

Abstract
Objective
Impaired endothelium-dependent arteriolar dilation in mice fed high salt is due to local oxidation of nitric oxide (NO) by superoxide anion (O2-). We explored the possibility that “uncoupled” endothelial nitric oxide synthase (eNOS) is the source of this O2-.
Methods
Levels of L-arginine (L-Arg), tetrahydrobiopterin (BH4) and O2- (hydroethidine oxidation) were measured in spinotrapezius muscle arterioles of mice fed normal salt (0.45%, NS) or high salt (4%, HS) diets for 4 weeks, with or without dietary L-Arg supplementation. The contribution of NO to endothelium-dependent dilation was determined from the effect of Nω-nitro-L-arginine methyl ester (L-NAME) on responses to acetylcholine (ACh).
Results
Arterioles in HS mice had lower [BH4] and higher O2- levels than those in NS mice. ACh further increased arteriolar O2- in HS mice only. L-Arg supplementation prevented the reduction in [BH4] in arterioles of HS mice, and O2- was not elevated in these vessels. Compared to NS mice, arteriolar ACh responses were diminished and insensitive to L-NAME in HS mice, but not in HS mice supplemented with L-Arg.
Conclusions
These findings suggest that eNOS uncoupling due to low [BH4] is responsible for O2- generation and reduced NO-dependent dilation in arterioles of mice fed a high salt diet.
Keywords: microcirculation, dietary salt, nitric oxide, tetrahydrobiopterin, superoxide anion, L-arginine, mouse
Introduction
High dietary salt intake, even in the absence of hypertension, can result in impaired arteriolar responses to hypoxia [20], transmural (myogenic) stretch [31], and vasoactive stimuli that normally rely on endothelial nitric oxide (NO) release [4,5,17-19]. Because metabolic, myogenic, and endothelium-dependent stimuli can all play a role in the moment-to-moment regulation of tissue blood flow, these salt-dependent changes in arteriolar function can have profound physiological relevance [6,21].
Our previous studies indicate that impaired NO-dependent responses in rats and mice fed high salt are due to oxidation of NO in the arteriolar wall by superoxide anion (O2-) or one of its immediate metabolites [17,18,32]. A strong candidate for the source of this O2- is endothelial nitric oxide synthase (eNOS), which can generate large amounts of O2- instead of NO if the normal flow of electrons from its heme core to L-Arginine (L-Arg) is diverted to molecular oxygen --a condition widely referred to as “uncoupling” [8,37]. In support of this possibility, we have recently found that in mice fed high salt, NOS inhibition completely abolishes steady-state arteriolar O2- accumulation and also prevents the increase in these O2- levels that can be triggered by ACh [32].
Tetrahydrobiopterin (BH4) is an essential cofactor for eNOS, and as such BH4 bioavailability plays a central role in the regulation of endothelial NO production. An increase in BH4 synthesis or availability leads to increased NO production [2,9,25], whereas a reduction in BH4 availability is one of the primary causes of eNOS uncoupling [8,37]. In the current study, we studied the relationships among dietary salt, vascular wall BH4 and O2- levels, and NO-dependent dilation to further investigate the contribution of uncoupled eNOS to the impaired endothelium-dependent dilation of arterioles after ingestion of a high salt diet.
Materials and Methods
All surgical and experimental procedures were approved by the West Virginia University Animal Care and Use Committee. Ten-week old inbred male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were placed on a whole-grain diet containing either 0.45% NaCl (normal salt, NS) or 4% NaCl (high salt, HS) (TD88311 and TD92100 diets; Teklad, Madison, WI). We have previously shown that these mice remain normotensive when placed on a high salt diet [30]. In subsets of mice from each dietary group, the drinking water also contained L-Arg (1.51% arginine-HCl). Dietary L-Arg supplementation by this method has been found to increase not only endothelial cell L-Arg levels, but also endothelial BH4 levels, leading to increased NO synthesis [13]. Other mice received drinking water containing L-alanine (2.55%) to serve as isonitrogenous controls for the mice receiving L-Arg. All mice were studied 4-5 weeks after being placed on their respective diets/drinking water regimens.
