

Abstract
Glucose stimulates rodent and human β-cell replication, but the intracellular signaling mechanisms are poorly understood. Carbohydrate response element-binding protein (ChREBP) is a lipogenic glucose-sensing transcription factor with unknown functions in pancreatic β-cells. We tested the hypothesis that ChREBP is required for glucose-stimulated β-cell proliferation. The relative expression of ChREBP was determined in liver and β-cells using quantitative RT-PCR (qRT-PCR), immunoblotting, and immunohistochemistry. Loss- and gain-of-function studies were performed using small interfering RNA and genetic deletion of ChREBP and adenoviral overexpression of ChREBP in rodent and human β-cells. Proliferation was measured by 5-bromo-2′-deoxyuridine incorporation, [3H]thymidine incorporation, and fluorescence-activated cell sorter analysis. In addition, the expression of cell cycle regulatory genes was measured by qRT-PCR and immunoblotting. ChREBP expression was comparable with liver in mouse pancreata and in rat and human islets. Depletion of ChREBP decreased glucose-stimulated proliferation in β-cells isolated from ChREBP−/− mice, in INS-1–derived 832/13 cells, and in primary rat and human β-cells. Furthermore, depletion of ChREBP decreased the glucose-stimulated expression of cell cycle accelerators. Overexpression of ChREBP amplified glucose-stimulated proliferation in rat and human β-cells, with concomitant increases in cyclin gene expression. In conclusion, ChREBP mediates glucose-stimulated proliferation in pancreatic β-cells.
β-Cells have an extraordinary intrinsic ability to detect and respond to changes in metabolic demand by altering β-cell mass: expansion by proliferation and/or neogenesis and contraction by cell death (1). To proliferate, β-cells must pass through strict cell cycle check points, and much progress has been made toward identification of the controlling elements of the cell cycle in β-cells (2). It is now appreciated, for example, that overexpression of groups or even single components of G0/G1-S phase cell cycle regulatory proteins, such as the D cyclins or their cdk partners, is sufficient to drive β-cell replication (3–5). Knockout and transgenic mouse models that remove or overexpress the cyclins or cdks have generally confirmed their critical role in β-cell proliferation and glucose homeostasis (2). Furthermore, dissection of the various physiologic processes that increase β-cell proliferation has led to the identification of a number of natural mitogens, including glucagon-like peptide 1, hepatic growth factor, parathyroid hormone-related protein, lactogens, and, the focus of the current study, glucose (6–9). What remains to be elucidated are the detailed molecular mechanisms by which natural mitogenic signals interact with the cell cycle regulatory machinery to promote β-cell proliferation.
Glucose increases β-cell proliferation in a variety of model systems, both in vitro and in vivo. In vitro, glucose stimulates β-cell proliferation in fetal and adult rat islets, in mouse islets, and in several rodent insulinoma β-cell lines (3,7,10). In vivo, glucose promotes β-cell proliferation in numerous models, including a high sucrose diet, recovery from hypoglycemia, and partial pancreatectomy (11–14). Alonso et al. (15) demonstrated that a 4-day intravenous infusion of 50% glucose into mice, which modestly increases blood glucose concentrations, leads to markedly increased β-cell proliferation as determined by 5-bromo-2′-deoxyuridine (BrdU) incorporation, consistent with earlier rodent infusion studies (16,17). Furthermore, human islets transplanted under the kidney capsule of immune-compromised, diabetic mice display increased BrdU incorporation that correlates with elevated circulating glucose concentrations (18). Recently, glucose was identified as a powerful systemic signal for mouse β-cell proliferation, wherein proliferation is proportional to β-cell glycolytic flux (9). How glucose metabolic flux translates to progression through the cell cycle is unknown.
Carbohydrate response element-binding protein (ChREBP; official name: Mlxipl) has emerged as the prototypical glucose-sensing transcription factor (19). Originally cloned from liver tissue, ChREBP upregulates genes involved in fatty acid synthesis in a glucose-dependent manner and is expressed in several metabolically relevant tissues, including hepatocytes, adipocytes, and pancreatic β-cells (19). It is noteworthy that although the role of ChREBP in the liver is clearly lipogenic, its physiological importance in the pancreatic β-cell is poorly understood. Given that ChREBP regulates glucose and lipid metabolism, it is perhaps not surprising that ChREBP was recently found to be crucial for cancer cell proliferation (20). In transformed cells, ChREBP promotes increased glucose flux, glycolysis over complete glucose oxidation, lipogenesis, and the production of reducing equivalents and other anabolic intermediates required for cell division.
In the current study, we determined the relative abundance of ChREBP in human and rodent β-cells and determined the effects of depletion and overexpression of ChREBP on glucose-stimulated β-cell proliferation. We found ChREBP abundance in rodent and human β-cells to be comparable with that of liver and that depletion of ChREBP blocked glucose-stimulated proliferation in INS-1–derived 832/13 rat insulinoma cells, in isolated rat β-cells, in β-cells isolated from ChREBP−/− mice, and, more importantly, in isolated human β-cells. In addition, depletion of ChREBP blunted glucose-dependent increases in several cell cycle regulators, including cyclin D2, cyclin A, cyclin E, cdk4, and cdk6. Furthermore, adenoviral overexpression of ChREBP amplified the glucose-stimulated proliferation of INS-1–derived 832/13 cells, with concomitant increases in the expression of cyclins A and E. Finally, overexpression of ChREBP amplified glucose-stimulated proliferation in isolated rat and human β-cells, with increases in cell cycle accelerator genes and decreases in cell cycle inhibitor genes. We conclude that ChREBP is an essential component of glucose-stimulated β-cell proliferation.
RESEARCH DESIGN AND METHODS
Cell culture and adenovirus.
INS-1–derived 832/13 rat insulinoma cells (21) were provided by Dr. Christopher Newgard (Sarah W. Stedman Nutrition and Metabolism Center, Duke University, Durham, NC). Cells were maintained as described previously (22). Wild-type ChREBP adenovirus was a gift from Howard Towle (University of Minnesota [23)]) and control green fluorescent protein (GFP) adenovirus was as described previously (24). Adenovirus was added to cells at a multiplicity of infection of 200 as determined by optical density at 260 nm and by plaque assay.
