

Summary
Type VI secretion systems (T6SSs) contribute to interactions of bacterial pathogens and symbionts with their hosts. Previously, we showed that Pseudomonas aeruginosa T6S is posttranslationally activated upon phosphorylation of Fha1, an FHA domain protein, by PpkA, a membrane-spanning threonine kinase. Herein, additional structural, enzymatic, and genetic requirements for PpkA-catalyzed T6SS activation are identified. We found that PpkA plays an essential structural role in the T6SS, and that this role is intimately linked to its ability to promote secretion and phosphorylate Fha1. Protein localization and protein-protein interaction studies show that a complex containing Fha1 and ClpV1 is recruited to the T6S apparatus in a phosphorylation-dependent manner. The mechanism of PpkA activation was also investigated. We identified critical PpkA autophosphorylation sites and showed that small molecule-induced dimerization of the extracellular domains of PpkA is sufficient to activate the T6SS. Finally, we discovered TagR, a component of the T6S posttranslational regulatory pathway that functions upstream of PpkA to promote kinase activity. We present a model whereby an unknown cue causes dimerization of the extracellular domains of PpkA, leading to autophosphorylation, recruitment of the Fha1-ClpV1 complex, phosphorylation of Fha1, and T6SS activation. Our findings should facilitate approaches for identifying physiological activators of T6S.
Introduction
P. aeruginosa is a versatile gram-negative opportunistic pathogen. The ability of this organism to draw upon diverse factors from its large genome allows it to effectively colonize and cause disease in a wide array of organisms, including humans (Stover et al., 2000). Depending on the state of its host, P. aeruginosa infection can proceed in either an acute or chronic course. An example of chronic P. aeruginosa infection of humans is found in the lungs of individuals with cystic fibrosis (CF). These patients typically battle a series of infections early in life, which become chronic in nearly all adult CF patients (Burns et al., 2001, Govan & Deretic, 1996). Although a number of explanations have been put forth, it remains unclear precisely why the CF lung is so well suited for colonization and proliferation of P. aeruginosa (Murray et al., 2007). Clearly, the result of this complex interaction is dictated by the interplay of a number of host and bacterial factors. Some pathways in P. aeruginosa that have been implicated in chronic infection fitness include biofilm formation (Lam et al., 1980, Singh et al., 2000, Parsek & Greenberg, 2005), quorum sensing (Schuster & Greenberg, 2006, Smith & Iglewski, 2003, Willcox et al., 2008), alginate production (May et al., 1991), and recently, T6S (Goodman et al., 2004, Mougous et al., 2006, Mougous et al., 2007, Potvin et al., 2003).
T6SSs are widely present among Gram-negative proteobacteria, where they have been reported to participate in numerous processes including biofilm formation, symbiont host range determination, and acute and chronic infection (Cascales, 2008, Filloux et al., 2008, Bingle et al., 2008, Yahr, 2006). In general, the mechanism(s) of action of T6S in these phenotypes is unknown. This lack of understanding stems in part from the dearth of known T6S substrates. So far, the release of just two proteins to the extracellular milieu, hemolysin co-regulated protein (Hcp) and valine-glycine repeat protein G (VgrG), has been attributed to most studied T6SSs. Several lines of evidence, including the X-ray crystallographic structure of Hcp1 from P. aeruginosa and its similarity to phage tail proteins (Mougous et al., 2006, Pell et al., 2009), the crystal structure of an E. coli VgrG homolog (Leiman et al., 2009), and the observation that Hcp and VgrG homologs are co-dependent for secretion (Pukatzki et al., 2006, Zheng & Leung, 2007), suggest that these proteins may be extracellular T6 structural components rather than classically-defined secreted substrates. VgrG1 from V. cholerae gains access to host cell cytoplasm, where it promotes cell rounding and cytoxicity via a C-terminal appended domain with actin crosslinking activity (Mougous et al., 2009, Pell et al., 2007).
T6S may play an important role in the chronic colonization of CF patient lungs by P. aeruginosa and Burkholderia cenocepacia (Aubert et al., 2008, Mougous et al., 2006). Several T6S genes were identified in independent signature tagged mutagenesis (STM) studies of these organisms using chronic lung infection models (Potvin et al., 2003, Hunt et al., 2004). Although the genome of P. aeruginosa encodes three apparently complete T6SSs at three unlinked loci termed Hcp secretion islands (HSIs), only one of these, HSI-I, has been implicated in virulence. Subsequent characterization of this locus has provided additional evidence that it may be important in chronic P. aeruginosa infections. For instance, HSI-I is co-regulated by the Gac/Rsm signaling pathway with other chronic virulence determinants such as the exopolysaccharide loci pel and psl (Goodman et al., 2004). This pathway appears to coordinate reciprocal expression of genes involved in acute and chronic virulence in P. aeruginosa. Hcp1, a protein secreted by the HSI-I-encoded T6SS (H1-T6SS) can be detected in the sputum of CF patients infected with P. aeruginosa, and serum antibody titers against Hcp1 are high in chronically infected patients (Mougous et al., 2006).
Regulation of the H1-T6SS is complex; the system is stringently regulated posttranscriptionally by the Gac/Rsm pathway and posttranslationally by threonine phosphorylation (Goodman et al., 2004, Mougous et al., 2006, Mougous et al., 2007). Previously, we identified three H1-T6SS proteins that participate in the posttranslational regulation of the secretion system. These proteins include PpkA, a type II membrane-spanning Hanks-type threonine kinase, PppA, a PP2C family protein phosphatase, and Fha1, a protein containing a Forkhead Associated (FHA) domain (Pallen et al., 2002, Motley & Lory, 1999). We showed that PpkA-catalyzed phosphorylation of Fha1 at Thr362, a process antagonized by PppA, initiates triggering of the H1-T6SS (Mougous et al., 2007). Phosphorylation of Fha1 was found to be highly dependent on conserved residues located in the FHA domain of Fha1 that are known to contact phospho-thr or phospho-ser residues in the activation loops of autophosphorylated kinases.
In the current study, we sought to further define the molecular determinants of H1-T6SS activation. We found that only the cytoplasmic domains of PpkA are essential for Fha1 phosphorylation and Hcp1 secretion. Next, fluorescent fusion proteins to ClpV1 and Fha1 were used to demonstrate that PpkA function is tightly linked to its ability to promote structural integrity of the secretion system. Also, we used fluorescence microscopy (FM) and tandem affinity purification (TAP) to show that ClpV1 and Fha1 reside in a stable complex that is recruited to the H1-T6S apparatus upon PpkA activation. The function of the extracellular domains of PpkA was probed using small molecule-induced dimerization. This method provided evidence that the extracellular domains of PpkA have the capacity to transmit extracellular binding events into H1-T6SS activation. A requisite for this activation was identified as autophosphorylation of PpkA at two sites, Thr158 and Thr161. Finally, we identified a protein, termed TagR, which serves as an additional modulator of the H1-T6SS posttranslational regulatory pathway. We showed that TagR acts upstream of PpkA to regulate Fha1 phosphorylation and Hcp1. We speculate that TagR may function as a PpkA co-receptor for the activation signal of the secretion system. Based on these data, we present a revised model for the mechanism of H1-T6SS activation.
Results
The extracellular portion of PpkA is dispensable for Hcp1 secretion and Fha1 phosphorylation
PpkA is a large (110 kDa), membrane-spanning protein that plays multiple roles in the H1-T6SS (Fig. 1A) (Mougous et al., 2007). The absence of PpkA blocks Hcp1 secretion and Fha1 phosphorylation, and it causes disruption of the structural integrity of the secretion system. To improve our understanding of the properties of this central component of the H1-T6S posttranslational activation pathway, we dissected the domains of PpkA and assayed their contribution to individual functions of the enzyme. Sites for PpkA truncation were chosen based on BLAST analysis, multiple sequence alignments of the protein with other T6S kinases (data not shown), and earlier studies of the protein. In accordance with the findings of others, we identified an N-terminal cytoplasmic kinase domain (residues 1-281), a proline-rich domain (residues 282-353), a transmembrane domain (residues 355-375), and a C-terminal perisplasmic domain with homology to von Willebrand Factor A (residues 613-803; Fig. 1A) (Motley & Lory, 1999, Whittaker & Hynes, 2002) (Zeng, 2004).
