure. Quantitative real-time PCR revealed that MCT1 mRNA increased significantly in hMSCs (left panel) and in CAFs (right panel) exposed to 15 mM lactate for 1 h when compared to untreated control cells (p<0.05 as determined by 2-tailed, paired, t-test, n=3). B MCT1 protein is induced in MSCs exposed to 15 mM lactate. Western blot analysis shows increase in expression of MCT1 in MSCs over time periods of 5 to 60 min of lactate exposure. C: Left and center panel: MDA-MB-231 cells and CAFs were exposed to 15 mM lactate for 30 min following which RNA was isolated from each cell line and reverse transcribed for subsequent amplification by RT-PCR using primers specific for MCT1, MCT2, MCT4, LDH-A, LDH-B, and PDH. Expression of LDH-A remained unchanged (not shown) and β-actin served as internal control. Right panel: MCF-7 cells were exposed to 15 mM lactate for 30 min following which RNA was isolated and reverse transcribed for subsequent amplification by RT-PCR using primers specific for MCT1 and using β-actin as a control. Bar graph shows quantitation of the resulting MCT1 mRNA expression using Quantity One® 1-D Analysis Software as per manufacturer’s protocol (Bio-Rad). Data were normalized to untreated controls.
Lactate is an important regulator of STAT3 and ERK activation in hMSCs
We have previously shown that STAT3 and ERK phosphorylation is important for activation of downstream pathways in hMSCs leading to enhanced migration and survival [27]. Therefore, we determined if lactate stimulation leads to activation of STAT3 and downstream effectors ERK 1/2. STAT3 activation was determined in hMSCs in response to 15 mM of lactate in a time-dependent manner by Western blot analysis. We found that p-STAT3 protein levels were induced in hMSCs treated with 15 mM lactate and not in the untreated control (Fig. 3 left panel). We observed a robust increase in phospho-ERK (Fig. 3 right panel) in response to lactate stimulation. There was no change in total ERK or STAT3 protein levels. Although transient, phosphorylation of STAT3 and ERK indicated that hMSCs were activated by exposure to lactate in a manner similar to that observed by SDF-1 in our previous investigation, in which subsequent activation of FAK and paxillin correlated with an increased migratory phenotype [27]. Thus, our studies indicate in addition to being a chemoattractant for hMSC migration, lactate is an activator of signaling pathways in hMSCs.
Fig. 3.
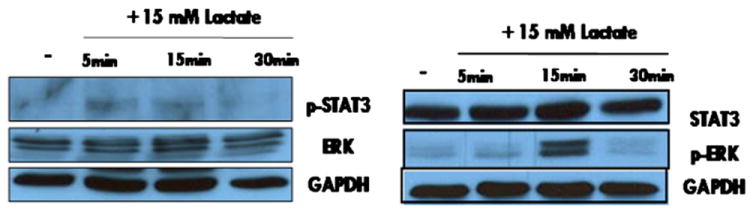
hMSCs were exposed to 15 mM lactate for 5, 15, and 30 min. Following each time point the lactate was removed. The reaction was terminated with ice-cold 1× PBS and hMSCs were lysed with 1× RIPA buffer. The protein concentration determined by Bradford assay and lysates ran on a 10% SDS-PAGE gel to analyze relative protein levels. Left panel: The protein levels of phosphorylated-STAT3 were induced in hMSCs upon exposure to 15 mM lactate. There was no change in the total expression levels of ERK. Right panel: 15 min exposure to 15 mM lactate activated phospho-ERK expression which reverted to basal levels after 30 min exposure to 15 mM lactate. There were no observable changes in the total protein levels of STAT3.