Assessment of arteriolar wall superoxide levels and endothelium-dependent dilation
Each mouse was anesthetized with sodium thiopental (50 mg/kg, i.p.) and placed on a heating pad to maintain a 37°C core temperature. The right spinotrapezius muscle was exteriorized for microscopic observation leaving all innervation and feed vessels intact and undisturbed. Throughout this surgery and subsequent procedures, the muscle was continuously superfused with an electrolyte solution (119 mM NaCl, 25 mM NaHCO3, 6 mM KCl and 3.6 mM CaCl2) warmed to 35°C and equilibrated with 95% N2-5% CO2 (pH=7.35-7.40). Superfusate flow rate was maintained at 4-6 ml/min to minimize equilibration with atmospheric O2 [7]. After muscle exteriorization, the mouse was transferred to the stage of an Olympus BHMJ microscope (Hyde Park, NY) fitted with a color CCD video camera (Dage-MTI, Michigan City, IN). Images were displayed on a high-resolution video monitor and stored on videotape for later analysis. Observations were made at final magnification of 1460X. For assessment of endothelium-dependent dilation, arteriolar inner diameters were measured during videotape replay with a video caliper (Cardiovascular Research Institute, Texas A&M University).
Arteriolar wall O2- levels were assessed by the hydroethidine (HE) oxidation assay [21,32]. Because it is not oxidized by H2O2, HOCl, ONOO- or O2, HE is considered to be relatively specific for the detection of O2- [3]. HE permeates cell membranes and, when oxidized by O2-, is converted to fluorescent ethidium bromide. HE (1 × 10-3 M) was added to the superfusate, and the muscle was continuously exposed to this solution for 30 minutes in complete darkness to prevent HE photo-destruction. In some preparations, the effect of increased eNOS activity on O2- production was assessed by also adding 1× 10-5 M ACh to the HE-containing superfusate. After this HE loading period, the muscle was rinsed with the normal superfusate for 15 minutes to remove extracellular HE before measuring fluorescence levels. Each vessel was selected for measurement using transillumination and standard brightfield microscopy, with the focal plane set at the center of the vessel. The inner (luminal) and outer edges of the vessel wall were clearly visible at the magnification we used (see above), but the practical resolution of our system under in-vivo conditions did not permit a clear distinction between the endothelium and adjacent smooth muscle layers. An image of the vessel was obtained during brief (1 s) epi-illumination of the muscle with a mercury lamp, using excitation and emission filters for detection of ethidium bromide fluorescence (480-550 nm bandpass, 590 nm barrier). Using a photometric window measuring 1 × 10 μm relative to the microscopic field, measurements of average pixel intensity (0-255 grey scale) were made at a series of 10 sites along each vessel wall. These measurements were then used to calculate a mean value for each vessel. Tissue autofluorescence at this wavelength was measured before HE exposure, and then subtracted from signals measured after HE exposure to determine specific ethidium bromide fluorescence intensities. All images were acquired, stored and analyzed with Metamorph 6.01 imaging software (Universal Imaging Corporation, Downingtown, PA).
