

Abstract
Tumor suppressor genes and oncogenes are both commonly altered during carcinogenesis. For oncogenes and other genes that drive growth, targeting mutated or activated forms (such as the EGFR-Her2/Nneu pathway) has been shown to be an effective anti-cancer approach. Pharmacologically targeting tumor suppressor genes has not been as fruitful, as many tumor suppressor genes are irreversibly silenced through somatic mutation or entirely deleted during carcinogenesis, thereby making it difficult to restore gene function. BRM, a key SWI/SNF complex subunit and a putative tumor suppressor gene, is inactivated in 15–20% of many solid tumor types. Unlike other tumor suppressor genes, the loss of BRM has been shown to be a reversible epigenetic change, rather than an irreversible genetic alteration. Using a high throughput drug screen, we identified a number of compounds that could effectively restore BRM expression and function. Two of these compounds, RH (RH02032) and GK (GK0037), were found to be such reactivating agents. Both compounds led to robust re-expression of BRM, induced downstream expression of BRM-dependent genes and inhibited BRM-dependent growth across a wide range of BRM-deficient cancer cell lines of different origins. We therefore show, for the first time, that pharmacologic reversal of epigenetic changes of the SWI/SNF chromatic remodeling complex subunit, BRM, is a potentially viable and novel therapeutic approach.
Keywords: SWI/SNF, epigenetic, tumor suppressor, target therapy, chromatin remodeling, cancer therapy
Introduction
Targeted cancer therapy is focused on blocking or interfering with key molecular alterations that give cancer cells a growth advantage and allow them to escape the normal control mechanisms. Both small-molecule inhibitors and antibodies have been devised to block the function of cell-surface receptors, such as HER2 and EGFR2, and inhibiting certain intracellular kinases, such as Bcr-Abl, have also been successful as targeted therapies (for example, imatinib). In general, these new cancer drugs have focused on blocking oncogenic molecular changes. Yet, cancer evolves not only because of unfettered growth mechanisms, but also because of the loss of proteins involved in growth-inhibition—that is, tumor suppressors. When oncogenes are amplified, altered or mutated, the resultant genes usually have acquired constitutive or enhanced growth-promoting properties that can serve as targets. In contrast, tumor suppressors are not simply mutated, but are deleted in the majority of cases, rendering them exceedingly difficult to target. However, in a subset of cases, certain tumor suppressor proteins are not mutated, but rather silenced, raising the possibility that their expression can be restored. Such suppression is often called epigenetic because the mechanism of silencing requires ancillary or additional proteins. In theory, although it may be feasible to restore epigenetically suppressed proteins, the practical application of such approaches has lagged far behind those that target oncogenic proteins.
Our pursuit to study the SWI/SNF complex involvement in cancer stemmed from the initial observation that the INI1(BAF47) proteins were likely bona fide tumor suppressors. In vivo murine knockout studies of this protein show that homogenous loss of BAF47 is highly tumorigenic—indeed, 100% of mice develop tumors within a median time of 10 weeks (Roberts et al., 2002). BAF47 is only one subunit of the SWI/SNF complex (Wang et al., 1996a, b), and in yeast the complex requires the bulk of subunits to be present (Carlson and Laurent, 1994); thus, we and others reasoned that other SWI/SNF units could be involved in cancer development. We found via western blot analysis in a variety of cancer cell lines that the SWI/SNF ATPase homologues Brahma (BRM) and Brahma-Related Gene 1 (BRG1) were missing in at least 10% of cancer cell lines and up to 30–40% of lung cancer cell lines (DeCristofaro et al., 2001; Reisman et al., 2003). Immunohistochemical staining of primary lung cancers showed that BRG1 is lost in 15% of lung cancers, whereas BRM is lost slightly more often, about 17–18% (Glaros et al., 2007) (unpublished data). Moreover, BRM has been found to be silenced in 10–20% of lung, breast, colon, esophageal, ovarian, bladder, prostate, gastric and head/neck tumors (Glaros et al., 2007; Yamamichi et al., 2007; Shen et al., 2008; Reisman et al., 2009), suggesting that this protein has a general role in cancer. Re-expression experiments showed that, like BAF47, restoring either BRG1 or BRM in deficient cell lines inhibits growth (Betz et al., 2002; Sansam and Roberts 2006). Moreover, BRG1 and BRM are known to bind to Rb and its family members, and BRG1 or BRM is specifically required for Rb-mediated growth inhibition (Dunaief et al., 1994; Strober et al., 1996; Reisman et al., 2002, 2009). Subsequent studies have linked SWI/SNF to anticancer cellular functions, such as DNA repair, cellular adhesion, differentiation and development (Peterson and Workman, 2000; Klochendler-Yeivin et al., 2002; Morrison and Shen, 2006; Simone, 2006; Reisman et al., 2009). Hence, the inactivation of SWI/SNF through the loss of one of its key ATPases, BRG1 or BRM, would likely spur the development of cancer. These studies together, therefore, indicate that BRM and BRG1 are likely tumor suppressors.