Metabolic fate of lactate in hMSCs and CAFs
Our previous studies have shown that hMSCs are recruited to the tumor microenvironment by tumor cell-conditioned medium and can become CAFs [20]. It was therefore of interest to determine whether lactate present in the tumor microenvironment is taken up by hMSCs as well as CAFs and the metabolic fate of lactate in these cells. Results of 14C-lactate uptake studies indicate that CAFs and hMSCs can take up lactate and this uptake is linear for up to 2 h at 37 °C (Fig. 4). On a per cell basis, CAFs took up significantly greater amounts of lactate as compared to hMSCs at both time points measured (Fig. 4A). Inhibition of lactate uptake in these cells by cyanohydroxycinnamic acid (CHC) indicated that the uptake was mediated by monocarboxylate transporters (Fig. 4B).
Fig. 4.
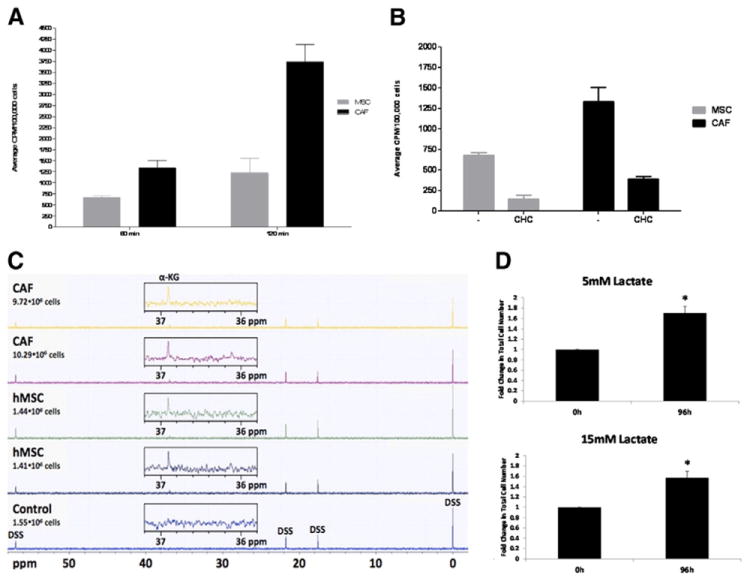
Lactate uptake in MSC and CAFs. Fig. 3A: CAFs exhibit higher uptake of lactate than hMSCs at 60 min and 120 min time points (p≤0.007 for 120 min time point and p<0.05 for 60 min time point, as determined by 2-tailed, unpaired, unequal variance t-test, n=3). 14C-lactate uptake was carried out in triplicate using hMSCs and CAFs. Numbers on Y-axis represent CPM per 1×105 cells. Inhibition of 14C-lactate uptake by CHC indicates that 14C-lactate transport occurs via MCTs. Inhibition studies were carried out by incubating hMSCs and CAFs with 10 mM CHC for 1 h prior to the uptake studies. Significant inhibition of lactate transport was observed in presence of CHC in both cell types (p<0.005). C. Lactate is taken up and metabolized by hMSCs and CAFs as demonstrated from 13C NMR spectra of cell extracts. Two samples each of hMSCs and CAFs were incubated with 10 mM sodium-l-lactate-13C-3 for 4 h followed by PCA extraction. Proton-decoupled 13C NMR spectroscopy revealed 13C labeling of a metabolite, assigned to α-ketoglutarate-13C-3(α-KG), based on its 13C chemical shift [28], for all 4 samples (2 each for hMSCs and CAFs) incubated with 13C-labeled lactate but not in the untreated hMSC sample (control). An additional signal, detected in all samples outside of the spectral region shown here, was assigned to . D. hMSCs can survive and grow in lactate only media for an extended period of time. hMSCs were plated in 6 well plates in α MEM and 24 h later media was changed to αMEM depleted of glucose. Lactate was supplemented at 5 or 15 mM and cells allowed to grow for another 96 h in the modified medium. Plates were harvested at the end of 96 h and viable cells counted (p<0.05, n=3).