Small interfering RNA duplex-mediated gene suppression.
SmartPool small interfering RNA (siRNA) duplexes (Dharmacon, Lafeyette, CO) were transfected into INS-1–derived cell lines for 48 h in the presence of Dharmafect Reagent 1 (Dharmacon). Duplexes were targeted to 19 to 21 bp regions of the rat ChREBP cDNA sequence (Cat. No. L-092970). A pool of duplexes with no known sequence homology or biological effect (siControl) was used as a control. Cells were cultured for 48 h in RPMI 1640 medium for INS-1 cells as described (21) with 5% FCS, followed by 6 h in low glucose (3 mmol/L). Glucose was then added to adjust the final glucose concentration to 15 mmol/L glucose for the high glucose groups, and the cells were cultured for an additional 18 h. For Accell siRNA treatment, dispersed rat or human islets cells were treated with Accell medium (Dharmacon) containing 2% FCS, 5.5 mmol/L glucose, and either control or species-specific ChREBP Accell siRNA (1 μmol/L) for 6 h, and then glucose was added to the high glucose group to adjust the concentration to 20 mmol/L. Cells were incubated for a total of 72 h. BrdU was added for the final 36 h for rat islets and 48 h for human islet preparations.
RNA isolation and RT-PCR.
RNA was isolated and quantitative RT-PCR was performed as described previously (25). Primer sequences can be found in Supplementary Table 1.
Immunoblot analysis.
Immunoblots were performed as described previously (25). Antibodies used included the following: ChREBP (NB400–136; Novus Biologicals, Littleton, CO), cyclin D2 (Ab3085; 1:1,000; Abcam, Cambridge, MA), cyclin A2 (c-4710; Sigma, St. Louis, MO), cyclin E1 (SC-481; Santa Cruz Biotechnology, Santa Cruz, CA), cdk-2 (SC-163; Santa Cruz), cdk4 (Ab3112; Abcam), cdk6 (Ab3126; Abcam), and β-actin (A2228; Sigma). All primary antibodies were used at a dilution of 1:1,000, with the exception of cyclin E, which was used at 1:500.
Isolation of rodent islets.
Islets were isolated from 8- to 10-week-old Wistar rats (Charles River Laboratories, Wilmington, MA) as described previously (25). Isolated rat islets were cultured in RPMI 1640 medium supplemented with 10% FCS, 5.5 mmol/L glucose, and penicillin-streptomycin for 24 h before further experiments. Islets were dispersed by trypsinization as described previously (26). Mouse islets were isolated from 8- to 10-week-old C57Bl/6 wild-type or ChREBP −/− mice (27), purchased from JAX Laboratories (stock no. 010537), and cultured as described previously (28). These animal studies were performed in compliance with, and with approval of, the University of Pittsburgh Institutional Animal Care and Use Committee.
Human islets.
Human cadaveric islets were obtained through National Institutes of Health- and Juvenile Diabetes Research Foundation (JDRF)-supported Integrated Islet Distribution Program (http://www.iidp.coh.org). The mean age of the donors was 44 years, the purity of the preparations was 88%, and the viability was 95%. Islets were cultured and dispersed by trypsinization as described previously (4).
Human liver.
Hepatocytes were isolated from five normal human liver donors. Human liver tissue was procured under an Institutional Review Board–approved protocol and with support from the liver tissue procurement and cell distribution system (Pittsburgh, PA). Hepatocytes were prepared by a three-step collagenase perfusion technique (29). Viability was >75% in all preparations. Protein and RNA were isolated as described earlier for Western blot and RT-PCR analysis.
Mouse histochemistry.
Mouse pancreata were removed, fixed overnight in Bouin's solution, and embedded in paraffin. Sections were placed onto glass slides and deparaffinized with xylene, and immunofluorescence was performed using an anti-insulin antibody (Invitrogen, Carlsbad, CA) at 1:100 dilution, an anti-ChREBP antibody at 1:1,000 dilution (Novus), and an anti-BrdU antibody (GE Healthcare, Piscataway, NJ) at 1:500 dilution. Visualization of staining was achieved using Alexa Fluor 488–conjugated secondary antibodies (1:250; Invitrogen). Nuclei were stained with DAPI, and imaging was performed as described above.
Cell proliferation and cell cycle analysis.
[3H]Thymidine incorporation measurements were performed essentially as described (7). In brief, INS-1–derived 832/13 cells were cultured in RPMI 1640 medium containing 11 mmol/L glucose and supplemented with 10% FBS. Twenty-four hours later, cells were serum-depleted with medium containing 2 mmol/L glucose. After an additional 24 h, cells were treated with fresh medium without FBS containing either 2 or 20 mmol/L glucose and [3H]thymidine (0.5 μCi/well; Amersham Pharmacia Biotech). [3H]Thymidine incorporation was stopped 4 h later; radioactivity was corrected for protein levels. Results are expressed as percentage of counts per minute incorporated per microgram of protein in control cells (100%). To analyze cell cycle progression, INS-1–derived 832/13 cells were fixed with 70% ethanol overnight at 4°C. Fixed cells were treated with 100 units/mL RNAase A (Sigma) in PBS and stained with 50 μg/mL propidium iodide (Sigma) at room temperature for 30 min. The cell cycle phase distribution of 10,000 cells was determined by flow cytometry using a BD LSR II flow cytometer (Becton Dickinson Biosciences, San Jose, CA). Analysis of data, including determination of gates, was performed using Modfit LT 3.0 software (Verity Software House, Topsham, ME) using gating as described (30). BrdU incorporation into isolated rat islet cells was determined as described by Cozar-Castellano et al. (3). All images were acquired using an Olympus Provis I fluorescence microscope or with a confocal Olympus Fluoview 1000 at the Center for Biologic Imaging Core Facility of the University of Pittsburgh.