Mutations are likely the prominent reason for loss of BRG1 expression, similar to many other tumor suppressors (Wong et al., 2000; Medina et al., 2008). In contrast, mutations are not associated with loss of BRM protein expression, and BRM is epigenetically silenced (Mizutani et al., 2002; Yamamichi et al., 2005; Glaros et al., 2007). Recently, several groups have shown that the suppression of BRM in cancer cell lines can be restored with histone deacetylase (HDAC) inhibitors (Bourachot et al., 2003; Yamamichi et al., 2005; Glaros et al., 2007). However, Bourachot et al. (2003) clearly demonstrated that HDAC inhibitors, when applied to cancer cells, induce the acetylated form of BRM, thereby inactivating the function of BRM. Thus, HDAC inhibitors can induce BRM and thereby demonstrate that BRM can be restored, but because they inhibit BRM function, they cannot be practically used to restore BRM in a clinical setting. As reactivation of BRM can inhibit growth and thus could be clinically advantageous, a major goal of our work has been to find novel small-molecule compounds that can restore both the expression and the function of BRM. This would represent that, potentially for the first time, a tumor suppressor pathway, rather than an oncogene, could be used as a targeted therapy. Here, we show the results of a high throughput drug screen identifying a number of compounds that not only restore BRM expression but its function as well. Moreover, we highlight the advantages of restoring BRM, as we found that ectopic as well as pharmacological restoration of BRM induced growth inhibition. Together, these findings demonstrate the initial step to showing to the feasibility of restoring an epigenetically suppressed protein, BRM. Like targeted therapy for oncogenes such as EGFR or HER2, restoration of epigenetically lost pathways could add to our arsenal of cancer fighting.
Results and discussion
Reversal of BRM silencing by infection of BRM-containing vectors resulted in growth inhibition
The first step toward assessing whether activating BRM could be used as a form of therapy is to show that there are compounds that can restore not only BRM expression, but also BRM function. Hence, an initial goal of our research was to identify agents that can reactivate BRM for therapeutic purposes. For these studies, we took advantage of the fact that the glucocorticoid receptor is SWI/SNF dependent. We stably integrated the MMTV (glucocorticoid-sensitive) promoter linked to the luciferase gene into the BRG1/BRM-deficient cell line, SW13. With the addition of the glucocorticoid agonist, dexamethasone, this reporter cell line yields high levels of luciferase only if and when the BRM gene is induced by a given compound. Hence, we used this reporter construct to screen and identify compounds that could pharmacologically restore a functional BRM protein. Further details of this high throughput screening are described elsewhere (Gramling and Reisman, 2011).