Preliminary metabolic NMR studies using sodium-l-lactate-3-13C indicated that lactate is taken up and metabolized by hMSCs and CAFs. Zoomed regions of 13C NMR spectra obtained from PCA extracts of one control hMSC sample and 2 cell samples each of hMSCs and CAFs exposed to 10 mM sodium-l-lactate-3-13C for 4 h are shown in Fig. 4C. In both, hMSCs and CAFs, a signal at 36.9 ppm, assigned to 13C-labeled alpha-ketoglutarate (C-3 position), based on the chemical shift [28] and the fate of the 13C-3 label of lactate [29], is visible for cells incubated with sodium-l-lactate-3-13C. Additional signals visible in the full 13C NMR spectra were from the concentration and reference standard DSS, and . hMSCs were able to survive and grow in media depleted of glucose but supplemented with 5 or 15 mM lactate (Fig. 4D), suggesting that they can utilize lactate as a metabolic source. CAFs showed similar survival and growth in glucose depleted but lactate supplemented medium (data not shown).
Discussion
The hypoxic tumor microenvironment plays a role in the alteration of glucose metabolism in cancer cells [30,31]. Metabolic reprogramming is one of the hallmarks of cancer favoring the suppression of normal OxPhos and the induction of glycolysis. In addition to providing a continuous energy source to meet the demands of tumors, enhanced rates of glycolysis may also protect cancer cells from hypoxia-induced apoptosis [31]. However, the molecular basis underlying the “glycolytic switch” in cancer cells remains unclear, but may be contributed to by epigenetic and oncogenic changes within the tumor microenvironment [1]. In this study, we took advantage of the altered tumor glucose metabolism to determine the consequence of hypoxia-induced lactate secretion. We examined whether hypoxia influences lactate secretion in tumors. The breast cancer cell lines MDA-MB-231 and MCF-7 showed strong induction of lactate secretion under hypoxic culture conditions. Lactate produced by these tumor cells is the end product of glycolysis and has been shown to favor tumor invasion [14,30-32]. Despite efforts to delineate the effects of lactate production and secretion on tumor progression, the exact role still remains elusive.
In our previous work, we established that human mesenchymal stem cells (hMSCs) specifically migrate towards the hypoxic tumor microenvironment via hypoxia-mediated IL-6 signaling [19]. In another study, we showed that hMSCs are a source of CAFs, contributing to tumor growth and survival [20]. Since tumors produce lactate by aerobic glycolysis and/or in response to hypoxia in the tumor microenvironment by anaerobic glycolysis, we focused our efforts on determining whether lactate can mediate hMSC migration. We observed that hMSCs migrate towards lactate in vitro. Furthermore, lactate exposure leads to activation of STAT3 signaling pathways in hMSCs. This suggested that, similar to IL-6 mediated activation, lactate has the capacity to recruit hMSCs towards tumors by activating signaling pathways to enhance their migration and survival. Signaling activity of lactate has been previously observed in macrophages where lactate boosted toll like receptor 4 (TLR4) signaling and NF-κB pathway-mediated gene transcription through MCTs and MD2 (a coreceptor for TLR4 signaling) upregulation [33]. As hMSCs express TLRs 1–6, it is quite likely that a signaling cascade through TLRs is operative in hMSCs following lactate exposure [34,35]. Thus, in addition to STAT3 signaling, other contributory pathways may be regulated by lactate to optimize hMSC migration toward tumors.
We have also observed that hMSCs express MCT1 upon exposure to lactate. MCT1 is a symporter protein involved in the bidirectional transport of lactic and pyruvic acid [17]. MCT1 is the most well studied and widely expressed of the 14 putative members of the monocarboxylate transporter (MCT) family [16,17]. It is now becoming clear that MCT1 interacts with several binding partners including emmprin (CD147) and CD44 to coordinate lactate efflux [36]. MCT1 and MCT4 are differentially expressed in a variety of malignant tumors [18]. Therefore, current studies utilizing small molecule inhibitors directed against MCTs or binding partners CD147/CD44 with the hopes of tumor regression are being explored [36,37].