To determine the effect of high salt intake on endothelium-dependent dilation, we evaluated arteriolar responsiveness to iontophoretically-applied acetylcholine (ACh; Sigma Chemical, St. Louis, MO) in each dietary group. Since L-alanine was found to have no effect on ACh-stimulated O2- generation (see Results), these experiments were conducted only on mice consuming normal or L-Arg-supplemented drinking water. A beveled micropipette (2-3 μm tip diameter) was filled with 0.025 M ACh in distilled water and positioned with its tip in light contact with the arteriolar wall. A retaining current of 70-100 nA was used to prevent passive diffusion of ACh from the micropipette tip during the control period, and net ejection currents of 5, 20, and 80nA (delivered in random order) were used for ACh application. For each level of ACh, the vessel was continuously observed during a 1-min control period, a 2-min application period, and a minimum 2-min recovery period. To determine the contribution of NO to these endothelium-dependent responses, the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) was then infused into the superfusate delivery line to produce an L-NAME concentration of 10-4 M in the muscle bathing solution. After a 10-min equilibration period, arteriolar responses to all levels of ACh were reassessed, with L-NAME superfusion continued throughout to ensure maximal NOS inhibition. Although we used NG monomethyl L-arginine (L-NMMA) to inhibit eNOS in our earlier study [32], we used L-NAME in the current study because it is more effective inhibitor of eNOS-mediated O2- production due to its ability to completely antagonize the transfer of electrons to oxygen as well as L-arginine [29].
In other mice from each group, the intrinsic responsiveness of arteriolar smooth muscle to NO was evaluated by iontophoretically applying the NO donor sodium nitroprusside (SNP, Sigma) to the arteriolar wall. Beveled micropipettes were filled with 0.05 M SNP in distilled water, retaining currents of up to 100 nA were used to prevent passive diffusion, and net ejection currents of 5, 10 and 20nA were used to apply SNP (1-min control, 2-min application, minimum 2-min recovery).
Assessment of vascular wall L-Arginine and biopterin levels
Arcade arterioles were quickly excised from the spinotrapezius muscle with the aid of a dissecting microscope, placed in a cryovial containing 50 μl of 5 mM dithioerythritol (deoxygenated using helium), and then snap frozen in liquid nitrogen. For each sample, vessels from the muscles of 6 mice were pooled to obtain enough tissue for analysis. To determine the effect of increased dietary salt intake and/or chronic L-Arg supplementation on local BH4 and arginine availability, arteriolar BH4 and arginine levels were measured by reverse-phase HPLC with fluorescence detection, as previously described [24]. To determine if any differences in arteriolar [BH4] among groups could be due to a change in BH4 metabolism, arteriolar [BH2] was also measured as biopterin level in base oxidation [24] for calculation of the [BH2]/[BH4] ratio in arterioles of each group. This ratio is widely used as an index of BH4 oxidation; the immediate product of BH4 oxidation is quinonoid-7,8-dihydrobiopterin (qBH2), but this species is extremely unstable and spontaneously rearranges to yield 7,8-dihydrobiopterin (BH2) and 7,8-dihydropterin [34]. Briefly, 10 μg of arteriolar tissue were homogenized in 0.1 ml of 0.1 M phosphoric acid containing 5 mM dithioerythritol and 15 μl of 2 M trichloacetic acid (TCA). BH4 standard (50 pmol/ml) or cell extract (50 μl) was mixed with 15 μl of 0.2 M TCA and 15 μl of acidic oxidizer (1% I2/2% KI in 0.2 M TCA) (acidic oxidation) or with 15 μl of 1 M NaOH and 15 μl of alkaline oxidizer (1% I2/2% KI in 3 M NaOH) (alkaline oxidation). After 1-h incubation at 25° C in the dark, excess iodine was removed by adding 25 μl of 20 mg/ml ascorbic acid. After neutralization, 50 μl of the solution was analyzed on a Phenosphere 5 ODS-1 column (4.6 mm × 25 cm, 5 um) using isocratic elution (flow rate of 1 ml/min) and fluorescence detection (excitation 350 nm and emission 440 nm). The mobile phase solvent was 5% HPLC-grade methanol, 95% HPLC-grade water, 7.5 mM sodium phosphate, pH 6.35 (15 min running time).
The amount of BH2 in tissue extracts was determined from the amount of biopterin measured after alkaline oxidation, and the amount of BH4 in tissue extracts was determined by subtracting the amount of biopterin measured after alkaline oxidation from the amount of biopterin measured after acidic oxidation [24].