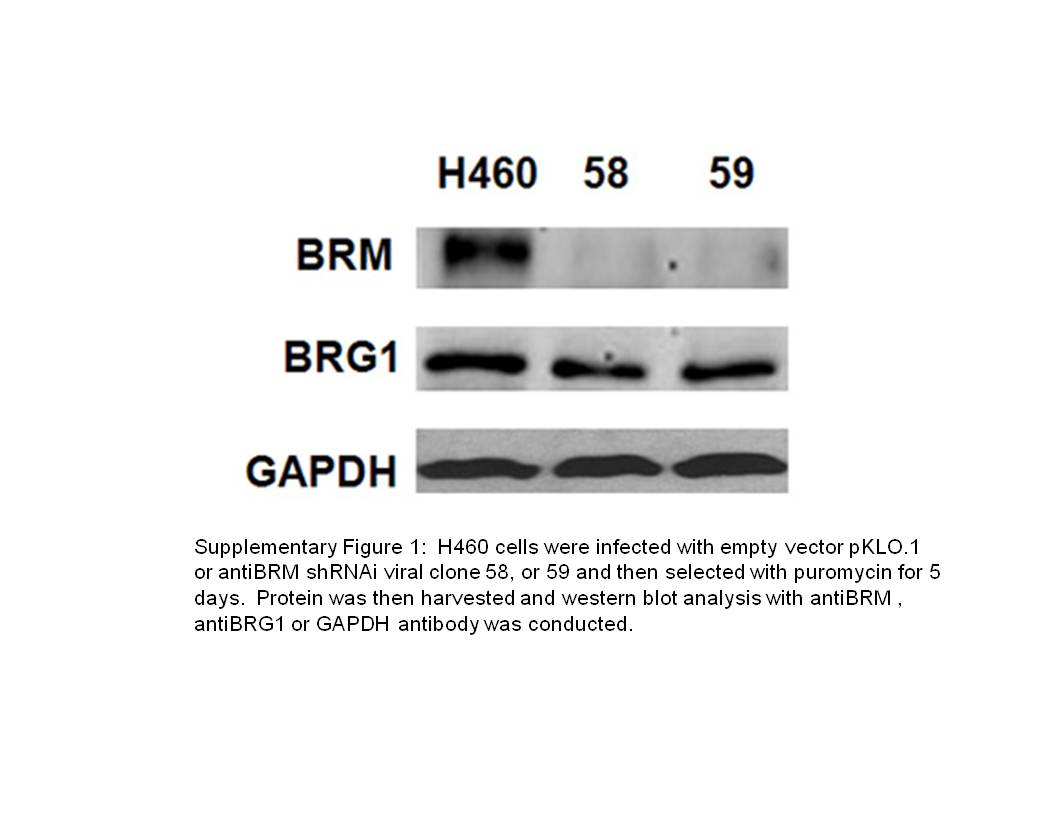
For this purpose, we developed a high throughput ‘BRM-functional’ cell-based assay to screen for BRM-reactivating agents from a number of drug libraries, including the ChemDiv, MayBridge and ChemBridge chemical libraries at the Life Science Institute at the University of Michigan and Michigan High Throughput Center (Kalamazoo, MI) (Gramling and Reisman, 2011). Approximately, 1% of the agents were screen-positive and further testing of a subset of these hits showed that two compounds (RH02032 [RH] and GK00397 [GK]) readily induced BRM, with relatively low toxicity (see Supplementary Figure 1 for the structures). As HDAC inhibitors maximally induce BRM protein and mRNA by 72 h (Glaros et al., 2007), we used a 72-h time point to conduct dose-response curves to determine the lowest concentration, providing the maximal induction of BRM mRNA (data not shown). At the optimal concentrations of these compounds (50 μM of RH and 200 μM of GK), we saw little-to-no toxicity over 72 h. In contrast, the induction of BRM proteins lagged behind the induction of BRM mRNA and required between 5–6 days to be maximally induced. During this timeframe, these concentrations of RH and GK did cause some toxicity (cell death), but this was minimized (~15–20%) if we used higher cell density and if the media (with compounds) was changed after 72 h. Application of these two compounds (50 μM of RH and 200 μM of GK) produced 10–12-fold induction of BRM mRNA over 72 h (as measured by quantitative PCR; Figure 1a) and robust induction of BRM protein levels (via western blotting; Figure 1b) after 5–6 days. The induction of both BRM mRNA and protein was essentially completely blocked by the introduction of anti-BRM short hairpin RNA interference (shRNAi) into these cell lines (Figures 1a and b). The efficiency of anti-BRM shRNAi can be seen in the knockdown of BRM (90–95%) of the BRM-positive cell line H460 (Supplementary Figure 1).
