

1. Protein folding problem in ageing
Proteins are among the predominant products of gene expression and contribute significantly to the shape and functionality of the cell. The status of the expressed proteome has an important role in the health of individual cells and the lifespan of the organism. In addition to cell-type- and tissue-specific regulation of protein expression, maintenance of the proteome depends on efficient protein folding homeostasis, or proteostasis, that monitors and ensures folding, assembly and targeting of newly-synthesized proteins, repair of damaged proteins, and clearance. Proteotoxic conditions arise by external stresses, or as a byproduct of normal cellular metabolic and signaling events during development and aging (Figure 1). This is in addition to the intrinsic variation in the proteome due to polymorphisms, which is further exacerbated by random errors that can occur at every step of protein biogenesis. Together, these factors contribute to a flux of metastable proteins that are at risk for misfolding and aggregation (1-4). In healthy young cells, these processes are balanced by the concerted action of molecular chaperones, detoxifying enzymes, degradation machinery, and adaptive stress responses (Figure 2) (5-7). In aging and disease, damaged proteins accumulate, leading to both loss-of-function and gain-of-function toxicity as these homeostatic mechanisms fail and contribute to pathology.
Figure 1.

Extrinsic and intrinsic factors detrimental to protein folding.
Figure 2. Proteostasis pathways.

Multiple interconnected pathways regulate expression of genes that contribute to maintenance of protein folding homeostasis during growth, development, and under various stress conditions. The complex genetic interactions among these pathways necessitate precise control over their activities. Many of these pathways also possess regulatory feedback mechanisms, which are not well characterized/understood.
Sources of protein damage – oxidative modifications
The ‘free radical theory’ (8) and the ‘oxidative stress theory’ (9, 10) of aging postulate that aging and many age-related diseases may be attributed to the generation of oxygen free radicals and reactive oxygen and nitrogen species (ROS and RNS), in excess of cellular antioxidants, resulting in oxidative damage to DNA, lipids and proteins. Early evidence connected many of the aging-associated diseases, including atherosclerosis, arthritis, muscular dystrophy, cataracts, pulmonary dysfunction, neurological disorders and cancer with oxidative damage (11). Levels of oxidized proteins increase exponentially in regions of aging brains, concomitant with a decrease in the activities of the enzymes glutamine synthetase and alcohol dehydrogenase, which are susceptible to oxidative damage (12). The decrease in glutamine synthetase activity was found to distinguish the brains of Alzheimer disease (AD) patients from age-matched individuals, leading to the suggestion that AD may represent a specific brain vulnerability to age-related oxidation. Furthermore, a functional connection between the accumulation of oxidized proteins and the phenotypic manifestations of cellular disfunction in aging has been suggested (13).
In addition to oxygen radicals, glucose, galactose, fructose, and many glycolytic intermediates participate in non-enzymatic protein glycosylation (glycation) and glycoxidation, contributing to age-related protein modifications. Some glycolytic intermediates can generate methylglyoxal (MG), which is a highly reactive glycating agent (14, 15) leading to the formation of advanced glycation end products (AGEs), that has been implicated in age-related diseases, including Alzheimer’s disease and complications associated with diabetes (15-17). A decrease in circulating glucose and reduction of MG, resulting from dietary restriction or fasting, may explain some of the health-improving effects of these treatments (18). Recently, an inactivating mutation in a glycolytic enzyme triosephosphate isomerase (Tpi) was shown to cause progressive motor impairment, severely reduced lifespan, and neurodegeneration in Drosophila, presumably by causing an accumulation of methylglyoxal and subsequent increase in generation of AGEs (19).
Loss of protein function due to oxidative modification can be due either to direct inhibition of activity of a protein (for example, covalent modification of a side chain in proximity of an active site), or to conformational changes or alteration of protein stability. Most modifications of the first type are irreversible, with the exception of cysteine and methionine modifications, which can be reversed by thiol transferases, thioredoxin, glutaredoxin, and methionine sulfoxide reductases (MsrA and MsrB). The detoxification of methylglyoxal and glyoxal is achieved by the glutathione-dependent glyoxalase system. These and other enzymes are part of the cellular proteostasis machinery that is activated in response to various protein damaging conditions. For example, MrsA and B allow recovery of protein function in specific cases (20, 21), and their overexpression confers protection against oxidative damage in yeast, Drosophila and human fibroblasts (21).
In addition to functional inactivation of proteins, free radicals can have a local effect on charged side chains, or cause cleavage of the polypeptide backbone, leading to a spectrum of consequences, from complete unfolding, to local conformational changes (22-24), to targeting for disposal. If not degraded, modified proteins often become cross-linked, leading to the accumulation of non-specific aggregated material, such as lipofuscin, ceroids and AGE-containing pigments. Nuclear and cytosolic oxidized proteins have been shown to be degraded by an ATP- and ubiquitin-independent 20S proteasome (25, 26), as well as by classical ubiquitin-26S proteasome system (27), whereas mitochondrial oxidized proteins are degraded by the ATP-dependent Lon protease (28). Interestingly, the 20S proteasome appears to be resistant to oxidative damage, while 26S proteasome is not (29). Chaperone-mediated autophagy represents an alternative protein degradation pathway that is induced under oxidative stress conditions (30). Under chronic oxidative stress conditions, there seems to be a damage cascade, whereby the initial appearance of modified proteins, which signifies the insufficiency of the repair and degradation arm of proteostasis machinery, results in further inhibition of both proteasomal and lysosomal functions (reviewed in (21, 24)), and thus in further accumulation of protein damage.
Sources of damaged proteins – misfolding
One source of damaged – often misfolded, mislocalized, or aggregated – proteins are errors inherent in protein biosynthesis and turnover. Variations in amino acid sequence, which may result from genetic mutations and polymorphisms, genomic instability, mistranslation, or incorporation of amino-acid analogues (such as certain antibiotics or plant metabolites, Figure 1), have the potential to affect folding pathways and the stability of the native state (2, 31-35). For example, coding polymorphisms are not rare and are estimated to occur at an average of two per coding sequence (36), while misincorporation during translation might cause up to 18% of expressed proteins to contain an amino acid substitution (37). Our ability to understand, let alone to predict, the consequences of missense mutations on the folding, stability or functionality of a given protein in vivo remains very limited, with data limited to a few model proteins studied in vitro, or to computational models. This is further complicated by the fact that the phenotypic expression of mildly destabilizing protein polymorphisms appears to depend on the robustness of the protein folding environment (1, 4, 38) and the capacity of the proteostasis network. Recent work on the evolution of protein sequences suggests that selection against the toxicity of misfolding due to mistranslation may represent an important evolutionary pressure, when considering highly expressed proteins (37). From a physiological perspective, this may be interpreted to indicate that the flux of misfolded or destabilized proteins in a cell bears a significant fitness cost, not only through the loss-of-function of the misfolded proteins, but also because of the toxic effects of induced aggregation and the consequences of inappropriate intermolecular interactions (2, 7, 39, 40), as well as abnormal engagement of molecular chaperones and degradative machinery. Because of these gain-of-function properties of destabilized proteins, their chronic presence may exceed the capacity of the proteostasis machinery to cope, leading to the disruption of folding homeostasis and cellular dysfunction. Indeed, introduction of even one protein with a strong tendency to misfold into a cell has been shown to interfere with the folding and function of proteins harboring mildly destabilizing amino acid substitutions (Figure 4) (3, 38).
Figure 4. Progressive disruption of cellular folding capacity by misfolded proteins.
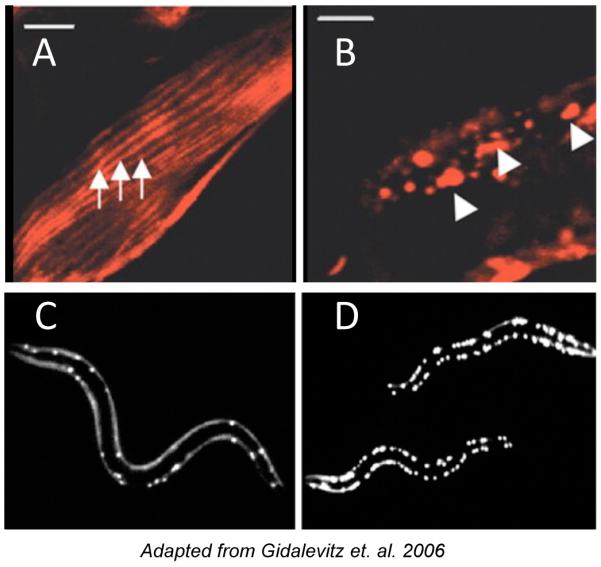
(A and B) Confocal images showing cellular localization of ts mutant paramyosin in the absence (A) or presence (B) of Q40-YFP. Arrows, normal muscle sarcomeres, arrowheads, abnormal paramyosin(ts) assemblies in the presence of Q40, both under permissive conditions.