Measurement of vascular wall GTP cyclohydrolase-1 expression
To determine if any differences in arteriolar wall [BH4] among groups could be due to differences in BH4 synthesis, we used Western analysis to determine the content of GTP cyclohydrolase-1 (GTPCH), the rate-controlling enzyme for BH4 biosynthesis [34]. Arterioles were quickly excised from the spinotrapezius muscle and snap frozen in liquid nitrogen. For each sample, vessels from the muscles of 5-6 mice were pooled to obtain enough tissue for Western blotting. Arteriolar protein was extracted and loaded on 9.5–16% polyacrylamide gels. Separated proteins were blotted onto nitrocellulose and the blots were blocked with 5% non-fat dried milk in Tris-buffered saline with 0.1% Tween 20 (blocking buffer). For analysis of GTPCH, primary antibody (prepared against purified rat recombinant GTPCH) was diluted 1:10000 in blocking buffer, and the secondary antibody, peroxidase-conjugated donkey anti-rabbit IgG, was used at 1:100000 in blocking buffer. Bands were visualized with a chemiluminescence signal enhancer and exposed to autoradiography film. Blots were digitized and optical density measurements made with BioRad Multi-Analyst software.
Results
After HE exposure, control ethidium bromide fluorescence was significantly greater in arterioles of untreated HS mice (mean pixel intensity: 210±5) than in those of untreated NS mice (mean pixel intensity: 155±2) (Figure 1). In NS mouse arterioles, L-Arg supplementation had no effect on control fluorescence, and ACh had no effect on fluorescence regardless of whether the mice were untreated or supplemented with L-Arg. In contrast, ACh significantly increased arteriolar fluorescence in untreated HS mice (to 234±5). In HS mice supplemented with L-Arg, control arteriolar fluorescence (153±3) was significantly lower than that in untreated HS mice, and not significantly different from the control values measured in NS mice (either untreated or L-Arg supplemented). Furthermore, ACh did not increase arteriolar fluorescence in HS mice supplemented with L-Arg (159±3).
Figure 1.
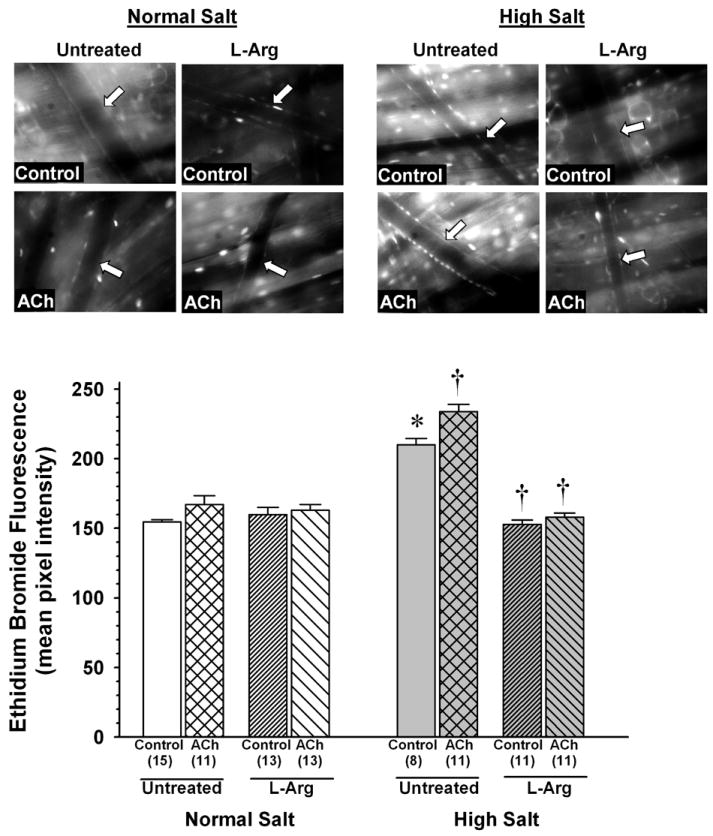
Arteriolar ethidium bromide fluorescence after exposure of the spinotrapezius muscle to hydroethidine in mice fed normal salt or high salt diet, with or without L-arginine (L-Arg) supplementation. Measurements were made in otherwise untreated muscles (Control) or in muscles also exposed to ACh (1×10-5M). Top: representative fluorescence images for each experimental group. Arrow in each image indicates position of vessel wall. Bottom: mean fluorescence values for each group. Values in parentheses represent number of vessels studied in each group. *P < 0.05 vs. Control value in untreated NS mice. † P < 0.05 vs. Control in same diet group.