Figure 1.

Pharmacological induction of BRM. (a, b) Induction of BRM pharmacologic application of compounds, RH and GK (Maybridge Trevillett, Tintagel, Cornwall, UK). Error bars reflect standard error of experiments repeated at least six times. (a) RNA was collected using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and isolated using Qiagen’s RNeasy kit, as per manufacturer’s instructions (Qiagen, Valencia, CA, USA). Complementary DNA was generated from 1 μg of DNase-free RNA using iScript (BioRad, Hercules CA, USA), as per manufacturer’s instructions. Real-time reverse transcriptase PCR was performed using RT Fast SYBR Green/ROX qPCR Master Mix (SABioscience, Valencia, CA, USA), with the following conditions: 95 °C, 10 min; 40 cycles (95 °C, 10 s and 60 °C, 30 s). Induction of BRM mRNA by the application of RH or GK for 72 h, measured by quantitative PCR, on the BRM-deficient C33A and SW13 cell lines. In the controls, anti-BRM shRNAi-infected SW13 and C33A daughter cell lines, BRM was not induced. Polr2A was used as the control for these experiments. (b) Cell lines were grown in RPMI with 5% fetal bovine serum supplemented with Pen/Strep and Glutamax (Invitrogen). These cell lines were authenticated by sequencing the p53 mutations and K-ras mutations in each cell line used compared with published results. C33A and SW13 cells were treated for 120 h with RH (50 μM) or GK (200 μM) and protein was isolated (Glaros et al., 2007). To perform western blot analysis, 80 μg of protein was used (Strobeck et al., 2002; Reisman et al., 2002, 2005). BRM expression was detected using polyclonal anti-BRM antibody, previously described in (Glaros et al., 2007), chemiluminescence (Amersham GE Healthcare, Piscataway, NJ, USA), and normalized to GAPDH expression (Trevigen, Gaitherburg, MD, USA). Induction of BRM protein by western blot analysis after the application of either GK or RH for 6 days. GAPDH was used as the loading control.
To show that the induced endogenous BRM is functional (and not inactive, as is the case of some HDAC inhibitors (Glaros et al., 2007)), we examined the C33A and SW13 BRM-negative cell lines treated with RH or GK for the induction of a number of BRM-dependent genes (DDX58, P8, XAF1 and LGAL). The BRM dependency of these genes was previously shown through microarray experiments on C33A and SW13 at 72 h after transfection with BRM (unpublished data). After 72 h of RH or GK exposure in BRM:BRG1-deficient cell lines (SW13 and C33A), DDX58, P8, XAF1 and/or LGAL genes were all induced a minimum of two ΔCT values (Figures 2a–d), or at least four- to five-fold. To demonstrate that this induction was due to BRM and not some other effect of these compounds, we stably infected both cell lines with anti-BRM shRNAi. We then repeated this experiment using SW13 and C33A infected with anti-BRM shRNAi and observed minimal induction of these genes, <1 ΔCT value or less than two-fold (Figures 2a–d), thereby demonstrating that the induction of these genes is BRM dependent. In turn, the induction of these genes shows that BRM is functional when induced by these compounds and that pharmacological restoration of BRM can occur. This is a novel advance, as no other agents to date have been able to restore BRM functionality. This discovery opens the door for possibly developing BRM as a form of targeted therapy.