Fig. 1. Identification of PpkA domains required for H1-T6S activation and Fha1 phosphorylation.
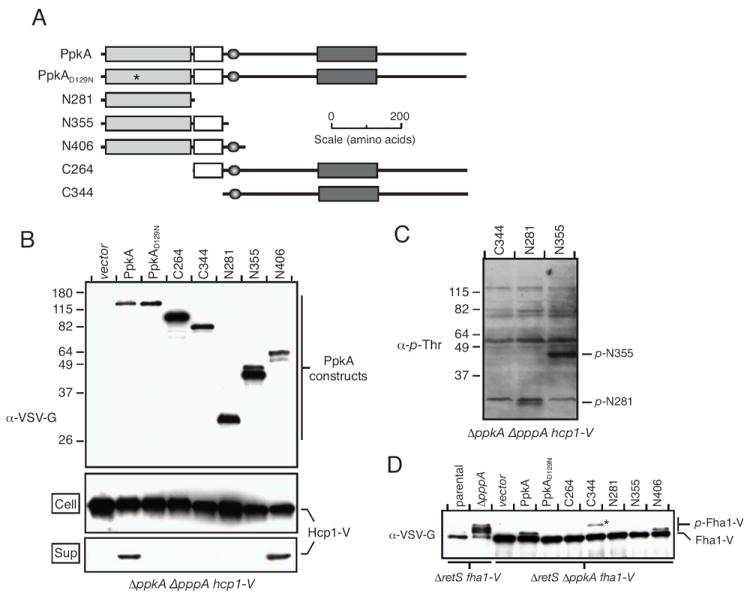
A. Overview of expression constructs and domain organization of PpkA: light grey, kinase; white, proline-rich; dark grey, von Willebrand factor A; shadowed oval, transmembrane.
B. The N-terminal 406 residues of PpkA are sufficient for H1-T6SS activation. Western blot analysis of PpkA truncation construct expression and their Hcp1 secretion complementation activity. Genotypes of strains used in each experiment are provided below the blot.
C. C-terminal PpkA truncations lacking the transmembrane domain can autophosphorylate. α-p-Thr-reactive bands corresponding to p-N281 and p-N355 are observed. C344, which lacks the kinase domain, is included as a control.
D. Domain requirements for Fha1 phosphorylation mirror those essential for Hcp1 secretion. Western blot analysis of Fha1 and p-Fha1 levels upon expression of PpkA truncations in (A). An asterisk highlights the position of an interfering band derived from VSV-G-tagged C344.
Full length PpkA and PpkA truncations were ectopically expressed as N-terminal fusions to the VSV-G epitope in P. aeruginosa ΔpppA ΔppkA hcp1-V (see Experimental Procedures). In this strain, the capacity of the proteins to complement the Hcp1 secretion defect of ΔppkA could be readily ascertained. The pppA mutation locks the H1-T6SS into a constitutively triggered state, where Hcp1 is actively secreted into the supernatant in a PpkA-dependent manner (Mougous et al., 2007). A chromosomal in-frame translational fusion of hcp1 to a DNA sequence encoding the VSV-G epitope is also included in this strain. Previously we showed that this fusion does not interfere with Hcp1 secretion, or regulation by PpkA and PppA (Mougous et al., 2006, Mougous et al., 2007). Note that in these studies, we refer to the presence of Hcp1 in culture supernatants as secretion. However, as stated in the preceding section, it remains unclear whether Hcp1 is a structural component or a secreted substrate of the system. Nonetheless, the extracellular release of this protein is a strong indicator of T6S activation that has been born out by numerous biochemical and genetic studies (Aubert et al., 2008, Dudley et al., 2006, Pukatzki et al., 2006, Schell et al., 2007, Suarez et al., 2007, Zheng & Leung, 2007, Wu et al., 2008).
As expected, the deletion of ppkA in ΔpppA hcp1-V abrogated Hcp1-V secretion (Fig. 1B). Importantly, this defect was complemented by expression of full-length PpkA. Complementation required the catalytic activity of the enzyme, since a ppkA allele encoding a protein containing a conservative amino acid substitution in a critical catalytic residue (D129N) was not able to return Hcp1 secretion (Fig. 1B). Not surprisingly, constructs lacking the kinase domain altogether were also not able to promote Hcp1 secretion (C264 and C344). Interestingly, the N-terminal 406 residues of PpkA (N406), but not the first 355 residues (N355), were able to complement the ΔppkA Hcp1 secretion defect (Fig. 1B). Amino acids 357-373 of PpkA encode a transmembrane helix (Zeng, 2004), therefore membrane anchoring of N406 is likely to account for the major difference in activity between these two constructs. We noted that the N355 protein reproducibly appeared as a doublet of bands in our assays. This may be due to proteolysis, an alternative initiation codon, or a combination of these factors. Importantly, full-length N355 protein is present at levels comparable to N406.
Membrane anchoring restricts proteins to two dimensions and increases their effective concentration, which can promote autophosphorylation. As discussed in detail below, autophosphorylation of PpkA is required for its function. Using α-phosphothreonine (α-p-Thr) Western blotting, we ruled out blunted autophosphorylation as a cause for the absence of activity in PpkA truncations lacking the transmembrane domain (Fig. 1C). We also ruled out expression level differences of the constructs as a potential confounding factor in this experiment (Fig. 1B).
Our prior work has shown that PpkA-catalyzed phosphorylation of Fha1 at T362 is essential for Hcp1 secretion. This reaction is efficiently catalyzed in vitro by the kinase domain of PpkA (Mougous et al., 2007). The finding that the same domain of PpkA is not sufficient for complementing the Hcp1 secretion defect of ΔppkA prompted us to test the in vivo Fha1 phosphorylation activity of each PpkA truncation. To measure Fha1 phosphorylation, we introduced our panel of PpkA expression constructs into ΔretS ΔppkA fha1-V. In this genetic background, phosphorylated Fha1 (p-Fha1) can be assayed by virtue of its retarded SDS-PAGE mobility. The ΔretS background is used to obtain maximal levels of constitutive PpkA-dependent Fha1 phosphorylation. As previously demonstrated, p-Fha1 was present in ΔretS fha1-V, and the presence of this species depended on ppkA (Fig. 1D). As a control, a deletion in pppA was introduced into ΔretS fha1-V. Since this strain lacks PpkA-antagonistic phosphatase activity, Fha1 becomes hyper-phosphorylated and is observed as higher apparent molecular mass species (Fig. 1D). Next, we determined which PpkA truncations had the capacity to complement the Fha1 phosphorylation defect of ΔppkA. Interestingly, we observed a direct correlation between activity in this assay and in the Hcp1 secretion complementation experiment described above; only wild-type ppkA and the N406 allele possessed the ability to generate p-Fha1 (Fig. 1D). Based on these data, we conclude that productive interaction of PpkA and Fha1 requires membrane anchoring of PpkA in addition to the catalytic activity of the enzyme. Furthermore, our results suggest that the extracellular domains of PpkA are not required for these basic functions of the protein.
PpkA function is tightly linked to structural integrity of the H1-T6SS
Fluorescent fusion protein experiments have proven useful for understanding the localization properties of H1-T6SS proteins. These studies provided evidence for a distinct subcellular assembly that constitutes the H1-T6S apparatus (Mougous et al., 2006, Mougous et al., 2007). Two fusion proteins, ClpV1-GFP and Fha1-mCherry (Fha1-mC), were shown by FM to co-localize to the secretory apparatus. The presence of other proteins, including TssM1 (see Fig. S1 for a description of updated HSI nomenclature), Hcp1, and PpkA, was inferred by their influence on ClpV1 localization. We hypothesized that the requirement for PpkA domains outside of its catalytic domain could reflect their role in maintaining structure of the H1-T6S apparatus.
To determine whether the function of PpkA truncations was linked to H1-T6S apparatus assembly, we measured the effect of their expression on ClpV1 localization. The N406 expression construct and positive (PpkA) and negative control (N281) vectors were introduced into ΔretS ΔppkA clpV1-gfp. Consistent with earlier data, punctate localization of ClpV1-GFP in ΔppkA was disrupted and the protein was distributed diffusely throughout the cell relative to the parental strain (Fig. 2A). Expression of full-length PpkA and N406 led to a return of ClpV1-GFP punctae resembling the parental strain, whereas expression of N281had no significant effect (Fig. 2A). Blinded quantification of ClpV1-GFP foci using randomly selected FM fields from each of these strains provided support for these observations (Fig. 2B). Additionally, flow cytometric analysis of total GFP levels in these cells showed that the effect was specific to localization and was not the result of overall lower levels of ClpV1-GFP (Fig. S2).