Induction of MCT1 in stromal cells and their ability to metabolize lactate suggest a possible role for these cells to participate in complementary metabolic pathways with tumor cells, i.e. by absorption of lactate extruded from the tumor cells followed by recycling of the absorbed lactate to pyruvate. This is supported by our observation that lactate stimulates MCT1 expression in stromal cells, but downregulates it in tumor cells (in both MDA-MB-231 and MCF7 cells), suggesting that lactate is exported from tumor cells and taken up by stromal cells. It has been proposed that stromal fibroblasts in the tumor microenvironment absorb lactate from the extracellular matrix (ECM) [15]. Once absorbed, lactate may be oxidized to pyruvate and can either be used by these stromal cells to regenerate pyruvate to sustain their own energy demands or may even be extruded to the ECM via MCT2 for subsequent consumption by tumor cells [15]. Collectively, this argues in support of the notion that stromal CAFs have the capacity to engage in metabolic cooperation within the tumor microenvironment to aid in tumor progression.
To directly investigate this, we first measured the expression of key MCTs in cancer cells CAFs. MDA-MB-231 cells express very low levels of MCT1 but express MCT2 and MCT4 which are pyruvate transporters [36]. Thus, lactate exported by MDA-MB-231 cells may be taken up by hMSCs which demonstrate induction of MCT1 in response to lactate. Indeed, CHC, an inhibitor of MCT1, attenuated lactate-mediated migration in hMSCs (Fig. 1D), implicates the functional importance of this transporter in the homing of hMSCs to tumor sites. Further, our uptake and metabolic studies indicate that hMSCs and CAFs can take up lactate from the surrounding medium and likely convert it to pyruvate which then enters the TCA cycle, producing NMR-detectable levels of α-ketoglutarate. Thus, it appears that enhanced expression of MCT1 on hMSCs prepares them for their role as CAFs in the tumor microenvironment for utilization of lactate. The CAFs convert the lactate to pyruvate presumably for their own metabolic needs. The increase in expression of LDH-B and PDH in CAFs, as seen by RT-PCR analysis, supports the idea that CAFs can absorb lactate from the microenvironment and convert it for use as an energy source.
The importance of cell–cell lactate shuttles for coupling glycolytic activities in one cell type with oxidative pathways in another neighboring cell type has been previously proposed [22,38,39]. Further, tumor-secreted lactate has been suggested to act as a paracrine molecule to affect signaling in the neighboring stroma [40]. Specifically, macrophages and L6 myoblast cells treated with lactate showed an increase in the transcription of MCT1, nuclear factor-κB (NF-κB), and lactate oxidation-promoting, mitochondrial biogenesis genes, indicating that lactate may directly modulate target cell metabolism through known signaling networks [41,42]. Studies investigating lactate signaling in macrophages are especially of interest as macrophages are resident in the tumor stroma, and are differentiated by tumors into tumor-associated macrophages, the immunological contemporaries of CAFs [42]. This suggests that additional activated stromal components may also undergo metabolic coupling with the neighboring cancer cells.
Thus, we hypothesized that lactate produced by a significant proportion of cancer cells is taken up by CAFs and utilized as an energy source. Our NMR spectroscopic measurements indicate that 13C-lactate is converted to 13C-αketoglutarate in stromal cells. We used an incubation time (with 13C-lactate) of 4 h in order to maximize our chances of detecting metabolic conversion of 13C-lactate in stromal cells. Previous studies using immunohistochemical techniques have suggested such metabolic cooperation between stromal fibroblasts and tumor cells [15]. In support of these and other studies demonstrating the functional significance of lactate in downstream stromal signaling [38,39], the current investigation offers first empirical evidence of lactate’s function as a paracrine modulator of metabolism in the tumor microenvironment (TME).