RESULTS
ChREBP is expressed at similar levels in pancreatic β-cells and liver.
ChREBP has mostly been studied in liver cells where it regulates the expression of genes involved in lipogenesis (19). Although it is known that ChREBP is expressed in pancreatic β-cells, its relative abundance, compared with liver, has not been well documented (25,31). In Fig. 1, we found that ChREBP mRNA was expressed at levels in rat islets and in rat INS-1–derived 832/13 cells that were comparable with the level found in primary rat hepatocytes (Fig. 1A, left). In addition, a similar equivalent abundance was found between ChREBP mRNA expression in primary human hepatocytes and in human islets (Fig. 1A, right). At the protein level, ChREBP expression was significantly higher in rat liver and hepatocyte preparations than in isolated rat islet samples (1.6 ± 0.4 vs. 0.6 ± 0.1 densitometric units after normalization to β-actin, P = 0.02, n = 3; Fig. 1B, top). By contrast, ChREBP protein expression was similar in human islets and human hepatocytes (1.2 ± 0.4 vs. 0.9 ± 0.1 densitometric units after normalization to β-actin, P = 0.44, n = 3; Fig. 1B, bottom). ChREBP protein expression appeared robust in insulin-positive cells isolated from both rat and human islets using immunofluorescence (Fig. 1C). Furthermore, immunostaining of sections of mouse pancreata revealed robust expression of ChREBP in both islets and surrounding acinar tissue. In conclusion, ChREBP is expressed at levels in rodent and human β-cells that are similar to the abundance of ChREBP in the liver.
FIG. 1.

ChREBP abundance in rodent and human β-cells is comparable with liver. A: Relative mRNA abundance from rat or human liver or pancreatic β-cells, using quantitative RT-PCR and species-specific primers. Results were normalized to β-actin and are the means of three independent experiments ± SEM. B: Relative abundance of ChREBP protein in immunoblots from whole protein extracts from liver, primary hepatocytes, or isolated islets from rats or humans. β-Actin is used as a loading control, and results are representative of three independent experiments. C: Immunofluorescence using species-specific antibodies against ChREBP in dispersed rat and human islets cells and in a section of a mouse pancreas using antibodies against ChREBP (red), insulin (green), and with a nuclear stain (DAPI). The far right column confirms that red ChREBP-dependent fluorescence is not a result of the nonspecific binding of secondary antibody. Results shown are representative of three independent experiments. (A high-quality digital representation of this figure is available in the online issue.)
ChREBP is required for glucose-stimulated rodent β-cell proliferation.
ChREBP plays an important role in the proliferation of transformed cells, where it redirects intermediary metabolism to provide the molecular building blocks and reducing equivalents required for cell division (20). Because ChREBP is abundant in β-cells (Fig. 1), and glucose is a natural and potent systemic driver of β-cell proliferation (9), we tested whether ChREBP is required for glucose-stimulated proliferation in isolated rat β-cells. A lipid-conjugated siRNA (Accell siRNA) (32) was used to knock down ChREBP expression in isolated dispersed rat islets. Treatment with siRNA targeted to ChREBP provided a 60–70% reduction in ChREBP mRNA, and a modest but reproducible decrease in ChREBP protein in dispersed isolated rat islet cells compared with control siRNA (Fig. 2A and B). Depletion of ChREBP completely blocked glucose-stimulated proliferation in rat β-cells (Fig. 2B and C). To confirm the role of ChREBP in glucose-stimulated β-cell proliferation, islets were isolated from control (C57BL/6) or ChREBP−/− mice on the same genetic background. As shown in Fig. 2E and F, although control β-cells displayed a robust glucose response as measured by BrdU incorporation, β-cells isolated from ChREBP−/− mice did not proliferate in response to glucose.
FIG. 2.

ChREBP is required for glucose-stimulated rat β-cell proliferation. Rat islets were isolated and dispersed by trypsinization and treated for 72 h in 5.5 mmol/L glucose with control or ChREBP-specific Accell siRNA. RNA was isolated and subjected to quantitative RT-PCR with primers specific for ChREBP and β-actin (A), or protein was isolated and subjected to immunoblotting with antibodies directed against either ChREBP or β-actin (B). Results in A are the means ± SE; n = 3; *P < 0.05; results in B are representative of two independent experiments with identical results. C: Dispersed rat islets cells treated with Accell siRNA were cultured on coverslips in either 5.5 or 20 mmol/L glucose for 72 h. BrdU was added for the last 36 h. Cells were fixed and stained with anti-insulin (green) and BrdU (red) antibodies, and nuclei were stained with DAPI (blue). D: Shown are the means ± SE of the percentage of BrdU-positive:insulin-positive cells, counted in a blinded fashion, and normalized to the low glucose control (average = 0.7%), from four independent experiments (*P < 0.05). At least 1,300 insulin-positive cells were counted for each treatment group. E: Dispersed islets from control (C57BL/6J) or ChREBP−/− mice on the same genetic background were cultured for 72 h in either 5.5 or 20 mmol/L glucose, and BrdU was added for the last 48 h. Cells were fixed and stained with anti-insulin (green) and BrdU (red) antibodies, and nuclei were stained with DAPI (blue). F: Results are the means ± SE of the percentage of BrdU-positive:insulin-positive cells, counted in a blinded fashion, and normalized to the low glucose control (average = 0.3%), from four control and four ChREBP−/− animals (*P < 0.05). An average of 1,988 ± 265 insulin-positive cells were counted for each treatment group. WT, wild type; n.s., not significant; Con, control; siCon, siControl. (A high-quality digital representation of this figure is available in the online issue.)