Fig. 2. Intracellular and transmembrane domains of PpkA are sufficient for ClpV1 and Fha1 localization to the H1-T6S apparatus.
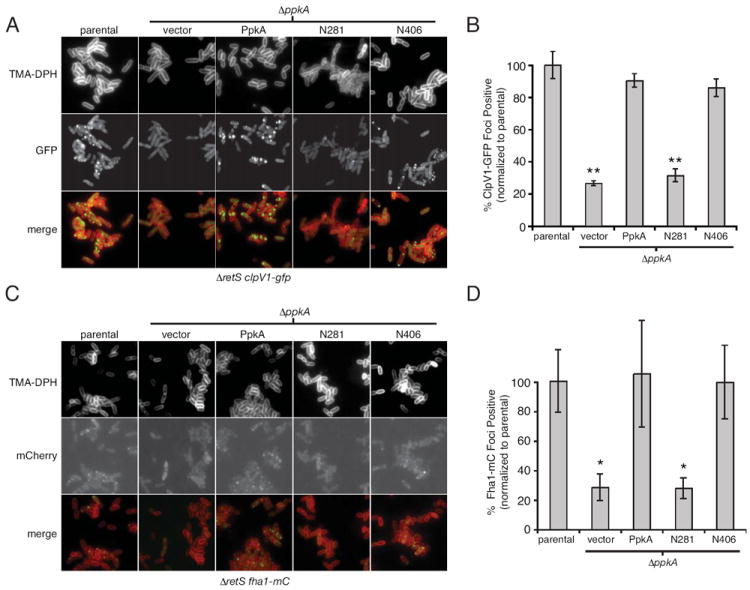
A and B. FM images (A) and quantitative analysis (B) of ClpV1-GFP localization in the indicated P. aeruginosa genetic backgrounds (parental strain is ΔretS clpV1-gfp). Data are normalized to the parental strain. See Experimental Procedures for explanation of quantification methods. TMA-DPH is a lipophilic dye that allows visualization of the bacterial membrane. *, P < 0.05; **, P < 0.005 C and D. Same as A and B, respectively, with ΔretS fha1-mC as the parental strain.
Given the shared biochemical pathway of PpkA and Fha1, we postulated that a likely structural role of PpkA could be to recruit Fha1 to the H1-T6S apparatus. In this scenario, the ClpV1 localization defect of ΔppkA might be indirectly mediated through a loss of Fha1 recruitment. Indeed, our prior work suggests such a hierarchy; we found that fha1 is required for proper ClpV1 localization, however clpV1 is not required for proper Fha1 localization (Mougous et al., 2007). To test the influence of PpkA on the localization properties of Fha1, we used FM to compare Fha1-mC localization in the presence and absence of PpkA. Physiologic regulation of the fusion protein was achieved by generating a chromosomally-encoded Fha1-mC fusion at the native fha1 locus in ΔretS (ΔretS fha1-mC). Interestingly, the absence of PpkA resulted in a dramatic loss of punctate Fha1-mC localization (Fig. 2C). Next, we asked whether the PpkA domain requirements for Fha1 localization mirror those required for ClpV1. Again we found that PpkA and N406, but not N281, were able to complement the Fha1-mC localization defect of ΔppkA (Fig. 2C and D). Blinded quantitative analysis of randomly selected FM fields was used to confirm these observations (Fig. 2D). These data support the hypothesis that N406 function is linked to essential structural roles played by the cytoplasmic and transmembrane domains of PpkA. This structural role appears to include the recruitment of Fha1 to the H1-T6S apparatus.
Fha1 resides in a stable complex with ClpV1
Matching PpkA domain requirements for Fha1 and ClpV1 recruitment to the H1-T6S apparatus led us to posit that these proteins reside in a single protein complex. We previously demonstrated that ClpV1 is strongly recruited to the apparatus when Fha1 becomes hyper-phosphorylated (in a strain lacking pppA) (Mougous et al., 2007), however Fha1 localization was not analyzed under these conditions. To obtain further evidence of Fha1 and ClpV1 association, we investigated whether these proteins co-localize in this highly activated state of the secretion system.
To gauge Fha1 and ClpV1 co-localization, we generated a strain in the ΔretS background carrying chromosomal in-frame translational fusions of DNA encoding mCherry and GFP to fha1 and clpV1, respectively (ΔretS fha1-mC clpV1-gfp). Consistent with our prior observations, Fha1-mC and ClpV1-GFP were found to co-localize to approximately 1-3 discrete foci within each cell (Fig. 3A) (Mougous et al., 2007). Introduction of the pppA deletion confirmed our earlier report that the PpkA-antagonistic activity of the phosphatase negatively regulates ClpV1-GFP recruitment to the T6S apparatus. The effect of ΔpppA on Fha1-mC recruitment was dramatic (Fig. 3A); average fluorescence intensity of Fha1-mC foci in ΔpppA was 2.0-fold that of the parental strain (Fig. 3B). Fha1-mC and ClpV1-GFP foci remained co-localized under these extreme conditions (Fha1 hyper-phosphorylation), indicating that Fha1 and ClpV1 localization is altered in a concerted fashion when the H1-T6SS progresses from the resting to activated state (Fig. 3A).
Fig. 3. ClpV1 and Fha1 interact in a stable protein complex.
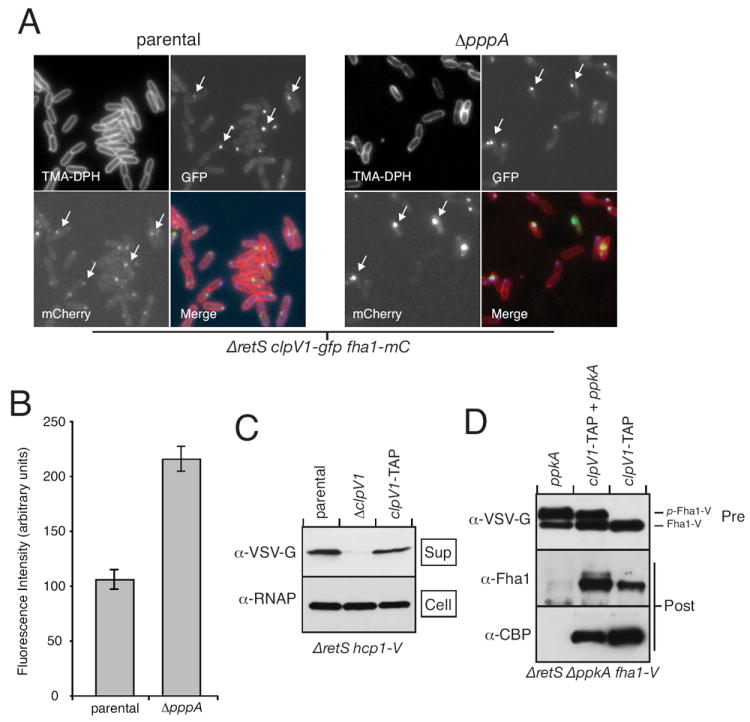
A. ClpV1 and Fha1 are hyper-recruited to the H1-T6S apparatus by deletion of pppA. Representative FM images of ClpV1-GFP (GFP) and Fha1-mC (mCherry) in the indicated P. aeruginosa genetic backgrounds. Arrows highlight the co-localization of foci containing these proteins.
B. Quantitative analysis of Fha1-mC foci intensity in the parental versus the ΔpppA background.
C. ClpV1 fusion to the TAP tag does not interrupt Hcp1 secretion. Western blot analysis of Hcp1 secretion from the indicated strains. An antibody specific to RNA polymerase (α-RNAP) is used in this and subsequent experiments as a loading control.
D. Fha1 associates with ClpV1-TAP. Western blot analysis of proteins before (Pre) and after (Post) TAPs from the indicated strains. α-CBP (calmodulin binding protein) recognized a TAP tag component that remains after the final purification step.