Based upon the results of our experiments, we propose the following model for a tumor–stromal CAF metabolic relationship (Fig. 5). Tumors utilize aerobic glycolysis to fuel their energetic and biosynthetic needs. Tumor cells convert the incoming glucose to pyruvate to yield ATP, but, rather than use it for oxidative phosphorylation, convert this pyruvate to lactate to produce more NAD + necessary for continued glycolysis. This results in the accumulation of lactate in the cell which the tumor cells excrete using MCT4. The stromal CAFs take up the secreted lactate via MCT1, convert it to pyruvate through increased expression of LDH-B, and, as supported by our NMR data, subsequently shunt it into the tricarboxylic acid cycle, thereby fueling their own energetic needs.
Fig. 5.
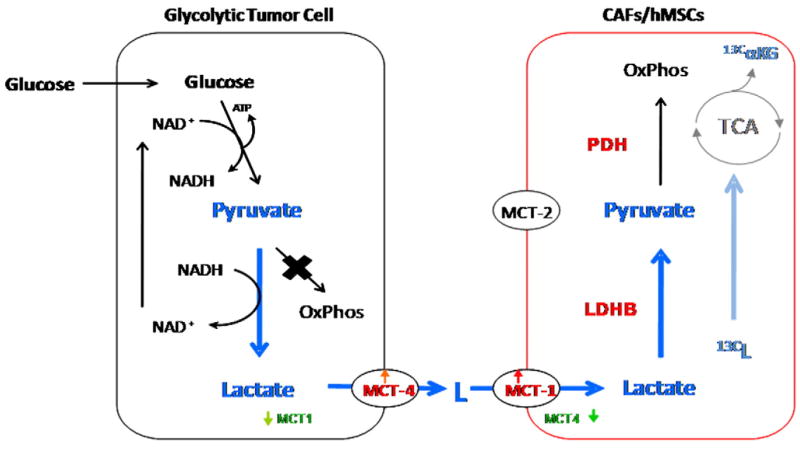
Model of metabolic cooperation between glycolytic tumor cells and stromal hMSCs/CAFs. Glucose is taken up by glycolytic tumor cells and converted to pyruvate to yield ATP at the cost of NAD+. As tumors preferentially utilize aerobic glycolysis over oxidative phosphorylation, this pyruvate is converted to lactate to replenish NAD+, thereby fueling further glycolysis. Tumor cells secrete the lactate via increased expression of lactate transporter MCT4. In response, MCT1 expression goes up in CAFs resulting in uptake of tumor-extruded lactate. Exposure to lactate increases expression of LDH-B in CAFs, resulting in the conversion of the influxed lactate to pyruvate. The pyruvate is shunted to the tricarboxylic acid cycle for ATP generation via oxidative phosphorylation, thereby satisfying the energetic needs of the CAFs.
To our knowledge this is the first in vitro model system demonstrating that stromal cells can utilize lactate produced by tumor cells. In future, varying incubation periods with 13C-lactate will allow us to investigate in detail the metabolic fate of lactate in CAFs. Based on the reaction mechanisms, it is likely that lactate is converted to α-ketoglutarate by the TCA cycle. However, it will be important to determine whether 13C-lactate is converted to 13C-α-ketoglutarate exclusively via the TCA cycle or whether this conversion may also occur in the cytoplasmic space via other metabolic conversion pathways. Additionally, our in vitro model system will further allow us to examine by NMR spectroscopy (of conditioned medium) whether stromal cells in turn secrete metabolites that can be used by the tumor cells as energy source. We will also examine the role of other factors present in tumor cell-conditioned medium that may contribute to efficient utilization of lactate by CAFs. Preliminary studies indicate that IL-8 and IL-6 may have a major role to play in regulation of the MCTs in both tumor cells as well as in CAFs.