(C and D) Images of Q40-YFP fluorescence in L2 larval stage animals expressing wild type (C) or ts mutant paramyosin (D) under permissive conditions.
Organismal consequences of misfolding due to biosynthetic errors can be gleaned from a sti mouse model, in which low levels of mischarged transfer RNAs due to an editing defect in alanyl-tRNA synthetase lead to neurodegeneration, with cerebellar Purkinje cell loss and ataxia (41). Neurodegeneration is also a consequence of a mutation of the tyrosyl-tRNA synthetase in a subtype of Charcot-Marie-Tooth neuropathy (42). The mouse sti mutation leads to the production of heterogeneous misfolded proteins, accompanied by increased expression of chaperones in the cytoplasm and the ER (41). This increase in the components of the proteostasis machinery indicates that adaptive transcriptional responses – the heat shock response and the unfolded protein response – are activated in the face of protein misfolding, as cells attempt to maintain folding homeostasis (7). However, the cellular dysfunction and degeneration in this mouse model together indicate that proteostasis is overwhelmed by the chronic elevation of misfolded proteins. A provocative question, then, is whether all misfolded and damaged proteins are recognized, refolded or cleared with equal efficiency, or whether certain proteins, such as those implicated in neurodegenerative diseases and other misfolding diseases of aging, are particularly challenging for the quality control machineries.
The lack of robustness of cellular folding homeostasis to chronic misfolding has significant implications for aging and age-related diseases. First, the aging of an organism is accompanied by an increased accumulation of damaged proteins, with contributions from both external stresses and physiological processes (Figure 1). Second, the fidelity of biosynthetic processes decreases, exacerbated by a decline in the functionality of repair enzymes (reviewed in (43)) and the dampening of the heat shock response lead to the compromise of protein quality control (reviewed in (44)). As accumulated misfolded proteins deplete essential components of the proteostasis machinery (45-47), and the quality control and repair mechanisms fail to respond and keep damaged proteins in check, the maintenance of the proteome becomes further compromised. With as much as 70% of rare missense alleles predicted to be mildly deleterious in humans (48), and approximately half of the genetic changes known to cause inherited disease (in OMIM and HGMD databases) being due to nonsynonymous changes (33), such compromise should manifest in gradual increase in cellular dysfunction and onset of disease. In this scenario, the strength and composition of the proteostatic buffer, the specific complement of mutations and polymorphisms in expressed genes, and the accumulation of molecular damage, all serve to set a dynamic threshold for the onset of dysfunction, both in a cell-specific, and an individual organism-specific manner.
Proteostasis regulation in aging and late onset diseases
Adaptive transcriptional stress responses such as the heat shock response and the unfolded protein response (UPR) serve to protect cells from extreme flux of misfolded and damaged proteins when cells or organism are exposed to an acute proteotoxic condition (reviewed in (49, 50)). Parallel to the sharply increased transcription/translation of molecular chaperones and components of degradative quality control machineries, mediated by the transcription factor HSF1, a generalized inhibition of protein synthesis allows the switch to refolding or degradation of existing proteins that become unstable during such proteotoxic shock (51). However, the fact that HSF1 (52, 53), the components of the UPR signaling (54-56), as well as some of the stress-inducible molecular chaperones are essential for life or development suggests that these adaptive stress responses are also necessary for the maintenance of proteostasis under normal, non-stress conditions. On the other hand, forced overexpression of molecular chaperones is deleterious to growth of normal cells (57, 58), while the transformed phenotype of cancer cells depends on HSF1 and on an increased expression of chaperones (59-61). It appears then that the precise regulation of proteostasis networks is essential for the health of the organism, while accumulating evidence points to its disregulation during aging as one of the main causes of cellular disfunction and disease onset.
The association with aging is one of the most distinctive characteristics of protein conformation diseases. This connection is particularly striking in neurodegeneration where, for each specific disease, age is the strongest predictor of disease onset even for the familial variants. On the other hand, age appears to have a modifying, rather than causative, influence on disease onset, as each disease has its characteristic age of onset, with Alzheimer’s disease and Parkinson’s disease being late onset, ALS occurring most frequently in early to mid-life, and Huntington’s disease exhibiting a strong positive correlation between age of onset and polyQ length polymorphism (62). Aging has been shown to be a potent enhancer of aggregation toxicity in C. elegans, in which length-dependent polyQ aggregation and toxicity phenotypes are enhanced during aging, but suppressed in animals rendered long-lived by mutations in the lifespan regulating insulin-like signaling (ILS) pathway (Figure 2) (63).
The modulatory effects of aging are most likely due to the functional and regulatory connections between aging and protein homeostasis. Activation of the kinase activity of IP3K (AGE-1), by downregulation of the insulin-like growth factor receptor DAF-2, in wild type C. elegans, initiates a signaling cascade that represses the fork head transcription factor DAF-16 (64, 65). De-repression of DAF-16 in age-1 or daf-2 mutant animals extends lifespan, while inactivating daf-16 mutations suppress longevity. The daf-2/age-1 effects on polyQ aggregation toxicity also require DAF-16 activation, revealing that the dual effects of daf-2/age-1 on longevity and polyglutamine toxicity share a common genetic pathway (ILS pathway, Figure 2) (66, 67). The modulation of protein misfolding and aggregation by the ILS pathway appears to be a general feature, and has also been observed in C. elegans expressing Aβ as a model for Alzheimer’s disease (68).
How do genetic pathways that regulate lifespan suppress proteotoxicity? The molecular interactions between these pathways are mediated, in part, by factors that detect and respond to misfolded proteins-- molecular chaperones, HSF1, DAF-16, and other transcription factors (Figure 2), while molecular chaperones have been shown to accumulate in the long-lived mutants of C. elegans. Downregulation of hsf-1 suppresses both the ILS-mediated lifespan extension and the protection against proteotoxicity. Moreover, hsf-1 downregulation leads to a decrease in normal lifespan and an accelerated aging phenotype in C. elegans, while overexpression of HSF1 extends lifespan (66, 67). The functional relationship between ILS and protein folding homeostasis can be demonstrated by the induction of both thermotolerance and life span extension not only by mutations in the ILS pathway, but also by a sublethal heat stress (69). Furthermore, cells from naturally long-lived (70) or lifespan mutant (71) rodents appear to be resistant to multiple proteotoxic stresses. The mechanistic relationship can be shown by the induction of stress resistance by ablation of cells making insulin-like ligands in wild type Drosophila (72), thus excluding indirect effects and adaptation to mutations. In C. elegans, inactivation of daf-16, hsf-1, hsp-1 (the major cytosolic Hsp70 chaperone), and small heat shock proteins accelerates polyQ protein aggregation (Figure 3C), supporting the proposition that the ILS could coordinately influence aging and proteostasis through the action of DAF-16 and HSF1 (66, 67). The regulation of ILS pathway and of HSF1 and other transcription factors (such as SKN-1 (73), the C. elegans Nrf2 homologue) in the context of adaptive stress responses must serve to integrate organismal growth and development with multiple networks that regulate different aspects of protein homeostasis (7, 66-68). We are only now beginning to understand how such integration could be achieved, with recent evidence showing that the ability of individual cells in an organism to respond to proteotoxic conditions is controlled by the activity of a subset of neurons (74)
Figure 3.

A) SOD1 mutant proteins aggregate in the body wall muscle cells of C. elegans.
Fluorescent micrographs of SOD1transgenic embryos (i–iv) and adult animals (ix through xii) showing the distribution of the SOD-YFP fluorescence (green) and Rhodamine-phalloidin stained myofilaments (red). WT SOD1 protein exhibits diffuse, if patchy, fluorescence, while all mutant strains contain discrete fluorescent foci as well as some diffuse fluorescence. Panels v through viii show G85R animals in all larval stages (L1 through L4).
B) Length-dependent aggregation of polyQ-YFP fusion proteins in C. elegans.
Epifluoresence micrographs of 3- to 4-day-old C. elegans expressing different lengths of polyQ-YFP (Q0, Q19, Q29, Q33, Q35, Q40, Q44, Q64, Q82).
C) RNAi for hsp-1 and hsf-1 induces aggregate formation of polyglutamine YFP proteins in a Q stretch length-dependent manner.
Fluorescence microscopy pictures of 4-day-old C. elegans adults expressing Q0-YFP or Q35-YFP. RNAi of either hsp-1 or hsf-1 results in premature onset of aggregation in Q35 animals.
D) HSP70 overexpression suppresses polyglutamine-induced neurodegeneration in Drosophila.