Next, we probed for a physical interaction between ClpV1 and Fha1 using TAP. Following the method of Dove and colleagues, we used a non-replicating vector and homologous recombination to generate a strain expressing a C-terminal fusion of ClpV1 to the TAP tag (ClpV1-TAP) at the native clpV1 locus (Vallet-Gely et al., 2005). Analysis of Hcp1 secretion in this strain showed that neither the function of ClpV1 nor other essential H1-T6S components was disrupted (Fig. 3C). To assess the contribution of PpkA to ClpV1-TAP interactions, we introduced a deletion of ppkA into this strain and performed TAPs in both the presence and absence of ectopically expressed PpkA. Western blot analysis of TAP eluents showed that Fha1 co-purified specifically with ClpV1-TAP (Fig. 3D). The association of these proteins did not strictly require PpkA, though the kinase reproducibly enhanced association. These data are consistent with our FM results and lead us to a model whereby ClpV1, and possibly other key H1-T6S components, are in complex with Fha1. Phosphorylation of Fha1 by PpkA recruits this complex to putative membrane-bound and periplasmic components of the apparatus.
PpkA autophosphorylation and dimerization contribute to triggering the H1-T6SS
The extracellular portions of many transmembrane kinases bind to a ligand and multimerize, which activates their cytoplasmic catalytic domain via autophosphorylation (Hubbard, 1999, Johnson et al., 1996, Schlessinger, 2000). Previously, we hypothesized that PpkA could act in an analogous manner to stimulate the H1-T6SS. Early experimental support for this hypothesis was provided by our observation that mutants in conserved phospho-ser/thr-interacting residues found in the FHA-domain of Fha1 cause a significant decrease in Hcp1 secretion (Mougous et al., 2007). Our finding that the extracellular domains of PpkA are not essential when the protein is ectopically expressed is also consistent with this hypothesis (Fig. 1). It is likely that PpkA activation is artificially induced by increased effective concentration, an effect well documented for similar kinases (Greenstein et al., 2007, Udo et al., 1995). Furthermore, the strain used in our truncation complementation studies lacks the antagonistic activity of PppA, thereby potentiating low-level activation.
Prior work has shown that PpkA is capable of autophosphorylation (Motley & Lory, 1999). The sites of phosphorylation, and the relevance to the activity of the enzyme, however, were not investigated. To test whether autophosphorylation of PpkA is important for T6S activation, we generated alanine substitutions at predicted sites of autophosphorylation within the activation loop of PpkA. Based on the conserved mechanism of Hanks-type kinases and the similarity of PpkA to a number of kinases that have had autophosphorylation sites determined experimentally, there was little ambiguity in these assignments (Fig. 4A). Three PpkA alleles, coding for PpkA-T158A, PpkA-T161A, and the double substitution mutant, were generated and introduced with the native enzyme into ΔpppA ΔppkA hcp1-V. Western blotting with α-p-Thr antibody was used to monitor levels of phosphorylated PpkA (p-PpkA) in the resulting strains. Neither the T158A nor T161A mutation had a significant effect on α-p-Thr reactivity, indicating that both sites are potential targets of autophosphorylation (Fig. 4B). This was confirmed by analysis of the double substitution mutant, which displayed a complete lack of autophosphorylation. Importantly, expression levels of the protein did not account for this observation. Function mirrored autophosphorylation; only the non-phosphorylated T158A/T161A PpkA mutant was incapable of complementing Hcp1 secretion and Fha1 phosphorylation. These data strongly suggest that autophosphorylation of PpkA is critical for H1-T6SS activation (Fig. 4B).
Fig. 4. Autophosphorylation and dimerization promote PpkA and H1-T6S activation.
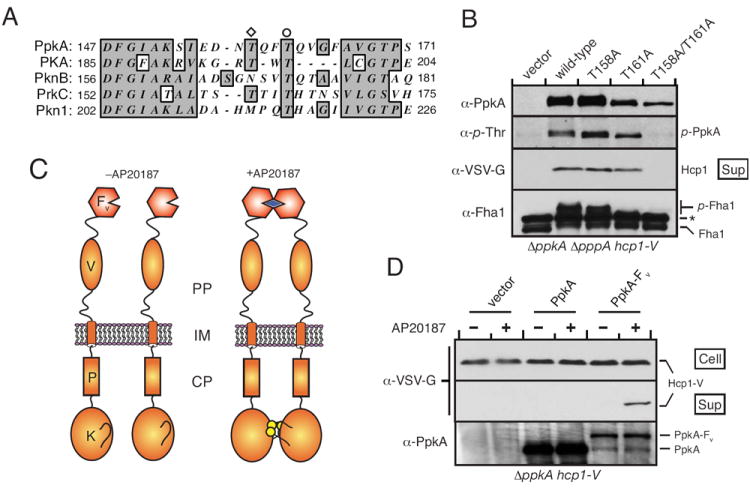
A. Partial multiple sequence alignment of PpkA with several characterized prokaryotic and eukaryotic kinases (PKA, NP_032880; PknB, NP_958950; PrkC, YP_001421154; Pkn1, YP_629724) highlighting conserved autophosphorylated residues, T158 (diamond) and T161 (circle).
B. Autophosphorylation of PpkA at two conserved Thr residues is required for Fha1 phosphorylation and Hcp1 secretion. Western blot analyses probing the activity and phosphorylation status of PpkA bearing either single or double alanine substitutions in T158 and T161. The band marked with an asterisk is a documented α-Fha1 cross-reactive species.
C. Schematic depiction of the topology of PpkA bearing a C-terminal fusion to Fv (PpkA-Fv) and the dimerization of this protein by AP20187. Intra-dimer autophosphorylation is implied, however inter-dimer autophosphorylation is also possible.
D. PpkA-Fv is specifically activated by AP20187. Where indicated, AP20187 (20 nM) was added to a ΔppkA strain containing empty vector or vector expressing either ppkA or ppkA-Fv. Western blot analyses of cell and supernatants samples from these strains indicated AP20187-dependent Hcp1 secretion only in PpkA-Fv-expressing cells.
In the absence of a known physiological ligand for PpkA, we turned to an artificial means for probing its mechanism of activation. We constructed a fusion of the C-terminus of PpkA to Fv (PpkA-Fv), an FK506 binding protein mutant, which homodimerizes in the presence of the rapamycin derivative AP20187 (Fig. 4C) (Amara et al., 1997, Yang et al., 2000). This system has been extensively used as a tool for achieving artificial inducible activation of kinases (Greenstein et al., 2007, Muthuswamy et al., 1999, Spencer et al., 1993), although to our knowledge it has never been applied to the study of a prokaryotic kinase in vivo. In these studies we avoided using either the retS or pppA mutations. In the absence of these mutations and an activation cue, PpkA levels are not sufficient to induce Hcp1 secretion (Mougous et al., 2007). We introduced the PpkA–Fv fusion expression construct and control constructs into ΔppkA hcp-V. As shown in Fig. 4D, AP20187 induced Hcp1 secretion in cells expressing PpkA-Fv, but the compound had no observable effect on cells expressing wild-type PpkA. Western blot analysis of the cellular fraction of these samples indicated that the effects of AP20187 on Hcp1 secretion were not caused by increased expression of Hcp1 or PpkA-Fv (Fig. 4D). Interestingly, PpkA-Fv levels were significantly lower than wild-type PpkA, further underscoring the extent of stimulation achieved by dimerization. These data suggest that PpkA activation under physiological conditions could be achieved by ligand recognition-induced dimerization followed by autophosphorylation. Due to the technical difficulty of detecting low levels of p-PpkA in the wild-type background, a direct in vivo link between PpkA dimerization and autophosphorylation has so far not been achieved.
Identification of TagR – a potentiator of PpkA activation
The signaling activity of eukaryotic transmembrane receptors and kinases can be affected by ligand recognition, however it can also be modulated by proteins that act as ligand co-receptors, or as inhibitors or potentiators of subsequent signaling (Fitzgerald et al., 2004, Ling et al., 2008, Polanska et al., 2008). Given the presence of a predicted protein-protein interaction motif (VWA) in the extracellular domains of PpkA and our data implicating these domains in ligand recognition, we hypothesized that PpkA signaling could be modulated by other components of the H1-T6SS. To this end, we pursued a bioinformatic approach to survey HSI-I ORFs for those encoding proteins or protein domains predicted to functionally interact with PpkA. This analysis led to the identification of tagR (type VI secretion associated gene R; PA0071), which encodes a single domain protein (DUF323, e value = 10-21) with closely related domains that are found fused to predicted periplasmic regions of prokaryotic ser/thr transmembrane kinases (see Discussion). Interestingly, tagR is part of a group of closely linked genes within HSI-I that flank ppkA and pppA. Like the ppkA and pppA, these genes are non-conserved components of T6S loci (Fig. 5A). These findings led us to question whether TagR could influence PpkA signaling.