Supplementation with L-alanine had no effect on arteriolar ethidium bromide fluorescence in NS mice, and in contrast to L-Arg, did not prevent the ACh-induced fluorescence increase in HS mice (data not shown). Because L-alanine did not mimic the effects of L-Arg in arterioles of HS mice, we concluded that the effect of L-Arg in these animals was specific and not due to elevated nitrogen intake. L-alanine supplementation was therefore discontinued for all subsequent experiments.
As shown in Figure 2, there was no difference between untreated NS and HS mice in arteriolar wall L-Arg content (97.5±3.0 and 93.3±3.8 fmol/mg, respectively), and L-Arg supplementation increased arteriolar L-Arg content by the same amount in both groups (NS: to 155.5±4.7 fmol/mg, a 59% increase; HS: to 145.4±3.1 fmol/mg, a 56% increase).
Figure 2.
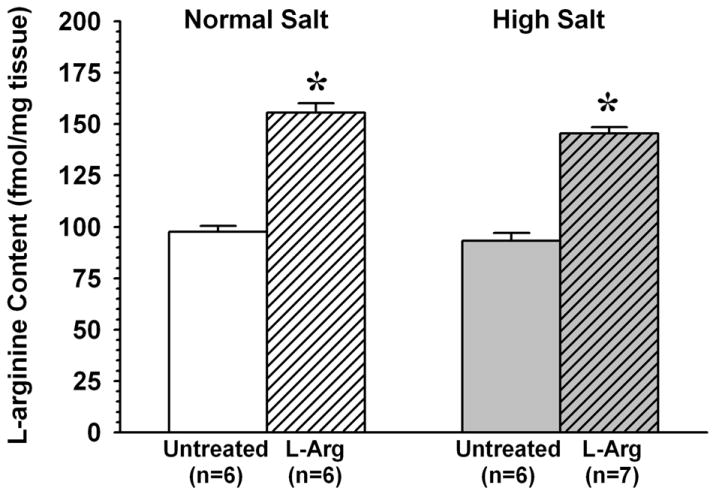
Effect of L-arginine supplementation (L-Arg) on arteriolar wall L-arginine levels in mice fed normal salt or high salt diet. * P < 0.05 vs. untreated group. In Figures 2-5, n= number of samples, with each sample comprised of muscle vessels pooled from 5-6 mice.
As shown in Figure 3, arteriolar wall BH4 content was significantly lower in untreated HS mice than in untreated NS mice (38.7±1.6 vs.70.0±3.1 fmol/mg). However, whereas L-Arg supplementation had no effect on arteriolar BH4 in NS mice, it increased BH4 content in HS mice to a level that was not significantly different from that in untreated NS mice (65.0±2.7 fmol/mg). The ratio of [BH2] to [BH4] was much higher in arterioles from untreated HS mice than in those from untreated NS mice (0.275±0.015 vs. 0.074±0.005) (Figure 4). However, whereas L-Arg supplementation had no effect on this ratio in NS mice, it returned the ratio in HS mice almost to the same level as that in NS mice (0.102±0.006). In contrast to biopterin levels, there was no difference between untreated NS and HS mice in arteriolar wall GTPCH expression, and L-Arg supplementation had no effect on this expression in either dietary group (Figure 5).