Eyes (i–iv) and retinal sections (v,vi) of flies expressing expanded polyglutamine protein and human HSPA1L are shown. i,v, Control fly expressing only the promoter transgene. ii,vi, Flies expressing HSPA1L, coding for Hsp70 protein. Eye structure appears grossly normal. More detailed analysis revealed abnormalities in nuclear position and photoreceptor rhabdomere morphology when HSPA1L is highly expressed. iii,vii, Flies expressing the expanded polyglutamine protein MJDtr-Q78. These flies have degenerate eyes that lack pigment and show severe loss of retinal structure. iv,viii, Flies expressing both MJDtr-Q78 and HSPA1L. Co-expression of HSPA1L ameliorates the degenerative effects of MJDtr-Q78. The eye appears normal externally. Internally, eye structure is largely restored, although photoreceptor rhabdomere specializations are not made.
In addition to the control of the heat shock response by neuronal signaling, there have been numerous observations in which the heat shock response is poorly or incompletely activated, including early in development (75). Of particular interest are studies of the deficiency of the heat shock response in the brain and during aging (76, 77). Restricted expression of heat shock genes has been observed in different regions of the brain, while neuronal cells in culture can exhibit selective induction of chaperone genes. Human neuroblastoma Y79 cells, for example, respond to heat shock by induction of many chaperones including Hsp90, but not Hsp70, despite activation of HSF1 (78). Intact primary hippocampal neurons from neonatal rat embryos express HSF2 but not HSF1 until later stages of development (79). Consequently, the heat shock response of primary hippocampal neurons is deficient, while astrocytes have a robust stress response. Similar observations have been made in primary motor neurons that exhibit a deficient heat shock response thought to be due to a defect in activation of HSF1 (80).
The involvement of proteostasis networks in conformational diseases and aging is underscored by a decrease in toxicity when individual molecular chaperones are overexpressed in various cell-based and animal models (reviewed in (81)). Overexpression of HSF1 and certain molecular chaperones has been shown to extend lifespan (66, 67, 82, 83). Furthermore, the ability to induce the heat shock response under conditions of environmental proteotoxic stress is predictive of the remaining post-stress lifespan (84, 85). Likewise, proteasomal adaptation (by modulation of substrate accessibility to the proteasome core) to environmental stress in C. elegans ensures both resistance to proteotoxic conditions and maintenance of life span under normal conditions, arguably through regulating degradation of misfolded proteins (86). Moreover, because both HSF-1 and DAF-16 are regulated by the NAD-dependent sirtuin, SIRT1 (87-89), in addition to the ILS-pathway, it is reasonable to suggest that regulation of proteostasis is intimately linked by multiple pathways to metabolic control and lifespan.
Late onset diseases are mainly protein folding diseases
Common features of protein conformation diseases are the accumulation of protein deposits - aggregates, inclusion bodies, and plaques, and a presumably consequential “gain-of-function” proteotoxicity (90, 91). These features, which are characteristic of misfolded protein species, are present in neurodegenerative diseases of aging such as Parkinson’s disease, amyotrophic lateral sclerosis (ALS), prion disease, Alzheimer’s disease and the family of disorders generally referred to as the CAG-repeat/polyglutamine-expansion diseases (Huntington’s disease (HD), Kennedy’s disease, spinocerebellar ataxias). Each disease has the distinctive characteristic of age-dependent onset, a progressive, usually fatal, clinical course, and selective neuronal vulnerability despite broader expression of the causative protein.
Despite the fact that the proteins that are now known to “cause” neurodegenerative diseases lack similarities in primary sequence (other than the polyQ tract in CAG-repeat expansion diseases), they all share the ability to form alternate unfolded or misfolded states that in turn aggregate and/or are toxic. Thus, misfolding and aggregation has been proposed to be the molecular underpinning of disease. This is supported by evidence from transgenic model systems including S. cerevisiae, C. elegans, and Drosophila (92-97), in which it has been possible to recapitulate many molecular, cellular, and behavioral phenotypes associated with neurodegenerative disease. The development of these non-mammalian models for protein conformational disease has been invaluable for the discovery of modifiers and pathways, underlying mechanisms of toxicity, and for the testing of small molecules (63, 98-101). Additionally, these models have solidified our understanding of the link between protein conformational disorders, molecular chaperones and proteostasis regulators, and aging (66, 68).
2. Invertebrate models of late onset conformational diseases
The link between protein misfolding and human neurodegenerative disease, and the fact that the machinery involved in proteosasis maintenance, is highly conserved amongst eukaryotes, has led to the widespread use of invertebrate models systems, such as C. elegans and Drosophila. Ultimately, modeling aspects of human neurodegenerative diseases in invertebrates allows for genetic manipulations, such as mutant screens/identification of modifiers, and aging studies that would be prohibitively time consuming if carried out in vertebrate animals.
These studies in C. elegans and Drosophila benefit from the sequenced genomes, relatively short life cycles, and abundant genetic tools of these well-characterized model organisms. C. elegans is particularly well suited for the study of neurodegenerative diseases of aging due to its a relatively simple (but still sufficiently complex) nervous system, its genetically defined aging pathway, and the relative ease to perform live cell imaging of fluorescent proteins for studies of disease-causing protein aggregation dynamics. Likewise, in Drosophila the photoreceptor neurons have proven to be highly amenable to these types of studies, as human proteins can be ectopically expressed, and the readout for proteotoxicity is revealed by effects on eye morphology.
To date, a relatively large number of invertebrate models of human neurodegenerative diseases have been generated. Importantly, these models have solidified our understanding that protein misfolding underlies the mechanism(s) of disease action. Furthermore, the invertebrate models have opened up new ways to study the molecular underpinnings of disease, and to identify genes and gene networks whose activities are involved, positively or negatively, in modulating aggregation/toxicity. We begin with an overview of the existing models, and discuss what has been learned from them generally and in what ways they recapitulate key aspects of human disease.
PolyQ
Huntington’s disease, and a number of related neurodegenerative diseases are caused genetically by expansions of polyQ-encoding CAG tracts in specific individual genes/proteins. Expressing these proteins, with WT or expanded polyQ tracts, in invertebrates has proven to be an effective way to study the molecular underpinnings of protein misfolding/toxicity associated with polyQ disorders. Specifically, SCA3/MJD (Figure 3D) (92) and HD (102) were modeled in Drosophila and shown to recapitulate many important aspects of disease including cell-type specific sensitivity to the expressed protein, nuclear inclusion formation, and late-onset cellular degeneration. These findings were furthered by the expression of polyQ proteins in C. elegans (Figure 3B, C). Huntington’s Disease (HD) models have been generated via the expression, in neuronal subsets, of an N-terminal fragment of the protein Huntingtin (Htt) with the wild type or a long polyglutamine (polyQ) tract (93, 97). Ultimately, it was shown that human Htt with expanded polyglutamine tract led to aggregate formation in, and apoptotic cell death of, ASH sensory neurons, in a sensitized genetic background (93). Additionally, Htt fused to YFP aggregated when expressed in PLM mechanosensory neurons with 128 glutamines and caused neuronal abnormalities but not death (97). Having such fluorescently tagged variants of disease-causing proteins in C. elegans has proven highly advantageous by allowing for the real-time monitoring of aggregation and using dynamic imaging techniques in this transparent organism (38, 63, 96, 103).
The overall similarities between various polyQ disorders suggested that the polyQ expansions are predominantly, if not solely, responsible for the observed disease pathologies. To address this, general polyQ models, in which only the polyQ tract, without any of its normal flanking protein sequences (but fused in some cases to YFP, or to another protein not usually containing a polyQ tract) were developed (Figure 3B) (63, 96, 104). The data from these models provided the first direct in vivo evidence that polyQ tracts themselves, independently of their protein context, aggregate and are toxic.
The significance of recapitulating key aspects of disease by expressing a single human protein in an invertebrate should not be underestimated. It confirmed that the mutant proteins are generally toxic even across species, and in different cell types. This, in turn, lead to the hypothesis that the highly conserved cellular quality control system, involving protein folding/clearance, is inherently lacking the ability to prevent damage by these polyQ-containing disease-causing proteins.