Fig. 5. TagR acts upstream of PpkA to promote H1-T6SS activation.
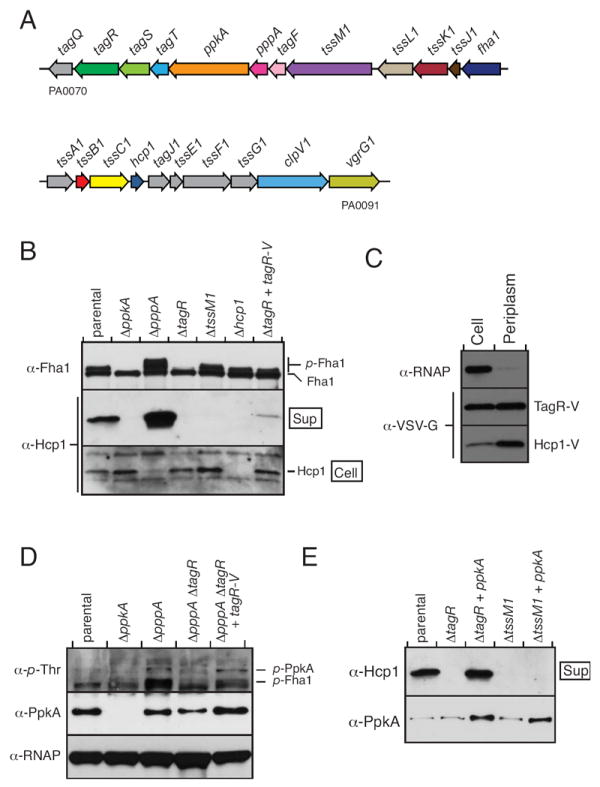
A. Overview of P. aeruginosa PAO1 HSI-I. See Fig. S2 for a description of updated HSI nomenclature.
B. TagR is required for Fha1 phosphorylation and Hcp1 secretion. Western blot analysis of Fha1 phosphorylation and Hcp1 secretion in the indicated P. aeruginosa strains (parental strain is ΔretS clpV1-gfp fha1-mC).
C. TagR is localized to the periplasm. Western blot analysis of TagR-V, Hcp1-V, and RNAP levels in the cell and periplasm fractions of P. aeruginosa PAO1 hcp1-V pPSV35CV-tagR (see methods). Equal proportions of cell and periplasm fractions were loaded so that relative protein distribution could be gauged directly.
D. TagR promotes PpkA autophosphorylation. Western blot analysis of p-PpkA levels and PpkA expression in the indicated strains. Parental genotype is same as (B).
E. PpkA expression overcomes the Hcp1 secretion defect of ΔtagR. Western blot analysis of Hcp1 secretion and PpkA expression in the indicated strains. Parental genotype is same as (B).
To investigate the function of TagR, we generated a deletion of tagR in ΔretS and assayed the effect of this deletion on two indicators of H1-T6SS activity: Hcp1 secretion and Fha1 phosphorylation. The deletion of tagR blocked Hcp1 secretion and significantly decreased Fha1 phosphorylation. Genetic complementation of ΔtagR via extrachromosomal ectopic expression of tagR fused to a sequence encoding the VSV-G epitope tag (tagR-V) returned Fha1 phosphorylation to approximately parental levels and partially restored Hcp1 secretion. The phenotype of ΔretS ΔtagR differed from a ΔretS strain containing a mutation in tssM1, which encodes a predicted structural component of the H1-T6SS. In ΔretS ΔtssM1, a similar Hcp1 secretion defect was observed, however p-Fha1 levels were not affected (Fig. 5B). These data are consistent with TagR performing a regulatory, rather than a structural role in the H1-T6S apparatus.
To gain insight into how TagR exerts its effect on H1-T6SS activity, we probed its localization using subcellular fractionation and Western blotting. As fractionation controls, we used RNA polymerase α (RNAP-α), a cytoplasmic protein, and Hcp1, which is localized to the periplasm when the H1-T6SS is not stimulated (Mougous et al., 2007). This experiment demonstrated specific localization of TagR to the periplasm (Fig. 5C). In support of these findings, sequence analysis of TagR identified a predicted N-terminal Sec signal in the protein. Of note, secreted TagR was not detected (data not shown).
The localization of TagR and our bioinformatic analysis linking the protein to ser/thr kinases led us to hypothesize that TagR regulates H1-T6S activity by influencing PpkA. Low levels of p-Fha1 in ΔtagR could reflect a function of TagR in promoting PpkA activation. As shown above, the activation state of PpkA is positively correlated to its autophosphorylation state (Fig. 4B); therefore, we investigated whether a deletion in tagR affects p-PpkA levels. Since PpkA is under native regulation in this experiment (PpkA alleles were over-expressed for Fig. 4B), only a faint band corresponding to p-PpkA was visualized in ΔretS (Fig. 5D). The additional deletion of pppA significantly improved the p-PpkA signal, thereby allowing us to assess the contribution of TagR. Intriguingly, deletion of tagR in ΔretS ΔpppA lowered levels of p-PpkA to approximately those found in ΔretS (Fig. 5D). Expression of tagR-V in ΔpppA ΔtagR fully complemented the p-PpkA phenotype. A band corresponding to p-Fha1 was also observed in this experiment. As expected, the abundance of p-Fha1 mirrored that of p-PpkA in the various genetic backgrounds (Fig. 5D). These data suggest that TagR is required for efficient PpkA autophosphorylation.
We reasoned that if TagR promotes HSI-I functionality by facilitating PpkA activation, then artificial activation of PpkA by over-expression of the enzyme should reverse the Hcp1 secretion defect of ΔtagR. On the contrary, PpkA over-expression in a strain lacking a H1-T6S structural component such as TssM1, should not complement Hcp1 secretion. To test this, PpkA levels were increased by introduction of a plasmid ectopically expressing the enzyme in ΔretS strains containing tagR and tssM1 deletions. PpkA expression fully complemented Hcp1 secretion in the ΔtagR background, yet it has no effect on Hcp1 secretion in the ΔtssM1 background (Fig. 5E). These results indicate that TagR functions upstream of PpkA. TagR could function independently of PpkA, or it could act in conjunction with the C-terminal domains of the kinase to efficiently recognize and signal in response to H1-T6SS activation cue(s).
Discussion
This study has significantly broadened our understanding of the mechanism and components of the H1-T6S posttranslational activation pathway. We showed that residues of PpkA extending from the N-terminal kinase domain through the transmembrane segment are essential for Fha1 phosphorylation and H1-T6S activation (Fig. 1). Interestingly, these domains were also found to be essential for ClpV1 and Fha1 localization to the H1-T6S apparatus (Fig. 2). Taken together with our earlier findings that phosphorylation of Fha1 by PpkA is not strictly required for localization of either Fha1 or ClpV1 to the apparatus, we postulate that a second function of PpkA is to facilitate docking of Fha1 onto the H1-T6S apparatus. These data imply that this structural role of PpkA supercedes its catalytic role (N281 cannot phosphorylate Fha1); therefore only docked Fha1 is a competent substrate for PpkA. Our ability to observe and isolate a stable protein complex containing Fha1 and ClpV1 – independent of PpkA – that is recruited to the H1-T6S apparatus upon activation further supports this hypothesis (Fig. 3).