Figure 3.
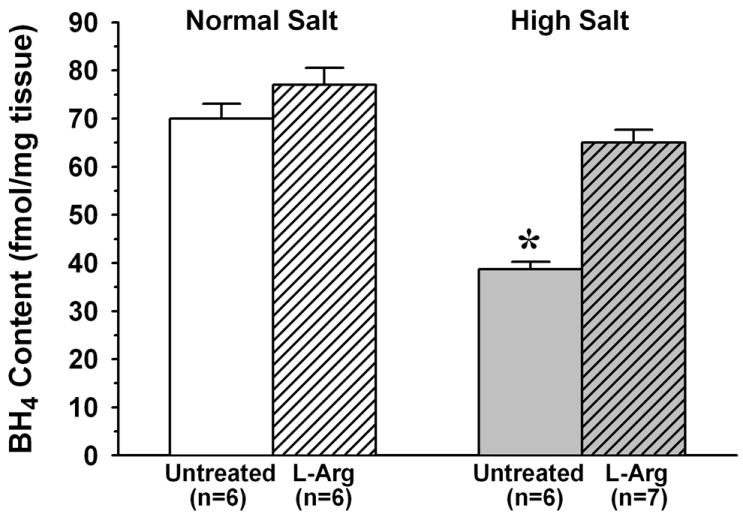
Arteriolar wall BH4 content in mice fed normal salt or high salt diet, with or without L-Arg supplementation. * P < 0.05 vs. untreated and L-Arg supplemented normal salt mice.
Figure 4.
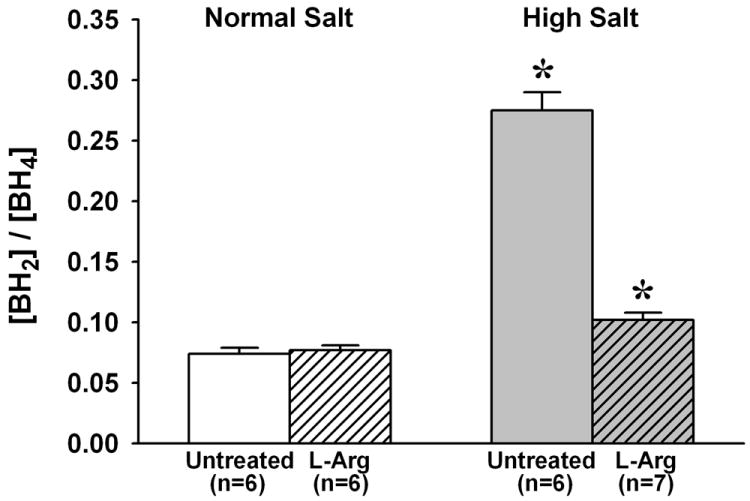
Ratio of BH2 content to BH4 content in arterioles from mice fed normal salt or high salt diet, with or without L-Arg supplementation. * P < 0.05 vs. untreated and L-Arg supplemented normal salt mice.
Figure 5.
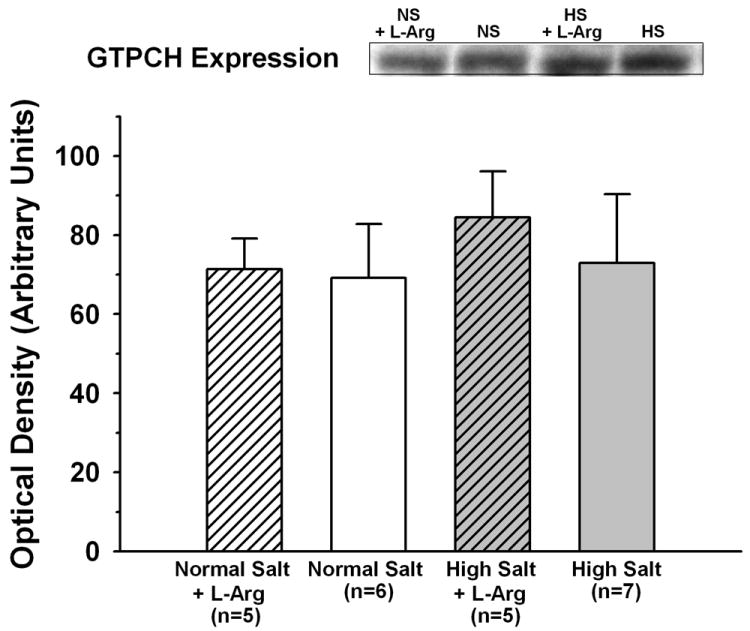
GTP cyclohydrolase-1 expression in arterioles from mice fed normal salt or high salt diet, with or without L-Arg supplementation.