Aβ
Alzheimer’s Disease (AD) is a relatively common neurodegenerative disorder affecting nearly 30% of all individuals greater than 85 years of age. It is characterized by the accumulation of β-amyloid plaques and neurofibrillary tangles. The major proteinaceous components of β-amyloid plaques in AD patients are Aβ peptides; specifically, Aβ1-40 and Aβ1-42, which are produced via proteolysis of APP, the amyloid precursor protein. To model Aβ aggregation/toxicity in an invertebrate model system, Aβ1-42 was expressed in C. elegans body wall muscle cells and was shown to form amyloid plaques that have biochemical characteristics similar to those found in the brains of AD patients (105). Additionally, Aβ1-42 and Aβ1-40 were expressed in Drosophila photoreceptor neurons (106). It was shown that Aβ1-42 was substantially more toxic than Aβ1-40 (106), consistent with the idea that Aβ1-42 has a higher amyloidogenic propensity.
tau
In addition to amyloid plaques, AD, and other neurodegenerative diseases are characterized by the formation of neurofibrillary tangles comprised of the protein tau. Expression of mutant tau in specific neuronal subtypes or under pan neuronal control in Drosophila resulted in neurodegeneration, but not neurofibrillary tangle/filamentous aggregate formation, as determined by EM, suggesting that the tangles themselves are not the source of mutant tau toxicity (107). Expression of tau in all C. elegans neurons resulted in motility defects indicative of tau proteotoxicity, providing further support to the idea that low levels of tau protein, not necessarily neurofibrillarly tau tangles or otherwise insoluble tau, are required for toxicity (108).
alpha-syncuclein
Familial forms of Parkinson’s Disease (PD) are caused by mutations in the protein alpha-synuclein (109, 110). Expression of alpha-synuclein in C. elegans neurons was shown to cause neurodegeneration of neuronal subsets (111). Expression of alpha-synuclein in body wall muscle cells resulted in age-dependent aggregation; a phenotype that was used to perform a genetic screen for suppressors of aggregation (see below) (112).
SOD1
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease that, in its inherited form, is often associated with mutation of superoxide dismutase 1 (SOD1). A large number of mutations in SOD1 have been identified and shown to be linked to ALS (113). Evidence suggests that mutations in SOD1 cause disease not by loss of SOD1 function, but instead by gain of toxic function by the mutant protein. Therefore, to model SOD1 protein misfolding/toxicity, these mutations have been expressed in a number of genetic systems including mouse, Drosophila and C. elegans. To model SOD1 protein misfolding in Drosophila, wild type human SOD1 (hSOD1), two mutant forms of SOD1 (G85R and A4V), and Drosophila SOD1 (dSOD1) were overexpressed in Drosophila motor neurons (114). It was shown that unlike dSOD1, wild type and mutant hSOD1 are toxic, but non-aggregating in Drosophila (114). This is in contrast to what was observed when hSOD1 was expressed in C. elegans body wall muscle cells (38), or neurons (115). Namely, expression of YFP-tagged SOD1 (G85R) under the control of a pan-neuronal promoter resulted in motility defects and distinct neuronal subtype-type-specific aggregation phenotypes that were confirmed by FRAP analysis (115). Likewise, expression of a number of different disease-associated SOD1 mutants in C. elegans body wall muscle cells resulted in the formation of SOD1 protein aggregates (Figure 3A) (38). Interestingly, the toxic effects of mutant forms of SOD1 were subtle in body wall muscle cells, but enhanced in the background of metastable ts proteins (38), consistent both with the finding that temperature sensitive proteins can exacerbate the toxic effects of polyQ protein expression (Figure 4) (3) and with the idea that disease phenotypes and/or proteotoxicity are highly sensitive to changes in the folding environment, presumably due to limited folding resources (see discussion below).
Why are these disease-causing proteins toxic to cells? In other words, why can’t cells, or more specifically, affected neurons, combat the toxic effect of their expression either by activating the machinery responsible for assisting in the folding of misfolded protein species or in clearing them via autophagy and/or proteasome-mediated degradation?
3. Failure of homeostasis in conformational disease of aging
Failure of adaptive stress responses
The cytoplasmic heat shock response and the ER and mitochondrial unfolded protein responses are the adaptive responses employed by cells to combat proteotoxic stress. The heat shock response is a multistep process that results in the immediate induction of transcription and translation of genes encoding molecular chaperones, proteases, and other proteostasis regulators, and in a generalized translational silencing. Although recent evidence indicates that additional signals may activate heat shock response (116-118), appearance of misfolded proteins is thought to serve as the predominant signal for its induction (119-123). Misfolded proteins are thought to titrate HSF1-associated chaperones, releasing the inhibition of HSF1and enabling it to trimerize and translocate into the nucleus. This results in the activation of Hsp gene transcription, rebalancing of proteostasis, and, finally, the attenuation of the heat shock response (reviewed in (124)). The main function of heat shock proteins during stress is to prevent inappropriate protein interactions and aggregation, mainly through binding to the exposed hydrophobic areas on a variety of cellular proteins, followed by the facilitation of refolding, or targeting to degradation during the recovery phase. Many of these proteins also function as molecular chaperones in the absence of stress, by guiding conformational transitions during synthesis, folding, translocation, assembly, and degradation of proteins (124-126). As chaperones affect other cellular functions, such as signaling (127), the proper regulation of chaperone expression is critical to the health of the cell.
The ability of cells to manage general/widespread protein misfolding during heat shock and other proteotoxic stress conditions suggests that the combination of the abundant expression of chaperones under basal conditions (50, 128), together with the adaptive stress responses, provides sufficient ‘folding capacity’ to buffer unexpected folding requirements. Thus, the accumulation of misfolded and aggregated proteins associated with the aging-related conformational diseases, and the misfolding-associated toxicity, indicate a failure of folding homeostasis.
Understanding the molecular mechanisms underlying this dysfunction is essential to both defining the mechanisms of toxicity, and finding targets for corrective interventions in conformational disease, and, perhaps, aging. Several possible, but not mutually exclusive, mechanisms are indicated by observations of dysregulation of proteostasis under these conditions. First, Hsp expression is often not induced in symptomatic cells, despite the accumulation of misfolded and aggregated proteins; moreover, decrease in specific chaperone expression has been noted (129-134). In C. elegans, intracellular accumulation of misfolded and aggregated polyQ proteins only sporadically activated HSP expression (96), and in fact required the downregulation of ILS signaling for modulation of its toxicity (66, 68). This points to a potential override of the cellular stress response at the organismal level (74), which traditionally has been considered to be cell-autonomous. Alternatively, it is possible that the accumulation of misfolded proteins in models of conformational diseases is either too gradual, or does not reach the threshold for heat shock activation. Second, molecular chaperones, components of degradative machinery and other proteins are often found trapped in aggregates, potentially mimicking hypomorphic phenotypes (96, 135, 136). Third, there is evidence that the accumulation of misfolded proteins leads to the inactivation of components of proteostasis networks, in particular the proteasome (45, 137, 138), and of the cell’s ability to induce a heat shock response (139), by the disease-associated misfolded proteins. The downregulation of specific chaperones and the inhibition of heat shock induction could potentially indicate that cells (or organisms) adapt to the chronic expression of misfolded proteins by actively preventing stress induction.
Whatever the specific mechanism of proteostasis dysfunction, it is clear that our ability to potentially adjust, if not correct, proteostatic networks, to cope with the chronic protein misfolding may be essential to our ability to manage, or even prevent, conformational diseases of aging (7).
Disruption of proteostasis by chronic misfolding
Our knowledge of how protein misfolding and the disruption of proteostasis translates into cell-specific and disease-specific toxicity is still quite limited. Evidence from C. elegans suggests that the toxicity associated with expression of expanded polyQ and mutant SOD1 proteins is caused, in part, by the global disruption of the cellular protein folding homeostasis, resulting in a destabilization or misfolding of various metastable proteins (3, 38). The potential sources of metastable proteins, as described above, range from expressed protein polymorphisms, to protein damage from environmental stresses, to what has been termed ‘activities of daily living’, for example cell signaling, glycolysis, or respiration. When temperature-sensitive (ts) metastable proteins were used to mimic naturally occurring mildly destabilizing polymorphisms, they acted both as sensors of cellular protein folding capacity, and contributors to cellular dysfunction. Expression of polyQ or mutant SOD1 in muscle or neuronal cells of C. elegans leads to the exposure of the temperature-sensitive phenotype at permissive conditions, mediated by misfolding and loss-of function of ts mutant protein present in the same cell (Figure 4A) (3, 38). Furthermore, the misfolding of ts proteins not only directed the specific phenotypes, but further increased aggregation of the polyQ proteins (Figure 4B). The misfolding of ts proteins was most likely due to the depletion, by the polyQ or mutSOD1 proteins, of folding resources, necessary for maintaining these ts proteins in their folded and functional conformations (140, 141). A recent finding that many of the modifiers of toxicity of polyQ-expanded ataxin-3 in Drosophila also rescue the generic toxicity of protein misfolding due to the reduced function of Hsp70 (142) strongly supports the disruption of proteostasis as a mechanism of toxicity. Furthermore, both the functionality of metastable proteins and the aggregation of polyglutamine expansions can be compromised by neuronally-mediated overexcitation of the muscle cells in C. elegans (101).