Expression of glycolytic genes in MDA-MB-231 cells, as well as our lactate uptake and NMR data, seem to suggest that the “reverse Warburg effect” may not be exclusively observed in all conditions of tumor–stroma interaction. Indeed, investigation into the role of attenuated expression of caveolin-1 in the stroma in regulating the reverse Warburg effect [30] does not address conventional aerobic glycolysis that may occur in tumors displaying robust caveolin-1 expression in the stromal components of their TME. This underscores the heterogeneity inherent to the microenvironment and may imply that the conventional and the reverse Warburg effect may occur simultaneously in dynamic equilibrium, in which activated CAFs co-reside with passenger constituents. Our study corroborates the important work of Koukourakis et al. regarding the positive role of oxidative tumor stroma in the metabolic support of growth and development of glycoytic tumors [15,43,44] and is further supported by the studies of Andreeff et al. showing that stromal cells promote aerobic glycolysis in leukemic cells by mitochondrial uncoupling, and, in turn, the cancer cells suppress glucose uptake in the stromal cells, thereby ensuring a more ready supply of glucose for the glycolytic tumors [45,46]. Our current studies are investigating the metabolic cooperation between tumor cells and stromal CAFs in greater detail.
Acknowledgments
We like to thank Ms. Natalia Kruchevsky for her help in the NMR sample preparation and Alister Martin for help with the migration assays.
Footnotes
Financial Support: We acknowledge NIH P01 CA94060 and CA83084 (JAK) and NJCCR-10-1964-CCR-EO (DB) for grant support.
References
- 1.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 6.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 7.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 8.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- 9.Gillies RJ, Gatenby RA. Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr. 2007;39:251–257. doi: 10.1007/s10863-007-9085-y. [DOI] [PubMed] [Google Scholar]
- 10.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Ristow M. Oxidative metabolism in cancer growth. Curr Opin Clin Nutr Metab Care. 2006;9:339–345. doi: 10.1097/01.mco.0000232892.43921.98. [DOI] [PubMed] [Google Scholar]
- 12.Schulz TJ, Thierbach R, Voigt A, et al. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J Biol Chem. 2006;281:977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- 13.Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92:329–333. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 14.Sonveaux P, Vegran F, Schroeder T, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. 2006;66:632–637. doi: 10.1158/0008-5472.CAN-05-3260. [DOI] [PubMed] [Google Scholar]
- 16.Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- 17.Halestrap AP, Price NT. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem J. 1999;343(Pt 2):281–299. [PMC free article] [PubMed] [Google Scholar]
- 18.Mathupala SP, Parajuli P, Sloan AE. Silencing of monocarboxylate transporters via small interfering ribonucleic acid inhibits glycolysis and induces cell death in malignant glioma: an in vitro study. Neurosurgery. 2004;55:1410–1419. doi: 10.1227/01.neu.0000143034.62913.59. discussion 9. [DOI] [PubMed] [Google Scholar]
- 19.Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res. 2010:1417–1424. doi: 10.1016/j.yexcr.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Mishra PJ, Humeniuk R, Medina DJ, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol. 2004;558:5–30. doi: 10.1113/jphysiol.2003.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks GA. Lactate shuttles in nature. Biochem Soc Trans. 2002;30:258–264. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 23.Ackerstaff E, Pflug BR, Nelson JB, Bhujwalla ZM. Detection of increased choline compounds with proton nuclear magnetic resonance spectroscopy subsequent to malignant transformation of human prostatic epithelial cells. Cancer Res. 2001;61:3599–3603. [PubMed] [Google Scholar]
- 24.Street JC, Mahmood U, Ballon D, Alfieri AA, Koutcher JA. 13C and 31P NMR investigation of effect of 6-aminonicotinamide on metabolism of RIF-1 tumor cells in vitro. J Biol Chem. 1996;271:4113–4119. doi: 10.1074/jbc.271.8.4113. [DOI] [PubMed] [Google Scholar]