Figure 2.

The induction of BRM-dependent genes. To show that BRM is functional when induced by either RH or GK, the induction of BRM-dependent genes was measured and demonstrated by quantitative PCR in the C33A and SW13 cell lines, which were treated first with either RH 50 μM (a, b) or GK 200 μM (c, d) for 72 h. The DDX58, P8 LGAL and/or XAF1 genes were significantly induced in the C33A (a, c) and SW13 (b, d) cell lines, although these genes were not significantly induced in the anti-BRM shRNAi-derived C33A and SW13 daughter cell lines. The standard error shown represents six experiments for each bar on the graph.
Previously published data have shown that introducing BRM or BRG1 into BRG1:BRM-deficient cell lines (SW13) arrests growth (Dunaief et al., 1994; Strober et al., 1996; Muchardt et al., 1998). However, these data are derived from only a few (~2) cell lines. To determine if the same is true in other BRM-deficient cells, we introduced BRM by viral infection into nine BRM-deficient cell lines and two (control) BRM-positive cell lines using pBABE-BRM. As a control, we also infected these cells with the empty vector version of pBABE. In each case, significant growth inhibition occurred when pBABE-BRM was re-introduced into the BRM-deficient parental line, with the vast majority of the cell lines having growth inhibition between 85–95% (Figure 3a; reference: pBABE (empty vector) infected daughter lines). In contrast, when pBABE-BRM was introduced in the BRM-positive cells (H460 and Calu-6), only ~20% growth inhibition was observed. In the control experiments, essentially no growth inhibition was observed when only pBABE (empty vector) was introduced into either BRM-negative or BRM-positive cells. These data and previously published data (Dunaief et al., 1994; Muchardt et al., 1998) show that restoration of BRM expression can lead to growth inhibition in BRM-deficient cancer cell lines.
Figure 3.

(a) The effect of restoring BRM expression on growth. We virally infected BRM-deficient cell lines, Panc-1, H1299, H522, SW13, A427, H23, H1573, H125 and C33A (white bars) and BRM-positive cell lines H460 and Calu-6 (black bars) with BRM, using pBABE-BRM and pBABE-empty vector. After infection, we selected for 48 h with puromycin to eliminate uninfected cells, and confirmed BRM re-expression in each cell line through quantitative PCR. We then measured growth inhibition by MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (using a BioTek plate reader, Biotek, Winooski, VT, USA) or by flow cytometry with trypan blue exclusion after either viral infection or pharmacological treatment with RH (50 μM) or GK (200 μM). For virally infected cell lines, we measured the number of cancer cells that were present 5 days after infecting with BRM (pBABE-BRM infected; Addgene, Cambridge, MA, USA), comparing them to the same cancer cells infected with empty vector (pBABE empty vector; Addgene). To eliminate uninfected cells, infected cultures were grown in the presence of 7.5 μg/ml of puromycin for 2–5 days after infection. In each case, BRM reexpression significantly inhibited the growth of BRM-deficient cell lines, whereas positive BRM cell lines were only mildly inhibited. The effect of RH and GK on growth inhibition. (b) The BRMinducing agents RH and GK also inhibited growth when applied for 3 days to the BRM-deficient cell lines C33A and SW13, whereas the anti-BRM shRNAi-infected SW13 and C33A daughter cell lines were not significantly inhibited with the application of RH and GK.