These findings parallel the evidence discussed above, that the selection against the toxicity of misfolding due to mistranslation exerts strong evolutionary pressure specifically on the highly expressed proteins (37), which we take to indicate that the flux of destabilized proteins in a cell bears a significant fitness cost, and that folding homeostasis is indeed not robust, at least when it comes to chronically misfolded proteins. Importantly for this proposition, the toxic interaction between the destabilized protein polymorphisms (ts proteins) and the disease-related polyQ or mutSOD1 was reversed not only by overexpression of HSF1, but also by the aging regulator DAF-16 (38).
The causative connection between proteostasis, aging, and cellular dysfunction was further illustrated in C. elegans by showing that, unlike in young animals, the ts proteins in older adults kept at the permissive conditions begin to gradually misfold and lose function, coincident with a reduced ability to activate the heat shock response and the unfolded protein response (143). Increasing the activity of either HSF1, or DAF-16, suppressed the misfolding of metastable proteins, and restored cellular proteostasis. Thus, the dysregulation of protein folding homeostasis may represent a set of early molecular events in aging, with an ability to amplify the protein damage cascade in age-related conformational diseases, while the complement of mutations and polymorphisms, together with the life history of an organism (environmental stress exposure, metabolic state, etc.), set the threshold for the onset of dysfunction and direct specific phenotypes.
Consistent with the proposal that a disruption in proteostasis is a key element of mechanism of toxicity in conformational diseases, genetic screens in invertebrate models described above have revealed proteostasis components as modifiers of aggregation/toxicity.
4. Modifiers of conformational disease
Genetic screens for modifiers of disease-related phenotypes
The versatility of C. elegans as a model system to study molecular processes involved in human disease has been demonstrated via the implementation of genome-wide RNA interference (RNAi) screens to identify genetic modifiers of disease-related phenotypes. Such screens have been facilitated by the availability of RNAi libraries, consisting of E. coli clones containing IPTG-inducible double stranded RNAs for the majority of C. elegans genes (144). Feeding of dsRNA-producing E. coli to C. elegans has proven to be a highly efficient method for targeted gene silencing, making high-throughput RNAi screens relatively straightforward (145).
One such study identified modifiers of polyQ protein aggregation in body-wall muscle cells in C. elegans (100). The genome-wide screen took advantage of a polyQ length at the threshold for aggregation (Q35), thereby allowing for a sensitized screen aimed at the identification of factors, which, when knocked down in the background of polyQ-YFP, led to the accumulation of visible protein aggregates. This screen identified 186 proteins that normally suppress age-dependent polyQ protein aggregation, including HSF-1 and Hsp-1 (Figure 3C). The authors found that the suppressors fall into five distinct biological classes, including RNA metabolism, protein synthesis, protein folding, protein trafficking, and protein degradation.
The identification of chaperones and factors involved in protein clearance was expected, and consistent with the results of a screen for genetic modifiers of polyQ protein toxicity in Drosophila, that uncovered homologs of Hdj1 and Tpr2, both J-domain-containing co-chaperones (99). Additionally, Hsp70 or Hdj1 overexpression significantly ameliorated toxicity of the polyglutamine-containing proteins ataxin-1 and ataxin-3 in Drosophila (Figure 3D) (146, Warrick et. al. 1999). Thus, chaperones have been found consistently as modifiers of aggregation and toxicity across different tissues and aggregation models. The C. elegans screen also uncovered six of the eight subunits of cytosolic chaperonin CCT, whose role as a suppressor of polyQ protein aggregation was previously unknown. This finding was later validated by using both S. cerevisiae and mammalian tissue culture cells expressing aggregation-prone Htt-polyQ proteins (147-149), supporting the idea that common mechanisms underlie polyQ protein aggregation/toxicity. These findings underscore the involvement of molecular chaperone activity in modulating the aggregation/toxicity of polyglutamine proteins. However, the identification of factors involved in other biosynthetic processes led to the conclusion that protein homeostasis is more complex than previously understood, and likely begins with gene expression, thus explaining the large fraction of modifiers involved in RNA and protein biosynthesis (100).
An independent RNAi screen was also performed in C. elegans for factors that normally suppress tau-induced motility defects (150). Wild-type and mutant tau protein become hyperphosphorylated, aggregate, and form neurofibrilllary tangles that are associated with neurodegeneration in patients suffering from Alzheimer’s disease (AD), and a number of related neurodegenerative diseases (151). Expression of tau in C. elegans neurons caused motility defects that were used as the basis to identify factors, via genome-wide RNAi screening, which, when absent, enhanced the motility (unc) phenotype (150). This analysis led to the identification of 75 suppressors of tau toxicity falling into the following functional categories: kinases, chaperones, proteases, and phosphatases, in addition to a number of genes whose function is unknown (150). Interestingly, the only RNAi hits in common between this and the polyQ screen described above are the Hsp70 molecular chaperone, hsp-1, and the heat shock transcription factor, hsf-1. Consistent with the idea that tau and polyQ proteins interact differentially with the cellular environment, a complementary study in a Drosophila tau model identified mostly kinases and phosphatases (152). Significantly, no HSPs, molecular chaperones, or components of the protein clearance machineries were identified (152).
A recent screen for suppressors of α-synuclein (α-syn) aggregation in body wall muscle cells revealed factors involved in vesicle trafficking and lipid metabolism (112). This screen, however, did uncover several regulators of life-span, including lagr-1 and sir-2.1, consistent with aging being a potent modifier of aggregation/toxicity. Interestingly, knock-down of Hsp70 had no effect on α-syn aggregation (112), suggesting that α-syn has distinctly different mechanisms of aggregation/toxicity than polyQ or tau proteins. In support of α-syn and polyQ having different mechanisms of aggregation, only one modifier was identified in C. elegans that was in common for both proteins (100, 112). Alternatively, since this was an aggregation screen that did not directly examine toxicity, it is possible that aggregation and toxicity of α-syn are uncoupled in the presence of Hsp70 RNAi, a finding that would be consistent with one from Drosophila (153).
The striking lack of overlap between modifiers of aggregation/toxicity for unique aggregation-prone proteins could presumably be due to different factors acting on different misfolded species, in addition to different factors acting in different tissue types, for example, body wall muscle cells as compared to neurons. Certainly, the identification of kinases and phosphatases in the screen for suppressors of tau toxicity provides support for the hypothesis that tau hyperphosphorylation is a prerequisite for disease. Consequently, these data also provide evidence that C. elegans and Drosophila models are valid system for the identification of factors which are capable of acting on particular human disease-causing proteins to suppress aggregation/toxicity.
In contrast to α-syn, a screen for modifiers of SOD1 aggregation and toxicity in C. elegans neurons yielded mostly molecular chaperones and other factors generally involved in protein quality control (115), more consistent with what was seen previously with respect to modifiers of polyQ protein aggregation/toxicity. Furthermore, the hits obtained by an RNAi screen aimed at the identification of suppressors of osmotic stress-induced gene expression overlapped to a great extent with those identified as suppressors of polyQ protein aggregation (154). In that study, the authors identified genes, including gpdh-1, that are upregulated in response to osmotic stress, which is known to cause generalized, non-specific protein misfolding. Although gpdh-1 expression does not respond to stresses other than osmotic stress (154), almost 30% of all the genes identified in this study overlap with those identified in the screen for suppressors of polyQ protein aggregation. Furthermore, 73% of the overlapping genes are predicted to fall into biological classes usually associated with the regulation of proteostasis, including RNA processing, protein synthesis, protein folding, and degradation (154). Finally, many of the modifiers of ataxin-3 toxicity in Drosophila were also able to rescue the toxic phenotypes due to the reduced function of HSP70, which also causes generalized protein misfolding (142). Ultimately, these data suggest that a core set of factors function generally in response to stress-induced protein damage, and others respond specifically to a particular stress, for example the stress of a misfolded, disease-causing protein, to tailor the response to the situation at hand.
However, the extent to which the molecular mechanisms of disease are conserved between aggregation-prone proteins is unknown. To address this, it will be necessary to express unrelated aggregation-prone proteins, such as Aβ, SOD1, tau, α-syn, and a polyQ-containing proteins, in the same cell type in a given model organism, for a direct comparison of modifiers of aggregation/toxicity. The expectation is that common modifiers would be those whose molecular function is in the general folding/clearance of misfolded proteins. On the other hand, modifiers that act on one or the other aggregation-prone protein would most likely be more closely associated with the specific function, or mode of disease progression, of a particular protein. For example, a screen performed in yeast expressing mutant Htt or α-synuclein, revealed almost entirely non-overlapping sets of genes, many with human homologs, acting as modifiers either of mutant Htt or α-synuclein toxicity (155). The authors speculate that their modifiers likely define mechanisms or pathways that are specific for particular disease-causing proteins, such as vesicle transport playing a role in α-synuclein toxicity (155).