Under control conditions in vivo, there were no significant differences among diet/treatment groups in resting arteriolar diameters (NS untreated: 31±3 μm, NS+L-Arg: 34±2 μm, HS untreated: 33±2 μm, HS+L-Arg: 29±1 μm). Iontophoretically-applied ACh caused arteriolar dilation in all groups, but for each current-dose of ACh, the magnitude of this dilation was significantly lower in untreated HS mice than in untreated NS mice (5±3% vs.15±4% for 5 nA ACh, 9±2% vs. 42±6% for 20 nA ACh, and 16±4% vs. 52±8 % for 80nA ACh) (Figure 6). However, although L-Arg supplementation had no significant effect on ACh responses in NS mice, it restored ACh-induced arteriolar dilation to normal in HS mice (12±4%, 36±6% and 48±8% dilation for 5, 20 and 80 nA ACh, respectively).
Figure 6.
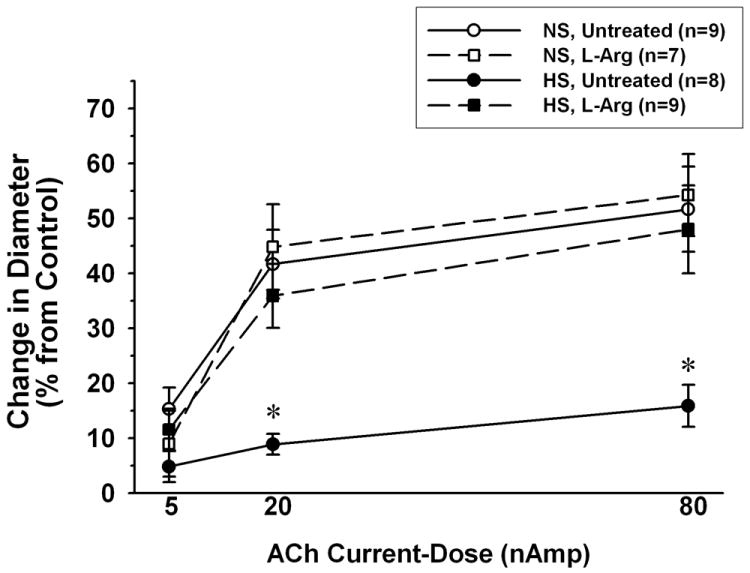
Arteriolar dilation in response to iontophoretic application of ACh in mice fed normal salt (NS) or high salt (HS) diet, with or without L-Arg supplementation. * P < 0.05 vs. both NS groups and the HS group supplemented with L-arginine. n= number of vessels (1 vessel studied per mouse).
Exposure to L-NAME had no significant effect on resting arteriolar diameters in any experimental group (NS untreated: 29±2 μm, NS+L-Arg: 34±2 μm, HS untreated: 29±2 μm, HS+L-Arg: 29±1 μm). However, as shown in Figure 7, L-NAME greatly reduced arteriolar dilation to all levels of ACh in NS mice, decreasing the magnitude of the dilation by 72-90%. L-Arg supplementation in these animals did not appreciably change the effect of L-NAME on responses to ACh, with the magnitude of dilation being decreased by 64-84%. In contrast to NS mice, arteriolar dilation to ACh was not sensitive to L-NAME in untreated HS mice. However, in HS mice supplemented with L-Arg, L-NAME reduced the magnitude of arteriolar responses by an amount similar to that found in NS mice (58-84%).