In addition to restoring BRM expression, we tested whether RH and GK could inhibit growth akin to the viral introduction of exogenous BRM. We evaluated the growth of BRM-deficient cells (C33A and SW13) upon exposure to RH and GK, and found that, indeed, RH/GK caused these cell lines to undergo growth arrest and flatten, akin to the experimental induction of the BRM gene (Figure 3b). Both cell lines may have also increased cellular adhesion, as the cells were much harder to remove from their plastic dishes after pharmacological intervention; this observation is not surprising since restoration of BRM can induce CD44, E-cadherin, CEACAM1 and a number of integrin adhesion molecules (unpublished data). When BRG1 is ectopically expressed, a similar inhibitory effect on growth is seen and similar cadres of adhesion proteins are induced (Dunaief et al., 1994; Strober et al., 1996; Liu et al., 2001; Hendricks et al., 2004). To test whether the observed growth arrest was due to BRM re-expression, we blocked expression by infecting these parental BRMdeficient cell lines with anti-BRM shRNAi (Figure 3). When BRM expression was inhibited by shRNAi, neither RH nor GK effectively inhibited growth when applied for 5 days. These data suggest that RH and GK inhibit cellular growth in BRM-deficient cell lines by inducing BRM expression; if BRM is abrogated, the growth inhibition seen with the application of RH, GK, or both, is also abrogated. Hence, pharmacological restoration of BRM seems to be a viable therapeutic avenue worth further experimental exploration.
As RH and GK become increasingly more toxic with prolonged exposures, these compounds, although valuable experimental tools, are probably not feasible drug candidates. Further work will be required to determine how and why BRM is suppressed, and part of this puzzle are two BRM polymorphisms, which have high morphology to MEF2-binding sites and correlate very tightly with BRM loss in cell lines and primary lung tumors. It is likely that these polymorphisms are functionally linked to the loss of BRM in cancers. Moreover, as these polymorphic sites are potentially correlated with lung cancer risk, BRM loss may be a precursor to cancer development. This observation (in turn) opens the door for pharmacological BRM restoration as a means to potentially modify cancer risk, in addition to being a primary form of targeted therapy for established cancers.
In summary, our results begin to lay the groundwork to pursue BRM reactivation as a form of clinical therapy. Restoring anticancer proteins may be a viable and reasonable therapeutic approach to complement the inhibition of oncogenic changes. We also showed, using two example compounds (RH and GK), that BRM can be effectively pharmacologically restored. In previous work, we have shown that BRM expression is absent (or suppressed) in a fifth of all tumor types (Reisman et al., 2009), which is a sufficiently large enough fraction (compared with subsets of lung, colorectal, and breast cancer patients that are defined by somatic alterations in EGFR, KRAS or HER2neu, respectively) to make such approaches prudent and economical. Moreover, we have sequenced both tumors and cell lines, and have not found any mutations (Glaros et al., 2007), thereby providing evidence that epigenetic silencing is the main method of suppression of BRM in cancer cells. Our data provide a foundation and basis for further study of BRM as a potential target for therapeutic intervention across many different tumor types.
Supplementary Material
{kind=link}
Acknowledgments
GL is funded by the Alan B Brown Chair in Molecular Genomics, CCO Chair in Experimental Therapeutics and Population Studies, and the Posluns Family Foundation; Reisman lab funding for this paper is from NCI: R03CA128038–02 and 1R01CA136683–01A1.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21:5193–5203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- Bourachot B, Yaniv M, Muchardt C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J. 2003;22:6505–6515. doi: 10.1093/emboj/cdg621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Laurent BC. The SNF/SWI family of global transcriptional activators. Curr Opin Cell Biol. 1994;6:396–402. doi: 10.1016/0955-0674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- DeCristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, Weissman BE. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol. 2001;186:136–145. doi: 10.1002/1097-4652(200101)186:1<136::AID-JCP1010>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058–7066. doi: 10.1038/sj.onc.1210514. [DOI] [PubMed] [Google Scholar]