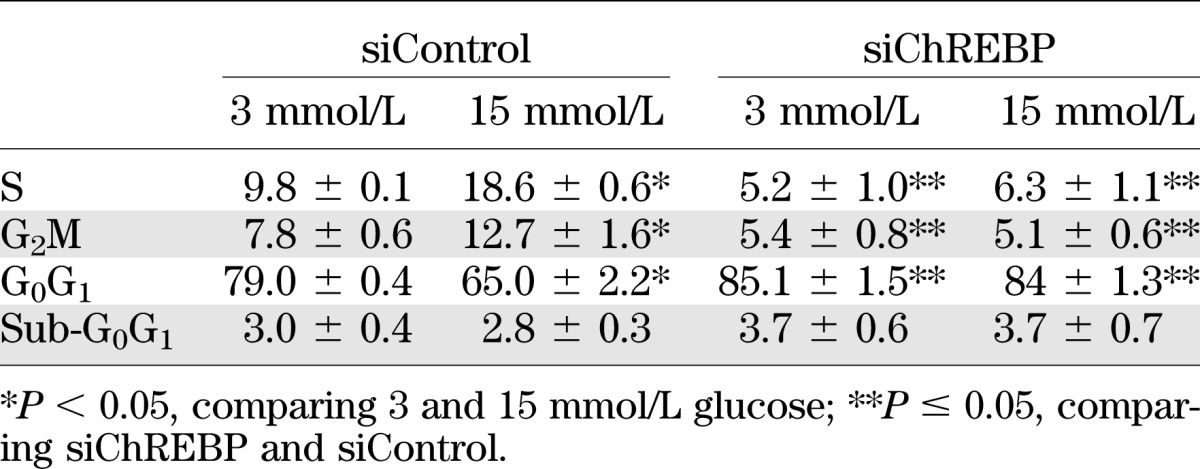
To explore the cellular mechanisms by which ChREBP contributes to glucose-stimulated β-cell proliferation, we tested whether ChREBP is required for glucose-stimulated proliferation in INS-1–derived 832/13 cells (a highly glucose-responsive clone of parental INS-1 cells [21]). As shown in Fig. 3A and B, a pool of siRNA duplexes directed against ChREBP significantly decreased ChREBP mRNA (65%) and protein (70%) levels. This decrease in ChREBP expression was sufficient, as we have described previously (33), to inhibit the glucose response of a standard glucose-responsive gene, Pklr (liver-type pyruvate kinase; Fig. 3C). It is noteworthy that this maneuver completely suppressed glucose-stimulated [3H]thymidine incorporation in 832/13 cells (Fig. 3D). These results were confirmed by fluorescence-activated cell sorter analysis. Depletion of ChREBP by siRNA decreased the percentage of cells in the S and G2/M phases of the cell cycle after glucose stimulation, while increasing the same relative percentage of cells in G0/G1 (Table 1). Furthermore, there was no increase in the sub-G0 fraction of cells, suggesting that depletion of ChREBP did not affect cell death or apoptosis. Thus, depletion of ChREBP was sufficient to block glucose-mediated gene expression and glucose-stimulated β-cell proliferation.
FIG. 3.

Depletion of ChREBP in INS-1–derived 832/13 cells inhibits glucose-stimulated proliferation. INS-1–derived 832/13 cells were transfected with either ChREBP-specific or control siRNA in 11 mmol/L glucose. After 48 h, media were switched to 3 mmol/L glucose for 6 h and then incubated in 3 or 15 mmol/L glucose for an additional 16 h. Total RNA or protein was extracted and used to determine relative levels of ChREBP by quantitative RT-PCR after normalization to β-actin (A) or by immunoblotting relative to β-actin (B), respectively. Pklr mRNA expression was determined in C. In addition, proliferation rates were determined by [3H]thymidine incorporation (D). Results are expressed as the means ± SE (n = 3 to 5; *P < 0.05; n.s., not significant).
TABLE 1.
Fluorescence-activated cell sorter analysis of INS-1–derived 832/13 cells after treatment with pooled siRNA directed against ChREBP and either 3 or 15 mmol/L glucose
ChREBP is required for the glucose-mediated induction of cell cycle regulators.
The abundance of several G1-S regulatory proteins was measured to determine the mechanism by which ChREBP influences the proliferation of INS-1–derived 832/13 cells. As shown in Fig. 4, mRNAs of cyclin D2 (ccnd2), cyclin A2 (ccna2), and cyclin E1 (ccne1) were stimulated by glucose in the presence of ChREBP, but not after depletion of ChREBP. In addition, mRNAs for cyclin D1 (ccnd1) and cdk2 showed nonsignificant trends toward glucose stimulation with dependence on ChREBP, whereas the abundance of mRNAs for Cdks 1, 4, and 6 were not affected by glucose. However, depletion of ChREBP significantly decreased cdk4 mRNA abundance in 15 mmol/L glucose (Fig. 4G). At the protein level, glucose significantly induced cyclins D2, A, and E, as well as cdk4 and cdk6 (Fig. 5). Furthermore, ChREBP was required for the glucose-mediated induction of cdk4, cdk6, and cyclins A and E, whereas depletion of ChREBP diminished both the basal and glucose-stimulated expression of cyclin D2. Thus, we conclude that ChREBP is required for the glucose-mediated regulation of cell cycle regulators, most likely at both the transcriptional and posttranscriptional levels, and ultimately for glucose-stimulated β-cell proliferation.
FIG. 4.

Depletion of ChREBP decreases mRNA expression of cell cycle regulators. INS-1–derived 832/13 cells were transfected for 48 h with either ChREBP-specific or scrambled siRNAs. Cells were pretreated in 3 mmol/L glucose for 6 h and then cultured in 3 or 15 mmol/L glucose for 16 h. Relative mRNA levels of the indicated genes were determined by quantitative RT-PCR and normalized to actin. Shown are the means ± SE (n = 3 to 4; *P < 0.05; n.s., not significant).
FIG. 5.

Depletion of ChREBP leads to decreased protein expression of cell cycle regulators. INS-1–derived 832/13 cells were transfected for 48 h with either ChREBP-specific or scrambled siRNAs. Cells were pretreated in 3 mmol/L glucose for 6 h and then cultured in 3 or 15 mmol/L glucose for 16 h. Immunoblot analysis was used to determine the relative protein expression of the indicated cell cycle regulators. Shown are representative autoradiograms for cyclins (A) and cyclin-dependent kinases (B). C–H: The results of densitometry from immunoblots as in A and B are presented as means from three to four separate experiments (± SE; *P < 0.05; #P < 0.05 compared with 3 mmol/L glucose control; n.s., not significant).