In addition to RNAi and overexpression screens, forward genetic screens have also been performed to identify modulators of polyQ protein aggregation/toxicity in C. elegans. One such screen revealed a novel gene, pqe-1, which normally functions to suppress the proteotoxicity of an Htt exon 1 fragment with an expanded polyQ tract (156). Another forward genetic screen was aimed at the identification of genes that normally function to suppress the aggregation of polyQ-YFP in body wall muscle cells (101). This screen uncovered mutations in unc-30 which is the transcription factor that regulates the synthesis of the neurotransmitter GABA (101). The findings described in (101) are of particular interest, because they demonstrate that the ability of an organism to manage proteotoxic stress is not a cell autonomous process as previously thought, but may be affected by cell-cell communication, for example via neuronal cholinergic signaling. Consistent with this, treating Q35-expressing C. elegans with small molecules, acting positively or negatively on neuronal signaling, suppressed or enhanced, respectively, polyQ protein aggregation (101) in post-synaptic cells. The identification, via chemical genetic screens, of additional small molecules that alleviate disease-causing protein aggregation/toxicity will be an important step in the development of pharmaceuticals.
Small molecule drug screens
The search for therapeutics to treat neurodegeneration is turning to the identification of small molecule proteostasis regulators. Such small molecules could function by enhancing the expression or activities of molecular chaperones, thereby effectively increasing the rates of folding of client proteins, or, alternatively, they could act by enhancing degradation/clearance (157) or by modulating protein translation rates.
A number of small molecule regulators of the heat shock response have been identified. These compounds include proteasome inhibitors, serine protease inhibitors, Hsp90 inhibitors, nonsteroidal anti-inflammatory drugs (NSAIDS), triterpenoids such as celastrol, and inhibitors of HSF1, including triptolide (49, 158). Protease inhibitors (DCIC, TPCK, and TLCK) and proteasome inhibitors (MG132, lactacystin) induce the heat-shock response by elevating the effective concentrations of misfolfed, damaged, or otherwise no longer needed, proteins that are normally targeted for degradation (159, 160). In contrast, other inducers of the heat shock response act as inhibitors of Hsp90. These include the fungal antibiotic radicicol, the benzoquinone ansamycins geldanamycin, and 17AAG. They activate HSF1, in part because Hsp90 is a negative regulator of HSF1 (61, 161, 162). NSAIDS, including sodium salicylate, have multiple properties. At higher concentrations they partially activate HSF1, while at lower concentrations they synergize with other stress conditions to induce the heat-shock response (163). Specifically, exposure of human tissue culture cells to sodium salicylate results in activation of HSF1 with respect to in vivo binding to the Hsp70 gene; however, transcriptional induction fails to occur. Salicylate-treated cells, however, are sensitized to stress and readily activate HS genes upon exposure to other mild stress conditions, often not sufficient themselves to activate the heat shock response. In a similar manner, indomethacin induces HSF1 DNA binding with full Hsp70 transcription upon exposure to a secondary stress (164). Of the inflammatory modulators, arachidonic acid, and the cyclopentenone prostaglandins, including PGA1, PGA2, and PGJ2, all induce the full complement of HSF1 activities (165, 166). The triterpenoid celastrol isolated from the Chinese plant Triptergium wilfordii represents an herbal medicine class of bioactive molecules that induces two protective stress responses, the heat shock response and the anti-oxidant response (167, 168). The effects of celastrol are rapid like heat shock; however, unlike the heat-shock response that self-attenuates, the celastrol induction of HS genes persists for an extended period (167). Consistent with small molecule inducers of HSF-1 acting to alleviate the toxic effects of misfolded disease-causing proteins, geranylgeranylacetone (GGA) (169) and celastrol (170) treatment of polyQ protein-expressing cells inhibited polyQ-associated cell death in tissue culture and mouse models of polyQ protein misfolding .
The modulation of protein translation rates also play an important role in inducible stress responses. A screen for small molecule inhibitors of ER stress-induced apoptosis in PC-12 cells yielded salubrinol, a selective inhibitor of eIF2α dephosphorylation (171). eIF2α is a translation initiation factor that, when in its phosphorylated state, either under conditions of heat stress or salubrinol treatment, is responsible for mediating a general decrease, but a selective increase in the translation of chaperones and other stress-associated proteins. Like small molecule inducers of HSF1, salubrinol, or other translational regulators might be expected to modulate the toxic effects of misfolded disease-associated proteins.
Most of the chemical genetic analyses that have yielded the small molecules described above have been performed in tissue culture cells. A limited number of studies aimed at the identification of small molecules that act in invertebrate models to alleviate the toxicity of misfolding-prone disease-associated proteins have been performed. More commonly, small molecules that were identified in tissue culture systems as having the properties of inducing the HSR have been tested in invertebrates. For example, it was shown that the treatment of Htt-expressing C. elegans with the antioxidant resveratrol reduced Htt toxicity in a manner dependent on the sirutuin, sir2.1 (172).
A small-scale chemical genetics analysis was performed with a C. elegans Htt-polyQ model, using a screening approach which circumvented the inherent problems associated with screening based on motility defects. The authors examined treated worms for lack of neuronal cell death by visualizing the loss, or lack thereof, of GFP fluorescence in ASH neurons of a sensitized line that rapidly undergoes polyQ-dependent neurodegeneration (173). These conditions were used to validate candidate compounds identified previously in a large-scale, tissue culture-based, screen (174, 175), and revealed two compounds, lithium chloride and mithramycin, which suppressed HD neurotoxicity in the C. elegans Htt-polyQ model (173). The use of this, and similar assays, should make it possible to rapidly screen large chemical libraries for their effect on toxicity in C. elegans models of neurodegenerative diseases.
Because C. elegans is a multicellular organism, we would expect that the successful implementation of large-scale chemical genetics screens will be highly effective in identifying novel therapeutic compounds, not previously identified in cell culture models, that act either cell autonomously or cell non-autonomously. Finally, fluorescent labeling of candidate molecules will be instrumental in elucidating the mode of drug action, and to determine whether the drug is acting directly or indirectly on the disease-causing proteins, and will be relatively straightforward in C. elegans due the easy visualization of fluorescent markers.
Summary
Taken together, the work discussed here highlights the utility of invertebrate models in the study of neurodegenerative diseases of aging. Recent findings with these model systems have strengthened our understanding of protein conformational diseases. Specifically, they have led to the proposal that protein folding homeostasis, while sufficiently robust to manage protein damage/misfolding caused by acute environmental stress such as heat shock, is apparently ineffective when faced with the chronic expression of an aggregation-prone disease-causing protein. This limited capacity of the cell to manage chronic misfolding is especially pronounced under conditions of additional stress on the proteome caused by the expression of metastable proteins, partially compromised in their folding and providing a relatively high demand for folding resources. To combat this inherent limitation for the purpose of treating patients, small molecule drugs are being sought that will enhance the ability of a cell, or organism, to deal with the expression of chronically misfolded proteins.
Acknowledgements
E.A.K was supported by an individual postdoctoral fellowship from the National Institutes of Health (NINDS); research in the laboratory of R.I.M. was supported by grants from the National Institutes of Health (NIGMS and NIA), the HDSA Coalition for the Cure, and the ALS Association.