- 25.Dietl K, Renner K, Dettmer K, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. 2010;184:1200–1209. doi: 10.4049/jimmunol.0902584. [DOI] [PubMed] [Google Scholar]
- 26.Rudrabhatla SR, Mahaffey CL, Mummert ME. Tumor microenvironment modulates hyaluronan expression: the lactate effect. J Invest Dermatol. 2006;126:1378–1387. doi: 10.1038/sj.jid.5700255. [DOI] [PubMed] [Google Scholar]
- 27.Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2009;27:857–865. doi: 10.1002/stem.23. [DOI] [PubMed] [Google Scholar]
- 28.Viswanatha TS, Johnson RE, Fisher HF. alpha-Ketoglutaric acid: solution structure and the active form for reductive amination by bovine liver glutamate dehydrogenase. Biochemistry. 1982;21:339–345. doi: 10.1021/bi00531a022. [DOI] [PubMed] [Google Scholar]
- 29.Martin G, Chauvin MF, Dugelay S, Baverel G. Non-steady state model applicable to NMR studies for calculating flux rates in glycolysis, gluconeogenesis, and citric acid cycle. J Biol Chem. 1994;269:26034–26039. [PubMed] [Google Scholar]
- 30.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 31.Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG. Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol. 2009;135:867–877. doi: 10.1007/s00432-009-0590-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 33.Samuvel DJ, Sundararaj KP, Nareika A, Lopes-Virella MF, Huang Y. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol. 2009;182:2476–2484. doi: 10.4049/jimmunol.0802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DelaRosa O, Lombardo E. Modulation of adult mesenchymal stem cells activity by toll-like receptors: implications on therapeutic potential. Mediators Inflamm. 2010;2010:865601. doi: 10.1155/2010/865601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomchuck SL, Zwezdaryk KJ, Coffelt SB, Waterman RS, Danka ES, Scandurro AB. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells. 2008;26:99–107. doi: 10.1634/stemcells.2007-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slomiany MG, Grass GD, Robertson AD, et al. Hyaluronan CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 2009;69:1293–1301. doi: 10.1158/0008-5472.CAN-08-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang J, Quinones QJ, Holman TL, et al. The H+-linked monocarboxylate transporter (MCT1/SLC16A1): a potential therapeutic target for high-risk neuroblastoma. Mol Pharmacol. 2006;70:2108–2115. doi: 10.1124/mol.106.026245. [DOI] [PubMed] [Google Scholar]
- 38.Boussouar F, Benahmed M. Lactate and energy metabolism in male germ cells. Trends Endocrinol Metab. 2004;15:345–350. doi: 10.1016/j.tem.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 39.Mangia S, Simpson IA, Vannucci SJ, Carruthers A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: evidence from modeling of measured lactate levels during visual stimulation. J Neurochem. 2009;109(Suppl 1):55–62. doi: 10.1111/j.1471-4159.2009.06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto T, et al. Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. FASEB J. 2007;21(10):2602–2612. doi: 10.1096/fj.07-8174com. [DOI] [PubMed] [Google Scholar]
- 42.Samuvel DJ, et al. Lactate boosts TLR4 signaling and NF-kappaB pathway-mediated gene transcription in macrophages via monocarboxylate transporters and MD-2 up-regulation. J Immunol. 2009;182(4):2476–2484. doi: 10.4049/jimmunol.0802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koukourakis MI, Giatromanolaki A, Bougioukas G, Sivridis E. Lung cancer: a comparative study of metabolism related protein expression in cancer cells and tumor associated stroma. Cancer Biol Ther. 2007;6(9):1476–1479. doi: 10.4161/cbt.6.9.4635. [DOI] [PubMed] [Google Scholar]
- 44.Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia. 2005;7(1):1–6. doi: 10.1593/neo.04373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69:2163–2166. doi: 10.1158/0008-5472.CAN-08-3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. The Warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008;68:5198–5205. doi: 10.1158/0008-5472.CAN-08-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]