Overexpression of ChREBP amplifies glucose-stimulated β-cell proliferation.
We tested whether overexpression of ChREBP, using an adenoviral vector, affected glucose-stimulated β-cell proliferation. As shown in Fig. 6A, treatment of INS-1–derived 832/13 cells with an adenovirus expressing ChREBP (34) increased the expression of ChREBP as determined by immunoblotting. The increased abundance of ChREBP led to a significant increase of both basal (3 mmol/L glucose) and glucose-stimulated (15 mmol/L glucose) proliferation in these cells as determined by [3H]thymidine incorporation (Fig. 6B). Furthermore, when ChREBP was overexpressed in dispersed rat islet cells (Fig. 6C), there was a dramatic amplification of glucose-stimulated proliferation, as determined by BrdU incorporation into β-cells (Fig. 6D and E). Although there was no effect on basal proliferation (5.5 mmol/L glucose), ChREBP overexpression led to a threefold amplification of glucose-stimulated β-cell proliferation (20 mmol/L glucose). In conclusion, ChREBP is a powerful mediator of glucose-stimulated proliferation in rat pancreatic β-cells.
FIG. 6.

Overexpression of ChREBP amplifies glucose-stimulated β-cell proliferation. Transduction of adenovirus expressing ChREBP in INS-1–derived 832/13 cells (A) or isolated rat islets (C) led to robust expression of ChREBP as determined by immunoblotting. Shown are blots representative of two independent experiments with identical results. B: INS-1–derived 832/13 cells were treated with adenovirus expressing ChREBP, or GFP as a control, for 2 h and cultured for an additional 22 h in 11 mmol/L glucose. Cells were then pretreated for 6 h in 3 mmol/L glucose, and after 16 h of culture in either 3 or 15 mmol/L glucose, the relative amount [3H]thymidine incorporation was determined. Results are expressed as means ± SE (n = 3 to 5; *P < 0.05). C–E: Dispersed rat islet cells were transduced with adenovirus expressing ChREBP or GFP for 2 h. Cells were cultured on coverslips in medium containing 5.5 mmol/L glucose for 24 h, at which time the high glucose group was adjusted to 20 mmol/L glucose and the cells were cultured for an additional 60 h. D: BrdU was added for the last 36 h, and cells were fixed and stained for insulin (green) or BrdU (red) and DAPI (blue). E: Cells were counted in a blinded fashion, and the results are expressed as a percentage of insulin:BrdU-positive cells (± SE; n = 4; *P < 0.05). At least 1,500 cells were counted for each treatment group. siCon, siControl. (A high-quality digital representation of this figure is available in the online issue.)
Overexpression of ChREBP alters the expression of cell cycle regulator genes.
In Fig. 7, we determined the effect of ChREBP overexpression on the abundance of the cell cycle control genes that were examined in Figs. 4 and 5. The culture conditions were different when INS-1–derived 832/13 cells were treated with adenovirus compared with when the cells were treated with the lipid-conjugated Accell siRNA (which uses a proprietary medium; see research design and methods). More specifically, in Figs. 4 and 5, glucose led to higher and significant changes in cyclins D2, A, and E under control conditions. By contrast, in Fig. 7, after control adenovirus treatment, cyclin D2 was significantly increased by 15 mmol/L glucose (Fig. 7A), but there were no significant changes noted for cyclins A and E (Fig. 7C and E) or the cdk proteins (Fig. 7B, D, and F). However, after treatment with the adenovirus expressing ChREBP, there was a significant increase in the glucose-stimulated expression of cyclins A and E (Fig. 7C and E), which paralleled the pattern of increased proliferation seen after ChREBP overexpression and glucose treatment as shown in Fig. 6B. Cyclin D2 was significantly increased by 15 mmol/L glucose after ChREBP overexpression but was not different than the control level in high glucose (Fig. 7A). Because overexpression of either cyclin A or E independently increases β-cell proliferation (3,35–37), it is likely that the ChREBP-mediated amplification of β-cell proliferation is the result of, at least in part, the glucose-stimulated increase of cyclins A and E. To confirm these findings, dispersed rat islet cells were transduced with adenoviruses expressing either GFP or ChREBP and cultured in 20 mmol/L glucose for 72 h. We found that in cells overexpressing ChREBP, numerous cell cycle accelerators (including cyclin A, cdk1, cdk2, cdk6, and c-Myc) were significantly increased and a number of cell cycle inhibitors (including p57, p15, p18, and p19) were significantly decreased (Supplementary Table 2).
FIG. 7.

Overexpression of ChREBP increases expression of cyclins A and E. INS-1–derived 832/13 were transduced with adenoviruses expressing either GFP or ChREBP as in Fig. 6 and were cultured for 18 h in 3 or 15 mmol/L glucose and processed for immunoblotting. A–F: Representative immunoblots of the indicated proteins are shown, with results of densitometry, normalized to β-actin shown above (means ± SE; n = 3 to 4; *P < 0.05).
ChREBP regulates glucose-stimulated β-cell proliferation in human β-cells.
Because human and rodent β-cells are often dissimilar and to test for the potential therapeutic application of ChREBP, it was important to test whether the effects of ChREBP depletion and overexpression in rat β-cells could be duplicated in human β-cells. A lipid-conjugated siRNA directed against human ChREBP decreased ChREBP mRNA expression by ∼60% (Fig. 8A). We found that when this siRNA was introduced into dispersed human islet cells, glucose-stimulated human β-cell proliferation was prevented (Fig. 8B and C), in agreement with our observations in rat β-cells (Fig. 2). Conversely, when ChREBP was overexpressed in dispersed human islet cells with a recombinant adenovirus (Fig. 8D), the glucose-stimulated increase in proliferation, as determined by BrdU incorporation, was significantly increased (Fig. 8E and F), paralleling our results with rat β-cells (Fig. 6). Thus, ChREBP is required for glucose-stimulated human β-cell proliferation and acts to amplify the glucose effect on β-cell proliferation when overexpressed in human β-cells.