References
- 1.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 2.Stevens FJ, Argon Y. Pathogenic light chains and the B-cell repertoire. Immunol Today. 1999;20:451–457. doi: 10.1016/s0167-5699(99)01502-9. [DOI] [PubMed] [Google Scholar]
- 3.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 4.Yeyati PL, Bancewicz RM, Maule J, van Heyningen V. Hsp90 selectively modulates phenotype in vertebrate development. PLoS Genet. 2007;3:e43. doi: 10.1371/journal.pgen.0030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michels AA, et al. Hsp70 and Hsp40 chaperone activities in the cytoplasm and the nucleus of mammalian cells. J Biol Chem. 1997;272:33283–33289. doi: 10.1074/jbc.272.52.33283. [DOI] [PubMed] [Google Scholar]
- 6.Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 7.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 8.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 9.Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 11.Stadtman ER, Oliver CN. Metal-catalyzed oxidation of proteins. Physiological consequences. J Biol Chem. 1991;266:2005–2008. [PubMed] [Google Scholar]
- 12.Smith CD, et al. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed N, Dobler D, Dean M, Thornalley PJ. Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J Biol Chem. 2005;280:5724–5732. doi: 10.1074/jbc.M410973200. [DOI] [PubMed] [Google Scholar]
- 15.Lo TW, Westwood ME, McLellan AC, Selwood T, Thornalley PJ. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J Biol Chem. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 16.Kalapos MP. Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett. 1999;110:145–175. doi: 10.1016/s0378-4274(99)00160-5. [DOI] [PubMed] [Google Scholar]
- 17.Kuhla B, et al. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol Aging. 2007;28:29–41. doi: 10.1016/j.neurobiolaging.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Hipkiss AR. On the mechanisms of ageing suppression by dietary restriction-is persistent glycolysis the problem? Mech Ageing Dev. 2006;127:8–15. doi: 10.1016/j.mad.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 19.Gnerer JP, Kreber RA, Ganetzky B. wasted away, a Drosophila mutation in triosephosphate isomerase, causes paralysis, neurodegeneration, and early death. Proc Natl Acad Sci U S A. 2006;103:14987–14993. doi: 10.1073/pnas.0606887103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moskovitz J. Roles of methionine suldfoxide reductases in antioxidant defense, protein regulation and survival. Curr Pharm Des. 2005;11:1451–1457. doi: 10.2174/1381612053507846. [DOI] [PubMed] [Google Scholar]
- 21.Friguet B. Oxidized protein degradation and repair in ageing and oxidative stress. FEBS Lett. 2006;580:2910–2916. doi: 10.1016/j.febslet.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 22.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 23.Hanson SR, Hasan A, Smith DL, Smith JB. The major in vivo modifications of the human water-insoluble lens crystallins are disulfide bonds, deamidation, methionine oxidation and backbone cleavage. Exp Eye Res. 2000;71:195–207. doi: 10.1006/exer.2000.0868. [DOI] [PubMed] [Google Scholar]
- 24.Breusing N, Grune T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol Chem. 2008;389:203–209. doi: 10.1515/BC.2008.029. [DOI] [PubMed] [Google Scholar]
- 25.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 26.Shringarpure R, Grune T, Mehlhase J, Davies KJ. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 27.Shang F, Nowell TR, Jr., Taylor A. Removal of oxidatively damaged proteins from lens cells by the ubiquitin-proteasome pathway. Exp Eye Res. 2001;73:229–238. doi: 10.1006/exer.2001.1029. [DOI] [PubMed] [Google Scholar]
- 28.Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol. 2002;4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 29.Reinheckel T, Grune T, Davies KJ. The measurement of protein degradation in response to oxidative stress. Methods Mol Biol. 2000;99:49–60. doi: 10.1385/1-59259-054-3:49. [DOI] [PubMed] [Google Scholar]
- 30.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schultz SC, Richards JH. Site-saturation studies of beta-lactamase: production and characterization of mutant beta-lactamases with all possible amino acid substitutions at residue 71. Proc Natl Acad Sci U S A. 1986;83:1588–1592. doi: 10.1073/pnas.83.6.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pakula AA, Sauer RT. Genetic analysis of protein stability and function. Annu Rev Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- 33.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 34.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat Rev Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 35.Suckow J, et al. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- 36.Sachidanandam R, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- 37.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gidalevitz T, Krupinski T, Garcia S, Morimoto RI. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 40.Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 41.Lee JW, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 42.Jordanova A, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 43.Martin I, Grotewiel MS. Oxidative damage and age-related functional declines. Mech Ageing Dev. 2006;127:411–423. doi: 10.1016/j.mad.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Calderwood SK, Murshid A, Prince T. The Shock of Aging: Molecular Chaperones and the Heat Shock Response in Longevity and Aging - A Mini-Review. Gerontology. 2009 doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI. Inefficient degradation of truncated polyglutamine proteins by the proteasome. Embo J. 2004;23:4307–4318. doi: 10.1038/sj.emboj.7600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 47.Suhr ST, et al. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- 50.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 51.Banerji SS, Theodorakis NG, Morimoto RI. Heat shock-induced translational control of HSP70 and globin synthesis in chicken reticulocytes. Mol Cell Biol. 1984;4:2437–2448. doi: 10.1128/mcb.4.11.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao X, et al. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. Embo J. 1999;18:5943–5952. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos SD, Saraiva MJ. Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience. 2004;126:657–663. doi: 10.1016/j.neuroscience.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 54.Zhang P, et al. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 56.Zhang K, et al. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elefant F, Palter KB. Tissue-specific expression of dominant negative mutant Drosophila HSC70 causes developmental defects and lethality. Mol Biol Cell. 1999;10:2101–2117. doi: 10.1091/mbc.10.7.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feder JH, Rossi JM, Solomon J, Solomon N, Lindquist S. The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev. 1992;6:1402–1413. doi: 10.1101/gad.6.8.1402. [DOI] [PubMed] [Google Scholar]
- 59.Nylandsted J, et al. Selective depletion of heat shock protein 70 (Hsp70) activates a tumor-specific death program that is independent of caspases and bypasses Bcl-2. Proc Natl Acad Sci U S A. 2000;97:7871–7876. doi: 10.1073/pnas.97.14.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 62.Duyao M, et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- 63.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morris JZ, Tissenbaum HA, Ruvkun G. A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- 65.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 66.Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 68.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 69.Lithgow GJ, White TM, Melov S, Johnson TE. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A. 1995;92:7540–7544. doi: 10.1073/pnas.92.16.7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salmon AB, Akha AA Sadighi, Buffenstein R, Miller RA. Fibroblasts from naked mole-rats are resistant to multiple forms of cell injury, but sensitive to peroxide, ultraviolet light, and endoplasmic reticulum stress. J Gerontol A Biol Sci Med Sci. 2008;63:232–241. doi: 10.1093/gerona/63.3.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salmon AB, et al. Fibroblast cell lines from young adult mice of long-lived mutant strains are resistant to multiple forms of stress. Am J Physiol Endocrinol Metab. 2005;289:E23–29. doi: 10.1152/ajpendo.00575.2004. [DOI] [PubMed] [Google Scholar]
- 72.Broughton SJ, et al. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci U S A. 2005;102:3105–3110. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tullet JM, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prahlad V, Cornelius T, Morimoto RI. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science. 2008;320:811–814. doi: 10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bienz M. Developmental control of the heat shock response in Xenopus. Proc Natl Acad Sci U S A. 1984;81:3138–3142. doi: 10.1073/pnas.81.10.3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sprang GK, Brown IR. Selective induction of a heat shock gene in fibre tracts and cerebellar neurons of the rabbit brain detected by in situ hybridization. Brain Res. 1987;427:89–93. doi: 10.1016/0169-328x(87)90049-0. [DOI] [PubMed] [Google Scholar]
- 77.Shamovsky I, Gershon D. Novel regulatory factors of HSF-1 activation: facts and perspectives regarding their involvement in the age-associated attenuation of the heat shock response. Mech Ageing Dev. 2004;125:767–775. doi: 10.1016/j.mad.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 78.Mathur SK, et al. Deficient induction of human hsp70 heat shock gene transcription in Y79 retinoblastoma cells despite activation of heat shock factor 1. Proc Natl Acad Sci U S A. 1994;91:8695–8699. doi: 10.1073/pnas.91.18.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marcuccilli CJ, Mathur SK, Morimoto RI, Miller RJ. Regulatory differences in the stress response of hippocampal neurons and glial cells after heat shock. J Neurosci. 1996;16:478–485. doi: 10.1523/JNEUROSCI.16-02-00478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Batulan Z, et al. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 82.Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell. 2003;2:131–139. doi: 10.1046/j.1474-9728.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 83.Yokoyama K, et al. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002;516:53–57. doi: 10.1016/s0014-5793(02)02470-5. [DOI] [PubMed] [Google Scholar]
- 84.Herndon LA, et al. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature. 2002;419:808–814. doi: 10.1038/nature01135. [DOI] [PubMed] [Google Scholar]
- 85.Yang J, Tower J. Expression of hsp22 and hsp70 transgenes is partially predictive of drosophila survival under normal and stress conditions. J Gerontol A Biol Sci Med Sci. 2009;64:828–838. doi: 10.1093/gerona/glp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yun C, et al. Proteasomal adaptation to environmental stress links resistance to proteotoxicity with longevity in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2008;105:7094–7099. doi: 10.1073/pnas.0707025105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 88.Westerheide SD, Anckar J, Stevens SM, Jr., Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- 90.Kakizuka A. Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet. 1998;14:396–402. doi: 10.1016/s0168-9525(98)01559-5. [DOI] [PubMed] [Google Scholar]