We previously showed that the ATP hydrolytic activity of a single AAA+ domain of ClpV1 is required for Hcp1 secretion (Mougous et al., 2006). Building on our understanding of the role of this ATPase in T6S function, Bonemann and colleagues demonstrated that TssB (VipA) and TssC (VipB) homologs from V. cholerae spontaneously assemble into a tubule-like structure, which can be disassembled by ClpV in an ATP-dependent manner. The authors proposed this structure and its remodeling is relevant to T6SS biogenesis. These findings, combined with our observation that Fha1 is constitutively associated with ClpV1 (Fig. 3D), lead us to propose that T6SS activation via phosphorylation could proceed by p-Fha1 allosteric activation of the ATP hydrolytic activity of ClpV1. Many T6SSs do not appear to utilize a threonine phosphorylation-based posttranslational regulatory system. Some of these, including the system in V. cholerae, nonetheless possess an apparent FHA domain protein (VC_A0112). In these instances, the activity of ClpV may be modulated by other factors, or by the FHA-domain protein responding to other inputs. There are also a multitude of T6SSs that do not encode any elements of the posttranslational regulatory pathway, including the FHA domain protein. It is therefore likely that the FHA domain protein and the posttranslational regulatory pathway are specialized adaptations that allow a given T6SS to respond to relevant stimuli.
By manipulating PpkA oligomerization with a small molecule, we demonstrated that dimerization of the extracellular domains of PpkA can promote activation of the H1-T6SS (Fig. 4C & D). Autophosphorylation of PpkA upon dimerization is likely key to the T6S activation process, as autophosphorylation-deficient mutants cannot phosphorylate Fha1 and cannot activate the secretion system (Fig. 4A & B). To our knowledge, these are the first data providing in vivo evidence that a prokaryotic transmembrane Hanks-type kinase initiates signaling by multimerization. It remains unclear whether PpkA activation is achieved by intra- versus inter-dimer autophosphorylation (Greenstein et al., 2007).
Guided by the growing list of shared properties of the H1-T6S posttranslational signaling pathway and more complex eukaryotic signaling pathways, we searched for additional factors that could modulate PpkA activity. This search identified TagR, which we found functions upstream of PpkA to promote efficient autophosphorylation of the kinase (Fig. 5). TagR is a member of the protein family that includes human sulfatase modifying factor SUMF1. Members of this enzyme family catalyze the oxidation of a cysteine residue to formyl glycine in the active site of sulfatases (Carlson et al., 2008, Cosma et al., 2003, Dierks et al., 2005, Dierks et al., 2003). Analysis of the sequence of TagR suggests that due to a lack of conserved catalytic residues, this protein is unlikely to perform this function in the H1-T6SS (data not shown). In many organisms, including Myxococcus xanthus and Plesiocystis pacifica, TagR-homologous domains are found at the C-termini of large ser/thr protein kinases. Given that TagR contains a Sec signal sequence and it seems to function upstream of PpkA, it is feasible that this protein functions as a co-receptor for the H1-T6S activation signal.
The genes encoding PpkA, PppA, and TagR are found in a cluster of genes that are not well conserved in other T6S loci (Fig. 5A, tag genes). It is tempting to speculate that the products of these uncharacterized genes also participate in posttranslational regulation of the H1-T6SS. Indeed, apparent orthologs of one of these genes, tagF, are translationally fused to apparent PppA orthologs in several T6S loci (Bingle et al., 2008). It is conceivable that TagF and TagF-like domains fused to phosphatases receive inputs that modulate the activity or specificity of their cognate phosphatases. This is well precedented, and even the norm for certain families of eukaryotic phosphatases (Moorhead et al., 2009). Two other genes in this cluster, tagS and tagT, appear to encode the minimal subunits of a functioning Lol-like lipoprotein transport/sorting system (56% and 40% identity with P. aeruginosa LolE and D, respectively) (Narita & Tokuda, 2006). The housekeeping copy of this system is essential in E. coli, where it uses ATP hydrolysis to sort lipoproteins destined for the outer membrane (Narita et al., 2004). TagS and TagT could be specialized to ensure the transport of a T6-specific lipoprotein. The importance of outer-membrane lipoprotein transport for T6S was recently demonstrated in entero-aggregative E. coli (Aschtgen et al., 2008), and HSI-I encodes at least two predicted lipoproteins (TagQ and TssJ1). T6S lipoproteins may serve a variety of roles, ranging from solely structural functions to mediating specific interactions with host cells.
Our current study of the posttranslational regulatory system that governs P. aeruginosa H1-T6SS activation has facilitated a number of insights into the mechanism, and functional and structural interactions of the system. By combining these data with our previous observations and the recent findings of others in the field, we have constructed a revised model for activation of the secretion apparatus (Fig. 6). Future investigations of posttranslational regulation of T6S in a broad host of organisms should lead to a greater mechanistic understanding of the many reported functions of this system.
Fig. 6. A model for structural organization and activation of the H1-T6SS.
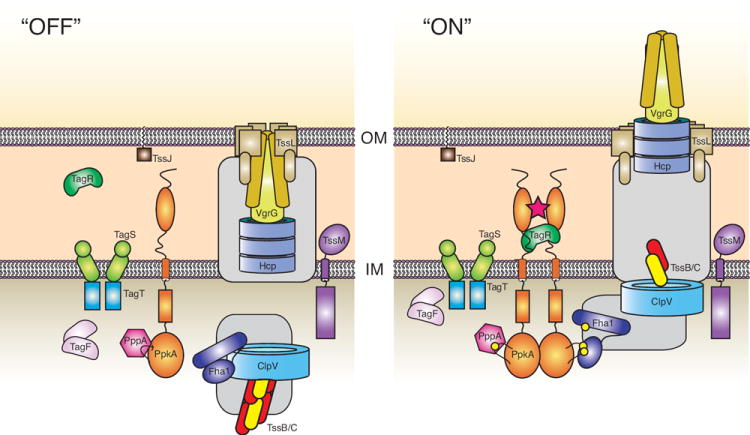
Proteins are colored according to the ORFs in Fig. 5A. Organization of the model is based on our current and previous work (Ballister et al., 2008, Mougous et al., 2006, Mougous et al., 2007), and recent reports from several other laboratories (Cascales, 2008, Filloux et al., 2008, Pukatzki et al., 2009). Most notably are the structure-function relationship of Hcp1 and VgrG (note – a secreted VgrG homolog has not been identified in Pseudomonas aeruginosa) to the T4 bacteriophage tail spike (Leiman et al., 2009, Pell et al., 2009, Filloux, 2009), the presence of the TssJ lipoprotein in the outer membrane (Aschtgen et al., 2008), and the remodeling of TssB/C oligomers by ClpV1 (Bonemann et al., 2009). The role of ClpV1 in system assembly and/or substrate translocation remains undetermined. Owing to a lack of published experimental data, we were unable to place several T6S proteins (gray boxes). The oligomeric state of TagF is based on the unpublished crystal structure of the protein (PDB entry 2QNU). TagS and TagT are configured in a manner consistent with their strong homology to the Lol lipoprotein ABC transport system (Narita & Tokuda, 2006). Sites of phosphorylation are represented by yellow spheres. The star represents a proposed signal that initiates the activation of the secretion system.
Experimental procedures
Bacterial strains, plasmids and growth conditions
The P. aeruginosa strains used in this study were derived from the sequenced strain PA01 (Stover et al., 2000). P. aeruginosa were grown on Luria-Bertani (LB) medium at 37°C supplemented with 30 μg ml-1 gentamicin, 25 μg ml-1 irgasan, 5% w/v sucrose, and 0.5 mM IPTG as required. E. coli SM10, used for conjugation with P. aeruginosa, was grown in LB medium containing 10 μg ml-1 gentamicin. Plasmids pPSV35 and pEXG2, used for inducible expression and gene deletion construction, respectively, have been reported previously (Rietsch et al., 2005). Plasmid pPSV35CV, also used for inducible expression (PpkA, PpkA truncations, PpkA autophosphorylation mutants) and complementation (TagR), was constructed by inserting a sequence encoding the VSV-G epitope into the XbaI site of pPSV35 using an oligonucleotide linker encoded by forward reading 5′-CTAGATATACAGATATTGAAATGAATAGATTAGGAAAATGAG and reverse reading 5′-CTAGCTCATTTTCCTAATCTATTCATTTCAATATCTGTATAT. Unless otherwise stated, the full-length ORF and native ribosome binding site were inserted in-frame with the XbaI site of this plasmid, thereby encoding a Ser-Arg linker before the VSV-G epitope. Expression was verified by α-VSV-G western blotting. Mutagenesis was performed using the quikChange site directed mutagenesis kit (Stratagene) following the instructions provided by the manufacturer. All mutations were verified by DNA sequencing.