FIG. 8.

ChREBP regulates glucose-stimulated proliferation in human β-cells. Isolated human islets were dispersed by trypsinization and treated with control or ChREBP-specific Accell siRNA in 5.5 mmol/L glucose, and after 72 h, the relative abundance of ChREBP mRNA was determined by quantitative RT-PCR (A) or protein was isolated and subjected to immunoblotting with antibodies directed against either ChREBP or β-actin (B). Results in A are the means ± SE (n = 3; *P < 0.05); results in B are representative of two independent experiments with identical results. C: Dispersed human islets cells treated with Accell siRNA were cultured on coverslips in either 5.5 or 20 mmol/L glucose for 72 h. BrdU was added for the last 48 h. Cells were fixed and stained with anti-insulin (green) and BrdU (red) antibodies, and nuclei were stained with DAPI (blue) (see Supplementary Fig. 1). At least 1,500 cells were counted for each treatment group. Shown are the means ± SE of the percentage of BrdU-positive:insulin-positive cells, counted in a blinded fashion, normalized to the low glucose control (average = 0.3%), from four independent experiments (*P < 0.05). D and E: Dispersed human islet cells were transduced with adenovirus expressing ChREBP or GFP and were treated exactly as described for isolated rat islet cells in Fig. 6, except BrdU was added 48 h before the end of the experiment. D: A representative immunoblot is shown with antibodies specific for either ChREBP or β-actin as indicated. Cells were fixed and stained with antibodies against insulin (green) and BrdU (red), with nuclei stained with DAPI (blue) (see Supplementary Fig. 1). E: Cells were counted in a blinded fashion, and the results are expressed as a percentage of insulin:BrdU-positive cells, normalized to the low glucose control, which averaged 0.3% (means ± SE; n = 4; *P < 0.05). At least 1,500 β-cells were counted for each condition in each of four independent experiments. Con, control; n.s., not significant.
DISCUSSION
People with type 1 or type 2 diabetes would benefit from therapies aimed at expanding β-cell mass (1). Thus, understanding how β-cells proliferate and the precise cellular and molecular mechanisms by which natural mitogens, such as glucose, stimulate the expansion of functional β-cells is a central focus of the diabetes research community. Glucose is a natural systemic mitogenic signal that drives rodent and human β-cell proliferation in vivo by increasing flux through glucokinase and glycolysis (9,15,18). An important gap in knowledge is an understanding of how glucose metabolism interacts with the cell cycle machinery to boost β-cell proliferation. In the current study, we found a direct correlation between the abundance of the glucose-sensing transcription factor, ChREBP, and the ability of rodent and human β-cells to proliferate in response to glucose. Depletion of ChREBP attenuated glucose-stimulated β-cell proliferation, and overexpression of ChREBP amplified the glucose effect on proliferation in rat and human β-cells. In addition, we found that the cell cycle regulators, cyclin D2, cyclin A2, and cyclin E1, all exhibited increased expression at both the mRNA and protein levels in a glucose- and ChREBP-dependent manner. Furthermore, when ChREBP was overexpressed, expression of cyclins A2 and E1 increased in high glucose, correlating with increased [3H]thymidine incorporation.
These observations provide a possible mechanistic explanation for the contribution of ChREBP to glucose-stimulated β-cell proliferation. Cyclins D2, E1, and A2 have been shown previously to drive β-cell proliferation (3,4,36). Cyclin D2 is crucial for postnatal β-cell proliferation (38), and it is upregulated by hyperglycemia in islets in vivo (15). Cyclin E is induced in response to overexpression of Nkx6.1 and is sufficient to drive proliferation of rat β-cells when overexpressed (36). Expression of cyclin A is stimulated by maneuvers that increase cAMP, leading to β-cell proliferation (37). Overexpression of cyclin A promotes β-cell proliferation independently as well. Thus, overexpression of any of at least three of the G1-S regulatory cyclins is sufficient to drive β-cell proliferation. In the current study, we found that a mitogenic glucose treatment increases the expression of cyclins D2, A, and E in a ChREBP-dependent manner, linking ChREBP to cell cycle regulation. Because ChREBP is a glucose-activated transcription factor, which is translocated to the nucleus in response to glucose (19), and is further dependent on glucose for transactivation after binding to DNA (39,40), it follows that ChREBP may directly activate the cyclin genes. We have shown previously that Myc is required for ChREBP recruitment and activity. In this study, overexpression of ChREBP led to a significant increase in c-Myc mRNA in high glucose, suggesting a potential feedback loop between these two transcription factors. Because Myc is a direct target of cyclins, and we have failed to observe ChREBP binding to regulatory regions of the cyclin genes (data not shown), it is possible that glucose stimulates β-cell proliferation, at least in part, via a pathway that includes glucose metabolism, activation of ChREBP and Myc, activation of cyclins, and progression through the cell cycle. Experiments are in progress to test this hypothesis.
The role that ChREBP plays in β-cells has been assigned to a variety of functions, including promotion of lipogenesis, promotion of glucolipotoxicity, and regulation of the response to hypoxia (31,41,42). The Kaestner laboratory recently demonstrated that the ChREBP gene is a direct target of Foxa1 and Foxa2, which are important for developing β-cells and for maintaining β-cell phenotype (43). With corroboration of the prevalence of ChREBP during islet development, ChREBP was found to be among the highest expressed transcription factors in Ngn3-positive islet progenitor cells (44). Global ChREBP knockout animals display a diminished capacity for hepatic lipogenesis, as expected for a transcription factor that activates hepatic glycolytic and lipogenic genes in response to carbohydrates (27). In addition, these animals display inappropriately low insulin concentrations in the face of modest fasting hyperglycemia. This suggests a defect in the β-cell, manifested either as a decrease in β-cell mass or as a defect in the ability of the β-cell to adequately secrete insulin in the face of hyperglycemia, although more extensive phenotypic analysis of the β-cell in ChREBP−/− mice has yet to be reported (27).