- 91.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 92.Warrick JM, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 93.Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci U S A. 1999;96:179–184. doi: 10.1073/pnas.96.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 95.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Satyal SH, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Parker JA, et al. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci U S A. 2001;98:13318–13323. doi: 10.1073/pnas.231476398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Warrick JM, et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 99.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 100.Nollen EA, et al. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garcia SM, Casanueva MO, Silva MC, Amaral MD, Morimoto RI. Neuronal signaling modulates protein homeostasis in Caenorhabditis elegans post-synaptic muscle cells. Genes Dev. 2007;21:3006–3016. doi: 10.1101/gad.1575307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jackson GR, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–642. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- 103.Brignull HR, Moore FE, Tang SJ, Morimoto RI. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J Neurosci. 2006;26:7597–7606. doi: 10.1523/JNEUROSCI.0990-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marsh JL, et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- 105.Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 107.Wittmann CW, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 108.Kraemer BC, et al. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nussbaum RL, Polymeropoulos MH. Genetics of Parkinson’s disease. Hum Mol Genet. 1997;6:1687–1691. doi: 10.1093/hmg/6.10.1687. [DOI] [PubMed] [Google Scholar]
- 110.Chase TN. A gene for Parkinson disease. Arch Neurol. 1997;54:1156–1157. doi: 10.1001/archneur.1997.00550210084017. [DOI] [PubMed] [Google Scholar]
- 111.Lakso M, et al. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem. 2003;86:165–172. doi: 10.1046/j.1471-4159.2003.01809.x. [DOI] [PubMed] [Google Scholar]
- 112.van Ham TJ, et al. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364:362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- 114.Watson MR, Lagow RD, Xu K, Zhang B, Bonini NM. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem. 2008;283:24972–24981. doi: 10.1074/jbc.M804817200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang J, et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hahn JS, Thiele DJ. Activation of the Saccharomyces cerevisiae heat shock transcription factor under glucose starvation conditions by Snf1 protein kinase. J Biol Chem. 2004;279:5169–5176. doi: 10.1074/jbc.M311005200. [DOI] [PubMed] [Google Scholar]
- 118.Thomson S, Hollis A, Hazzalin CA, Mahadevan LC. Distinct stimulus-specific histone modifications at hsp70 chromatin targeted by the transcription factor heat shock factor-1. Mol Cell. 2004;15:585–594. doi: 10.1016/j.molcel.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 119.Ananthan J, Goldberg AL, Voellmy R. Abnormal proteins serve as eukaryotic stress signals and trigger the activation of heat shock genes. Science. 1986;232:522–524. doi: 10.1126/science.3083508. [DOI] [PubMed] [Google Scholar]
- 120.Hiromi Y, Okamoto H, Gehring WJ, Hotta Y. Germline transformation with Drosophila mutant actin genes induces constitutive expression of heat shock genes. Cell. 1986;44:293–301. doi: 10.1016/0092-8674(86)90763-4. [DOI] [PubMed] [Google Scholar]
- 121.Parsell DA, Sauer RT. Induction of a heat shock-like response by unfolded protein in Escherichia coli: dependence on protein level not protein degradation. Genes Dev. 1989;3:1226–1232. doi: 10.1101/gad.3.8.1226. [DOI] [PubMed] [Google Scholar]
- 122.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 123.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 124.Morimoto RI, Kline MP, Bimston DN, Cotto JJ. The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 1997;32:17–29. [PubMed] [Google Scholar]
- 125.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 126.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 127.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 128.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 129.Kieran D, et al. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- 130.Katayama T, et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J Biol Chem. 2001;276:43446–43454. doi: 10.1074/jbc.M104096200. [DOI] [PubMed] [Google Scholar]
- 131.Cowan KJ, Diamond MI, Welch WJ. Polyglutamine protein aggregation and toxicity are linked to the cellular stress response. Hum Mol Genet. 2003;12:1377–1391. doi: 10.1093/hmg/ddg151. [DOI] [PubMed] [Google Scholar]
- 132.Hay DG, et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 133.Zourlidou A, et al. Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: chronic neurodegeneration does not induce Hsp27 activation. Hum Mol Genet. 2007;16:1078–1090. doi: 10.1093/hmg/ddm057. [DOI] [PubMed] [Google Scholar]
- 134.Wen FC, et al. Down-regulation of heat shock protein 27 in neuronal cells and non-neuronal cells expressing mutant ataxin-3. FEBS Lett. 2003;546:307–314. doi: 10.1016/s0014-5793(03)00605-7. [DOI] [PubMed] [Google Scholar]
- 135.Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stenoien DL, et al. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 137.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 138.Nishitoh H, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tatzelt J, et al. Scrapie prions selectively modify the stress response in neuroblastoma cells. Proc Natl Acad Sci U S A. 1995;92:2944–2948. doi: 10.1073/pnas.92.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Van Dyk TK, Gatenby AA, LaRossa RA. Demonstration by genetic suppression of interaction of GroE products with many proteins. Nature. 1989;342:451–453. doi: 10.1038/342451a0. [DOI] [PubMed] [Google Scholar]
- 141.Brown CR, Hong-Brown LQ, Welch WJ. Correcting temperature-sensitive protein folding defects. J Clin Invest. 1997;99:1432–1444. doi: 10.1172/JCI119302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–1964. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fraser AG, et al. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- 145.Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2 doi: 10.1186/gb-2000-2-1-research0002. RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ghosh S, Feany MB. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum Mol Genet. 2004;13:2011–2018. doi: 10.1093/hmg/ddh214. [DOI] [PubMed] [Google Scholar]
- 147.Kitamura A, et al. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol. 2006;8:1163–1170. doi: 10.1038/ncb1478. [DOI] [PubMed] [Google Scholar]
- 148.Behrends C, et al. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 149.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kraemer BC, Burgess JK, Chen JH, Thomas JH, Schellenberg GD. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet. 2006;15:1483–1496. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- 151.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 152.Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–1242. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 154.Lamitina T, Huang CG, Strange K. Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc Natl Acad Sci U S A. 2006;103:12173–12178. doi: 10.1073/pnas.0602987103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003;302:1769–1772. doi: 10.1126/science.1090389. [DOI] [PubMed] [Google Scholar]
- 156.Faber PW, Voisine C, King DC, Bates EA, Hart AC. Glutamine/proline-rich PQE-1 proteins protect Caenorhabditis elegans neurons from huntingtin polyglutamine neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:17131–17136. doi: 10.1073/pnas.262544899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 158.Westerheide SD, Kawahara TL, Orton K, Morimoto RI. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J Biol Chem. 2006;281:9616–9622. doi: 10.1074/jbc.M512044200. [DOI] [PubMed] [Google Scholar]
- 159.Mathew A, Mathur SK, Morimoto RI. Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol Cell Biol. 1998;18:5091–5098. doi: 10.1128/mcb.18.9.5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rossi A, Elia G, Santoro MG. Activation of the heat shock factor 1 by serine protease inhibitors. An effect associated with nuclear factor-kappaB inhibition. J Biol Chem. 1998;273:16446–16452. doi: 10.1074/jbc.273.26.16446. [DOI] [PubMed] [Google Scholar]
- 161.Bagatell R, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6:3312–3318. [PubMed] [Google Scholar]
- 162.Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther. 2004;3:1021–1030. [PubMed] [Google Scholar]
- 163.Jurivich DA, Sistonen L, Kroes RA, Morimoto RI. Effect of sodium salicylate on the human heat shock response. Science. 1992;255:1243–1245. doi: 10.1126/science.1546322. [DOI] [PubMed] [Google Scholar]
- 164.Lee BS, Chen J, Angelidis C, Jurivich DA, Morimoto RI. Pharmacological modulation of heat shock factor 1 by antiinflammatory drugs results in protection against stress-induced cellular damage. Proc Natl Acad Sci U S A. 1995;92:7207–7211. doi: 10.1073/pnas.92.16.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Amici C, Sistonen L, Santoro MG, Morimoto RI. Antiproliferative prostaglandins activate heat shock transcription factor. Proc Natl Acad Sci U S A. 1992;89:6227–6231. doi: 10.1073/pnas.89.14.6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jurivich DA, Sistonen L, Sarge KD, Morimoto RI. Arachidonate is a potent modulator of human heat shock gene transcription. Proc Natl Acad Sci U S A. 1994;91:2280–2284. doi: 10.1073/pnas.91.6.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Westerheide SD, et al. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 168.Trott A, et al. Activation of Heat Shock and Antioxidant Responses by the Natural Product Celastrol: Transcriptional Signatures of a Thiol-targeted Molecule. Mol Biol Cell. 2008 doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Katsuno M, et al. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang YQ, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med. 2007;85:1421–1428. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Boyce M, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 172.Parker JA, et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- 173.Voisine C, et al. Identification of potential therapeutic drugs for huntington’s disease using Caenorhabditis elegans. PLoS ONE. 2007;2:e504. doi: 10.1371/journal.pone.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Varma H, Cheng R, Voisine C, Hart AC, Stockwell BR. Inhibitors of metabolism rescue cell death in Huntington’s disease models. Proc Natl Acad Sci U S A. 2007;104:14525–14530. doi: 10.1073/pnas.0704482104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Varma H, et al. Selective inhibitors of death in mutant huntingtin cells. Nat Chem Biol. 2007;3:99–100. doi: 10.1038/nchembio852. [DOI] [PubMed] [Google Scholar]