Construction of P. aeruginosa chromosomal mutations
All deletions were in-frame and were constructed by allelic replacement using pEXG2. Deleted alleles were generated from PCR-amplified sequences flanking the gene of interest using splicing by overlap extension (SOE) (Horton et al., 1993) as described previously (Mougous et al., 2006). TagS and tagR overlap by 8 nucleotides; therefore, the deletion of tagR includes only codons 22 through the penultimate codon so as to avoid disrupting the function of the protein encoded by tagS. The resulting plasmids were transformed into E. coli SM10 and conjugated into appropriate strains. Homologous recombinants were selected as described by Ferrández et al. (Ferrandez et al., 2002). Mutant candidates were screened by PCR. Strains containing chromosomal fluorescent fusions to gfp and mCherry were generated by allelic replacement. The construction of clpV1-gfp was described previously (Mougous et al., 2006). To generate fha1-mCherry, a sequence encoding a short linker (Ser-Arg-Ala3) followed by mCherry (Shaner et al., 2004) was inserted after the penultimate codon of fha1, thus recapitulating our prior fusion of Fha1 to this protein (Mougous et al., 2007).
Preparation of proteins and Western blotting
Overnight cultures of strains used for Western blots were back-diluted into 2 ml LB (1:1000) with appropriate additives. The samples were grown at 37°C with shaking and harvested by centrifugation (10,000 r.p.m. for 3 min) at mid-log phase. The cell pellets were re-suspended in 100 μl of a buffer containing 0.5M NaCl, 50mM Tris 7.5 and 10% Glycerol, and mixed 1:2 with SDS-PAGE sample loading buffer. To collect extracellular proteins, 1.4 ml of the supernatant from each sample was taken from the initial centrifugation step and subjected to a second centrifugation step in order to further contaminating bacterial cells. Trichloroacetic acid (100% w/v) was added to the supernatant at a final concentration of 10% v/v and centrifuged at 4°C for 30 min at 13,500 r.p.m. The protein pellets were washed once with acetone and re-suspended in 20 μl 2x SDS-PAGE loading buffer. Western blotting was carried out as described previously (Mougous et al., 2006). For anti-phosphothreonine Western blotting, 3% BSA in Tris-buffered saline containing 0.05% Tween 20 was used for blocking and the primary antibody was used at a 1:200 dilution (Zymed Laboratories). α-CBP was obtained from Millipore.
Subcellular fractionation
A 2 ml culture was grown at 37°C with aeration in LB supplemented with IPTG (0.25mM) to an OD600 of 1.0. The sample was centrifuged (6,000 r.p.m. for 3 min) and the supernatant was processed as described above. The periplasmic fraction was isolated using PeriPreps™, as described by the manufacturer (Epicentre Biotechnologies).
Tandem Affinity Purification
Cells were grown at 37°C with aeration in 50 ml of LB supplemented with carbenicillin (300 μg/mL), gentamicin (30 μg/mL) and IPTG (0.5 mM) in 250 ml flasks to an OD600 of 1.0. Cells were harvested by centrifugation at 4°C and TAP was then performed essentially as described by Rietsch et al. (Rietsch et al., 2005, Puig et al., 2001). Minor differences with this protocol were the addition of lysozyme (1 mg/mL) and Triton X-100 (1% w/v) to Buffer 1 (TAP lysis buffer).
Fluorescence microscopy and statistical analysis
Strains were grown in a similar manner as described for Western blotting. Identically prepared mid log-phase cultures were harvested by gentle centrifugation, washed with phosphate-buffered saline, and resuspended to an optical density (600 nm) of 5 with PBS containing 0.5 mM TMA-DPH (Molecular Probes). 3 μl of the cell suspension was spotted on the surface of agarose pads (1.0% agarose PBS). Images were acquired with a Nicon 80i microscope equipped with 100× PlanApochromat objective (numerical aperture, 1.4), and were recorded with a CoolSnap HQ camera (Photometrics). The imaging system was operated with Metamorph 6.3r2 software. Filter sets were purchased from Chroma Technology Corp. All images that were compared with each other were manipulated (brightness and contrast adjustments and resizing of files) identically. For statistical analysis of foci, cells were chosen (69 ≤ n ≤ 466) and counted in a blinded fashion from three randomly selected phase contrast fields. Foci in the selected cells were then enumerated using GFP and/or mCherry images. Statistical significance was assessed using the unpaired Student’s t-test. For fluorescence intensity measurements, phase-contrast images were again used as a reference point to select well-isolated single cells (n = 30) within three microscopic fields. Fluorescence images were then analyzed using Metamorph software to record maximal pixel brightness in foci-positive cells.
FKBP homodimerization
To generate PpkA-Fv, the sequence encoding Fv (FKBP-Phe36Val) was amplified from plasmid pC4-Fv1E (from ARGENT Homodomerization Kit) and inserted after the VSV-G coding sequence in pPSV35CV-ppkA. Overnight cultures were diluted 1:1000 into 2 ml LB medium containing gentamicin, IPTG [200 μM], and AP20187 [20nM] where indicated. Cultures were incubated with shaking and harvested in log phase at equivalent optical density. Cellular and extracellular fractions were prepared and Western blotting was performed as described above.
Supplementary Material
Acknowledgments
The authors wish to thank Jason Rush for guidance in the sequence analysis of TagR, Carrie Harwood and members of the Harwood laboratory for helpful suggestions and assistance with fluorescence microscopy, members of the Mougous laboratory for valuable scientific insights, and Ariad™ for generously providing regulated homodimerization reagents.
References
- Amara JF, Clackson T, Rivera VM, Guo T, Keenan T, Natesan S, Pollock R, Yang W, Courage NL, Holt DA, Gilman M. A versatile synthetic dimerizer for the regulation of protein-protein interactions. Proc Natl Acad Sci U S A. 1997;94:10618–10623. doi: 10.1073/pnas.94.20.10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschtgen MS, Bernard CS, De Bentzmann S, Lloubes R, Cascales E. SciN is an outer membrane lipoprotein required for Type VI secretion in enteroaggregative Escherichia coli. J Bacteriol. 2008 doi: 10.1128/JB.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert DF, Flannagan RS, Valvano MA. A Novel Sensor Kinase-Response Regulator Hybrid Controls Biofilm Formation and Type VI Secretion System Activity in Burkholderia cenocepacia. Infection and immunity. 2008 doi: 10.1128/IAI.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballister ER, Lai AH, Zuckermann RN, Cheng Y, Mougous JD. In Vitro Self-Assembly of Tailorable Nanotubes from a Simple Protein Building Block. Proc Natl Acad Sci U S A. 2008;105:3733–3738. doi: 10.1073/pnas.0712247105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingle LE, Bailey CM, Pallen MJ. Type VI secretion: a beginner’s guide. Current opinion in microbiology. 2008;11:3–8. doi: 10.1016/j.mib.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Bonemann G, Pietrosiuk A, Diemand A, Zentgraf H, Mogk A. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. The EMBO journal. 2009;28:315–325. doi: 10.1038/emboj.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, Hiatt P, McCoy K, Castile R, Smith AL, Ramsey BW. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis. 2001;183:444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- Carlson BL, Ballister ER, Skordalakes E, King DS, Breidenbach MA, Gilmore SA, Berger JM, Bertozzi CR. Function and structure of a prokaryotic formylglycine-generating enzyme. The Journal of biological chemistry. 2008;283:20117–20125. doi: 10.1074/jbc.M800217200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales E. The type VI secretion toolkit. EMBO reports. 2008;9:735–741. doi: 10.1038/embor.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell. 2003;113:435–444. doi: 10.1016/s0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- Dudley EG, Thomson NR, Parkhill J, Morin NP, Nataro JP. Proteomic and microarray characterization of the AggR regulon identifies a pheU pathogenicity island in enteroaggregative Escherichia coli. Mol Microbiol. 2006;61:1267–1282. doi: 10.1111/j.1365-2958.2006.05281.x. [DOI] [PubMed] [Google Scholar]