A recent report by Salpeter et al. (45) demonstrated that the glucose activation of cyclin D2 in β-cells requires glucose-stimulated Ca2+ influx. In our study, glucose stimulation of cyclin D2 requires ChREBP. These two observations are not necessarily in conflict for at least two reasons. First, there may be multiple signaling pathways activated by glucose metabolism that ultimately result in increased cyclin expression and activity. In addition to calcium signaling, the insulin/IGF1/IRS2 signaling pathway, the cAMP signaling pathway, and the repression of Hif1β have all been implicated in glucose stimulation of β-cell proliferation (41,46–48). We note that glucose increases Myc expression (25), and cyclin genes are direct targets of c-Myc (49) as well as calcineurin/NFAT (50). Thus, it is possible that all of these pathways play an important role in the activation of cyclins and the progression of cell cycle in β-cells. Second, these pathways may operate at different time scales (Ca2+ influx happens within seconds to minutes; activation of ChREBP-dependent transcription takes minutes to hours) or have different cellular targets of regulation (e.g., transcription vs. protein turnover). A future challenge will be to determine the relationship between ChREBP-dependent signaling and other signaling pathways. Another important challenge will be to determine whether ChREBP is required for any other mitogenic signals, such as growth factors, recovery from pancreatectomy, pregnancy, or high-fat diet. Clearly, much more must be learned about ChREBP in the β-cell.
In conclusion, the glucose-sensing transcription factor ChREBP is required for glucose-stimulated β-cell proliferation. These results provide a platform for numerous experimental approaches to test the feasibility of using ChREBP as a target to increase β-cell proliferation, mimicking and accentuating a natural β-cell mitogen.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the following grants: National Institutes of Health (NIH) R56DK065149; American Diabetes Association ADA 7-11-BS-128; Juvenile Diabetes Research Foundation (JDRF) 17-2011-598 (D.K.S.); NIH DK077096 (A.G.-O.) and NIH DK078060 (R.C.V.); and JDRF 34-2008-630, JDRF 1-2008-39, NIH DKR0155023, and NIH DK U-01 89538 (A.F.S.).
No potential conflicts of interest relevant to this article were reported.
M.R.M. and D.K.S. designed the research and wrote the manuscript. M.R.M., P.Z., M.K.B., C.C., R.E.S., K.T., and R.G. performed the experiments. All authors contributed to the discussion. M.R.M., P.Z., L.C.A., R.M.O., A.F.S., R.C.V., A.G.-O., and D.K.S. reviewed and edited the manuscript. D.K.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Dr. Howard Towle (University of Minnesota) for the ChREBP-expressing adenovirus and Chris Newgard for INS-1–derived 832/13 cells (Stedman Nutrition and Metabolism Center, Duke University).
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-0802/-/DC1.
REFERENCES
- 1.Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 2008;9:193–205 [DOI] [PubMed] [Google Scholar]
- 2.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev 2006;27:356–370 [DOI] [PubMed] [Google Scholar]
- 3.Cozar-Castellano I, Harb G, Selk K, et al. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes 2008;57:3056–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiaschi-Taesch NM, Salim F, Kleinberger J, et al. Induction of human β-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 2010;59:1926–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiaschi-Taesch N, Bigatel TA, Sicari B, et al. Survey of the human pancreatic β-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human β-cell replication and function in vivo. Diabetes 2009;58:882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buteau J, Spatz ML, Accili D. Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic β-cell mass. Diabetes 2006;55:1190–1196 [DOI] [PubMed] [Google Scholar]
- 7.Vasavada RC, Wang L, Fujinaka Y, et al. Protein kinase C-zeta activation markedly enhances β-cell proliferation: an essential role in growth factor mediated β-cell mitogenesis. Diabetes 2007;56:2732–2743 [DOI] [PubMed] [Google Scholar]
- 8.Cozar-Castellano I, Weinstock M, Haught M, Velázquez-Garcia S, Sipula D, Stewart AF. Evaluation of β-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes 2006;55:70–77 [PubMed] [Google Scholar]
- 9.Porat S, Weinberg-Corem N, Tornovsky-Babaey S, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab 2011;13:440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Assmann A, Ueki K, Winnay JN, Kadowaki T, Kulkarni RN. Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol 2009;29:3219–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Del Zotto H, Gómez Dumm CL, Drago S, Fortino A, Luna GC, Gagliardino JJ. Mechanisms involved in the beta-cell mass increase induced by chronic sucrose feeding to normal rats. J Endocrinol 2002;174:225–231 [DOI] [PubMed] [Google Scholar]
- 12.Koiter TR, Wijkstra S, van Der Schaaf-Verdonk CJ, Moes H, Schuiling GA. Pancreatic beta-cell function and islet-cell proliferation: effect of hyperinsulinaemia. Physiol Behav 1995;57:717–721 [DOI] [PubMed] [Google Scholar]
- 13.Liu YQ, Han J, Epstein PN, Long YS. Enhanced rat beta-cell proliferation in 60% pancreatectomized islets by increased glucose metabolic flux through pyruvate carboxylase pathway. Am J Physiol Endocrinol Metab 2005;288:E471–E478 [DOI] [PubMed] [Google Scholar]
- 14.Liu YQ, Montanya E, Leahy JL. Increased islet DNA synthesis and glucose-derived lipid and amino acid production in association with beta-cell hyperproliferation in normoglycaemic 60 % pancreatectomy rats. Diabetologia 2001;44:1026–1033 [DOI] [PubMed] [Google Scholar]
- 15.Alonso LC, Yokoe T, Zhang P, et al. Glucose infusion in mice: a new model to induce β-cell replication. Diabetes 2007;56:1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes 1989;38:49–53 [DOI] [PubMed] [Google Scholar]
- 17.Jetton TL, Everill B, Lausier J, et al. Enhanced beta-cell mass without increased proliferation following chronic mild glucose infusion. Am J Physiol Endocrinol Metab 2008;294:E679–E687 [DOI] [PubMed] [Google Scholar]