- Gramling S, Reisman D. Discovery of BRM targeted therapies: novel reactivation of an anticancer gene. Lett Drug Des Discov. 2011;8:93–99. doi: 10.2174/157018011793663840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks KB, Shanahan F, Lees E. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol. 2004;24:362–376. doi: 10.1128/MCB.24.1.362-376.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler-Yeivin A, Muchardt C, Yaniv M. SWI/SNF chromatin remodeling and cancer. Curr Opin Genet Dev. 2002;12:73–79. doi: 10.1016/s0959-437x(01)00267-2. [DOI] [PubMed] [Google Scholar]
- Liu R, Liu H, Chen X, Kirby M, Brown PO, Zhao K. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell. 2001;106:309–318. doi: 10.1016/s0092-8674(01)00446-9. [DOI] [PubMed] [Google Scholar]
- Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–622. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- Mizutani T, Ito T, Nishina M, Yamamichi N, Watanabe A, Iba H. Maintenance of integrated proviral gene expression requires Brm, a catalytic subunit of SWI/SNF complex. J Biol Chem. 2002;277:15859–15864. doi: 10.1074/jbc.M112421200. [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Shen X. Chromatin modifications in DNA repair. Results Probl Cell Differ. 2006;41:109–125. doi: 10.1007/400_008. [DOI] [PubMed] [Google Scholar]
- Muchardt C, Bourachot B, Reyes JC, Yaniv M. ras transformation is associated with decreased expression of the brm/SNF2alpha ATPase from the mammalian SWI-SNF complex. EMBO J. 1998;17:223–231. doi: 10.1093/emboj/17.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Workman JL. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr Opin Genet Dev. 2000;10:187–192. doi: 10.1016/s0959-437x(00)00068-x. [DOI] [PubMed] [Google Scholar]
- Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- Reisman DN, Strobeck MW, Betz BL, Sciariotta J, Funkhouser W, Jr, Murchardt C, et al. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene. 2002;21:1196–1207. doi: 10.1038/sj.onc.1205188. [DOI] [PubMed] [Google Scholar]
- Reisman DN, Sciarrotta J, Bouldin TW, Weissman BE, Funkhouser WK. The expression of the SWI/SNF ATPase subunits BRG1 and BRM in normal human tissues. Appl Immunohistochem Mol Morphol. 2005;13:66–74. doi: 10.1097/00129039-200503000-00011. [DOI] [PubMed] [Google Scholar]
- Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–566. [PubMed] [Google Scholar]
- Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- Sansam CG, Roberts CW. Epigenetics and cancer: altered chromatin remodeling via Snf5 loss leads to aberrant cell cycle regulation. Cell Cycle. 2006;5:621–624. doi: 10.4161/cc.5.6.2579. [DOI] [PubMed] [Google Scholar]
- Shen H, Powers N, Saini N, Comstock CE, Sharma A, Weaver K, et al. The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res. 2008;68:10154–10162. doi: 10.1158/0008-5472.CAN-08-1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone C. SWI/SNF: the crossroads where extracellular signaling pathways meet chromatin. J Cell Physiol. 2006;207:309–314. doi: 10.1002/jcp.20514. [DOI] [PubMed] [Google Scholar]
- Strobeck MW, Reisman DN, Gunawardena RW, Betz BL, Angus SP, Knudsen KE, et al. Compensation of BRG-1 function by Brm: insight into the role of the core SWI-SNF subunits in retinoblastoma tumor suppressor signaling. J Biol Chem. 2002;277:4782–4789. doi: 10.1074/jbc.M109532200. [DOI] [PubMed] [Google Scholar]
- Strober BE, Dunaief JL, Guha, Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins1. Mol Cell Biol. 1996;16:1576–1583. doi: 10.1128/mcb.16.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996a;15:5370–5382. [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996b;10:2117–2130. doi: 10.1101/gad.10.17.2117. [DOI] [PubMed] [Google Scholar]
- Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–6177. [PubMed] [Google Scholar]
- Yamamichi N, Inada K, Ichinose M, Yamamichi-Nishina M, Mizutani T, Watanabe H, et al. Frequent loss of Brm expression in gastric cancer correlates with histologic features and differentiation state. Cancer Res. 2007;67:10727–10735. doi: 10.1158/0008-5472.CAN-07-2601. [DOI] [PubMed] [Google Scholar]
- Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24:5471–5481. doi: 10.1038/sj.onc.1208716. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.