- Ferrandez A, Hawkins AC, Summerfield DT, Harwood CS. Cluster II che genes from Pseudomonas aeruginosa are required for an optimal chemotactic response. J Bacteriol. 2002;184:4374–4383. doi: 10.1128/JB.184.16.4374-4383.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux A. The type VI secretion system: a tubular story. The EMBO journal. 2009;28:309–310. doi: 10.1038/emboj.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux A, Hachani A, Bleves S. The bacterial type VI secretion machine: yet another player for protein transport across membranes. Microbiology (Reading, England) 2008;154:1570–1583. doi: 10.1099/mic.0.2008/016840-0. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD-2 complex. Microbes and Infection. 2004;6:1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004;7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiological reviews. 1996;60:539–574. doi: 10.1128/mr.60.3.539-574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein AE, Echols N, Lombana TN, King DS, Alber T. Allosteric activation by dimerization of the PknD receptor Ser/Thr protein kinase from Mycobacterium tuberculosis. The Journal of biological chemistry. 2007;282:11427–11435. doi: 10.1074/jbc.M610193200. [DOI] [PubMed] [Google Scholar]
- Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, Pease LR. Gene splicing by overlap extension. Methods in enzymology. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Structural analysis of receptor tyrosine kinases. Progress in biophysics and molecular biology. 1999;71:343–358. doi: 10.1016/s0079-6107(98)00047-9. [DOI] [PubMed] [Google Scholar]
- Hunt TA, Kooi C, Sokol PA, Valvano MA. Identification of Burkholderia cenocepacia genes required for bacterial survival in vivo. Infection and immunity. 2004;72:4010–4022. doi: 10.1128/IAI.72.7.4010-4022.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Lam J, Chan R, Lam K, Costerton JW. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infection and immunity. 1980;28:546–556. doi: 10.1128/iai.28.2.546-556.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc Natl Acad Sci U S A. 2009;106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2008 doi: 10.1016/j.gene.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Ma AT, McAuley S, Pukatzki S, Mekalanos JJ. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell host & microbe. 2009;5:234–243. doi: 10.1016/j.chom.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May TB, Shinabarger D, Maharaj R, Kato J, Chu L, DeVault JD, Roychoudhury S, Zielinski NA, Berry A, Rothmel RK, et al. Alginate synthesis by Pseudomonas aeruginosa: a key pathogenic factor in chronic pulmonary infections of cystic fibrosis patients. Clinical microbiology reviews. 1991;4:191–206. doi: 10.1128/cmr.4.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead GB, De Wever V, Templeton G, Kerk D. Evolution of protein phosphatases in plants and animals. The Biochemical journal. 2009;417:401–409. doi: 10.1042/BJ20081986. [DOI] [PubMed] [Google Scholar]
- Motley ST, Lory S. Functional characterization of a serine/threonine protein kinase of Pseudomonas aeruginosa. Infection and immunity. 1999;67:5386–5394. doi: 10.1128/iai.67.10.5386-5394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougous JD, Gifford CA, Ramsdell TL, Mekalanos JJ. Threonine phosphorylation post-translationally regulates protein secretion in Pseudomonas aeruginosa. Nature cell biology. 2007;9:797–803. doi: 10.1038/ncb1605. [DOI] [PubMed] [Google Scholar]
- Murray TS, Egan M, Kazmierczak BI. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Current opinion in pediatrics. 2007;19:83–88. doi: 10.1097/MOP.0b013e3280123a5d. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Gilman M, Brugge JS. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Molecular and cellular biology. 1999;19:6845–6857. doi: 10.1128/mcb.19.10.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita S, Matsuyama S, Tokuda H. Lipoprotein trafficking in Escherichia coli. Archives of microbiology. 2004;182:1–6. doi: 10.1007/s00203-004-0682-4. [DOI] [PubMed] [Google Scholar]
- Narita S, Tokuda H. An ABC transporter mediating the membrane detachment of bacterial lipoproteins depending on their sorting signals. FEBS letters. 2006;580:1164–1170. doi: 10.1016/j.febslet.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Pallen M, Chaudhuri R, Khan A. Bacterial FHA domains: neglected players in the phospho-threonine signalling game? Trends Microbiol. 2002;10:556–563. doi: 10.1016/s0966-842x(02)02476-9. [DOI] [PubMed] [Google Scholar]
- Parsek MR, Greenberg EP. Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol. 2005;13:27–33. doi: 10.1016/j.tim.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Pell LG, Kanelis V, Donaldson LW, Howell PL, Davidson AR. The phage lambda major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc Natl Acad Sci U S A. 2009;106:4160–4165. doi: 10.1073/pnas.0900044106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanska UM, Fernig DG, Kinnunen T. Extracellular interactome of the FGF receptor-ligand system: Complexities and the relative simplicity of the worm. Dev Dyn. 2008;238:277–293. doi: 10.1002/dvdy.21757. [DOI] [PubMed] [Google Scholar]
- Potvin E, Lehoux DE, Kukavica-Ibrulj I, Richard KL, Sanschagrin F, Lau GW, Levesque RC. In vivo functional genomics of Pseudomonas aeruginosa for high-throughput screening of new virulence factors and antibacterial targets. Environmental microbiology. 2003;5:1294–1308. doi: 10.1046/j.1462-2920.2003.00542.x. [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods San Diego, Calif. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A. 2007;104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pukatzki S, McAuley SB, Miyata ST. The type VI secretion system: translocation of effectors and effector-domains. Current opinion in microbiology. 2009;12:11–17. doi: 10.1016/j.mib.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2005;102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MA, Ulrich RL, Ribot WJ, Brueggemann EE, Hines HB, Chen D, Lipscomb L, Kim HS, Mrazek J, Nierman WC, Deshazer D. Type VI secretion is a major virulence determinant in Burkholderia mallei. Mol Microbiol. 2007;64:1466–1485. doi: 10.1111/j.1365-2958.2007.05734.x. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schuster M, Greenberg EP. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol. 2006;296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- Smith RS, Iglewski BH. P. aeruginosa quorum-sensing systems and virulence. Current opinion in microbiology. 2003;6:56–60. doi: 10.1016/s1369-5274(03)00008-0. [DOI] [PubMed] [Google Scholar]
- Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Suarez G, Sierra JC, Sha J, Wang S, Erova TE, Fadl AA, Foltz SM, Horneman AJ, Chopra AK. Molecular characterization of a functional type VI secretion system from a clinical isolate of Aeromonas hydrophila. Microb Pathog. 2007 doi: 10.1016/j.micpath.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udo H, Munoz-Dorado J, Inouye M, Inouye S. Myxococcus xanthus, a gram-negative bacterium, contains a transmembrane protein serine/threonine kinase that blocks the secretion of beta-lactamase by phosphorylation. Genes & development. 1995;9:972–983. doi: 10.1101/gad.9.8.972. [DOI] [PubMed] [Google Scholar]
- Vallet-Gely I, Donovan KE, Fang R, Joung JK, Dove SL. Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2005;102:11082–11087. doi: 10.1073/pnas.0502663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox MD, Zhu H, Conibear TC, Hume EB, Givskov M, Kjelleberg S, Rice SA. Role of quorum sensing by Pseudomonas aeruginosa in microbial keratitis and cystic fibrosis. Microbiology (Reading, England) 2008;154:2184–2194. doi: 10.1099/mic.0.2008/019281-0. [DOI] [PubMed] [Google Scholar]
- Wu HY, Chung PC, Shih HW, Wen SR, Lai EM. Secretome analysis uncovers an Hcp-family protein secreted via a type VI secretion system in Agrobacterium tumefaciens. J Bacteriol. 2008;190:2841–2850. doi: 10.1128/JB.01775-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahr TL. A critical new pathway for toxin secretion? N Engl J Med. 2006;355:1171–1172. doi: 10.1056/NEJMcibr063931. [DOI] [PubMed] [Google Scholar]
- Yang W, Rozamus LW, Narula S, Rollins CT, Yuan R, Andrade LJ, Ram MK, Phillips TB, van Schravendijk MR, Dalgarno D, Clackson T, Holt DA. Investigating protein-ligand interactions with a mutant FKBP possessing a designed specificity pocket. Journal of medicinal chemistry. 2000;43:1135–1142. doi: 10.1021/jm9904396. [DOI] [PubMed] [Google Scholar]
- Zeng L. PhD Thesis: Retrieved August 05, 2008 from University of Florida Libraries - Electronic Theses & Disserations. 2004. Pseudomonas aeruginosa Pathogenecity and Antibiotic Resistance. [Google Scholar]
- Zheng J, Leung KY. Dissection of a type VI secretion system in Edwardsiella tarda. Mol Microbiol. 2007;66:1192–1206. doi: 10.1111/j.1365-2958.2007.05993.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.