- 18.Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 2011;54:572–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr 2007;27:179–192 [DOI] [PubMed] [Google Scholar]
- 20.Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Natl Acad Sci USA 2009;106:21660–21665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000;49:424–430 [DOI] [PubMed] [Google Scholar]
- 22.Pedersen KB, Zhang P, Doumen C, et al. The promoter for the gene encoding the catalytic subunit of rat glucose-6-phosphatase contains two distinct glucose-responsive regions. Am J Physiol Endocrinol Metab 2007;292:E788–E801 [DOI] [PubMed] [Google Scholar]
- 23.Ma L, Robinson LN, Towle HC. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem 2006;281:28721–28730 [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Ocana A, Takane KK, Reddy VT, Lopez-Talavera JC, Vasavada RC, Stewart AF. Adenovirus-mediated hepatocyte growth factor expression in mouse islets improves pancreatic islet transplant performance and reduces beta cell death. J Biol Chem 2003;278:343–351 [DOI] [PubMed] [Google Scholar]
- 25.Zhang P, Metukuri MR, Bindom SM, Prochownik EV, O’Doherty RM, Scott DK. c-Myc is required for the CHREBP-dependent activation of glucose-responsive genes. Mol Endocrinol 2010;24:1274–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ricordi CRC. Methods in pancreatic islet separation. In Methods in Cell Transplantation C R, Ed. Austin, TX, R.G. Landes, 2000, p. 433-443 [Google Scholar]
- 27.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA 2004;101:7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.García-Ocaña A, Vasavada RC, Cebrian A, et al. Transgenic overexpression of hepatocyte growth factor in the β-cell markedly improves islet function and islet transplant outcomes in mice. Diabetes 2001;50:2752–2762 [DOI] [PubMed] [Google Scholar]
- 29.Strom SC, Pisarov LA, Dorko K, Thompson MT, Schuetz JD, Schuetz EG. Use of human hepatocytes to study P450 gene induction. Methods Enzymol 1996;272:388–401 [DOI] [PubMed] [Google Scholar]
- 30.Harb G, Vasavada RC, Cobrinik D, Stewart AF. The retinoblastoma protein and its homolog p130 regulate the G1/S transition in pancreatic β-cells. Diabetes 2009;58:1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.da Silva Xavier G, Rutter GA, Diraison F, Andreolas C, Leclerc I. ChREBP binding to fatty acid synthase and L-type pyruvate kinase genes is stimulated by glucose in pancreatic beta-cells. J Lipid Res 2006;47:2482–2491 [DOI] [PubMed] [Google Scholar]
- 32.Deering TG, Ogihara T, Trace AP, Maier B, Mirmira RG. Methyltransferase Set7/9 maintains transcription and euchromatin structure at islet-enriched genes. Diabetes 2009;58:185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burke SJ, Collier JJ, Scott DK. cAMP opposes the glucose-mediated induction of the L-PK gene by preventing the recruitment of a complex containing ChREBP, HNF4alpha, and CBP. FASEB J 2009;23:2855–2865 [DOI] [PubMed] [Google Scholar]
- 34.Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP.Mlx in regulating hepatic glucose-responsive genes. J Biol Chem 2005;280:12019–12027 [DOI] [PubMed] [Google Scholar]
- 35.Guthalu Kondegowda N, Joshi-Gokhale S, Harb G, et al. Parathyroid hormone-related protein enhances human β-cell proliferation and function with associated induction of cyclin-dependent kinase 2 and cyclin E expression. Diabetes 2010;59:3131–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schisler JC, Fueger PT, Babu DA, et al. Stimulation of human and rat islet β-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Mol Cell Biol 2008;28:3465–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song WJ, Schreiber WE, Zhong E, et al. Exendin-4 stimulation of cyclin A2 in β-cell proliferation. Diabetes 2008;57:2371–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kushner JA, Ciemerych MA, Sicinska E, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol 2005;25:3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collier JJ, Zhang P, Pedersen KB, Burke SJ, Haycock JW, Scott DK. c-Myc and ChREBP regulate glucose-mediated expression of the L-type pyruvate kinase gene in INS-1-derived 832/13 cells. Am J Physiol Endocrinol Metab 2007;293:E48–E56 [DOI] [PubMed] [Google Scholar]
- 40.Li MV, Chang B, Imamura M, Poungvarin N, Chan L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 2006;55:1179–1189 [DOI] [PubMed] [Google Scholar]
- 41.Noordeen NA, Khera TK, Sun G, et al. Carbohydrate-responsive element-binding protein (ChREBP) is a negative regulator of ARNT/HIF-1β gene expression in pancreatic islet β-cells. Diabetes 2010;59:153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Wollheim CB. ChREBP rather than USF2 regulates glucose stimulation of endogenous L-pyruvate kinase expression in insulin-secreting cells. J Biol Chem 2002;277:32746–32752 [DOI] [PubMed] [Google Scholar]
- 43.Gao N, Le Lay J, Qin W, et al. Foxa1 and Foxa2 maintain the metabolic and secretory features of the mature beta-cell. Mol Endocrinol 2010;24:1594–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soyer J, Flasse L, Raffelsberger W, et al. Rfx6 is an Ngn3-dependent winged helix transcription factor required for pancreatic islet cell development. Development 2010;137:203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salpeter SJ, Klochendler A, Weinberg-Corem N, et al. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic β-cells through glycolysis and calcium channels. Endocrinology 2011;152:2589–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Folli F, Okada T, Perego C, et al. Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS ONE 2011;6:e28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hussain MA, Porras DL, Rowe MH, et al. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Mol Cell Biol 2006;26:7747–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demozay D, Tsunekawa S, Briaud I, Shah R, Rhodes CJ. Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet β-cells. Diabetes 2011;60:2892–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jansen-Dürr P, Meichle A, Steiner P, et al. Differential modulation of cyclin gene expression by MYC. Proc Natl Acad Sci USA 1993;90:3685–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006;443:345–349 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.