Figure 7.
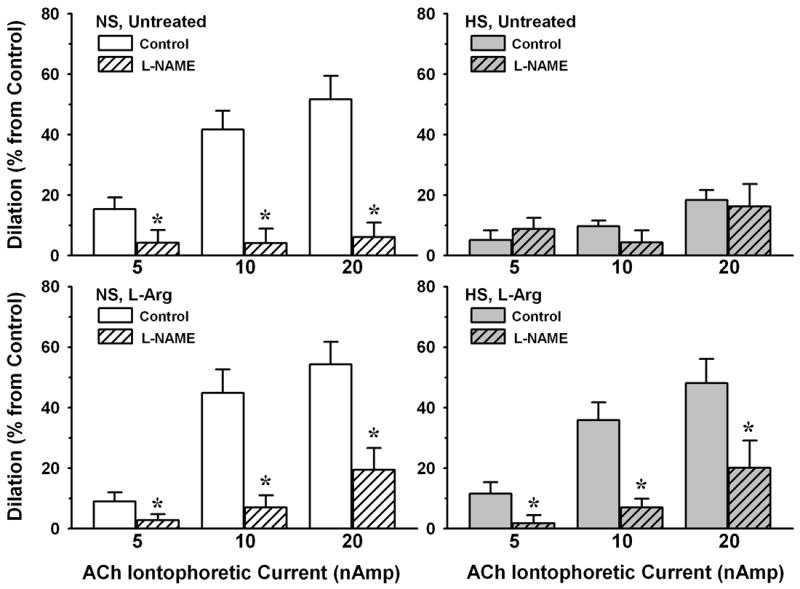
Magnitude of responses to ACh, under normal superfusate (Control) and in the presence of Nω-nitro-L-arginine methyl ester (L-NAME, 10-4M), in mice fed NS or HS diet, with or without L-Arg supplementation. Number of vessels for each group same as in Figure 6. * P < 0.05 vs. Control.
As shown in Figure 8, iontophoretic application of SNP caused a dose-dependent dilation of arterioles in all groups, but there were no significant differences among any of the groups in the magnitude of this dilation for any level of SNP application.
Figure 8.
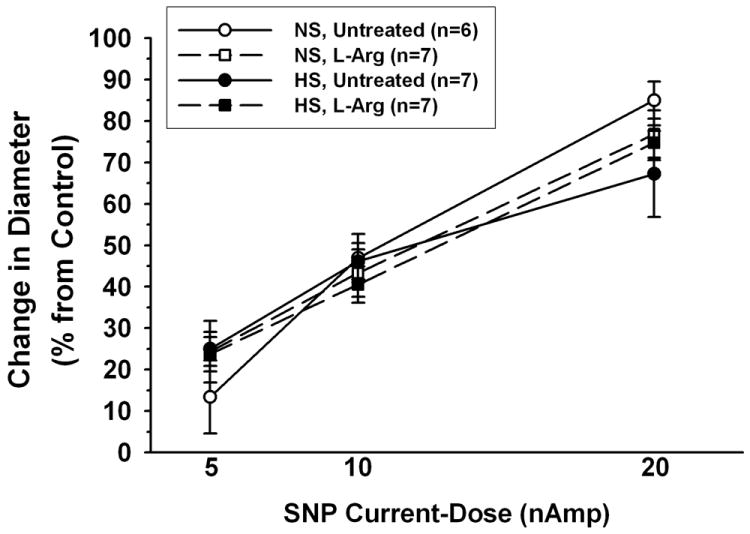
Arteriolar dilation in response to iontophoretic application of sodium nitroprusside (SNP) in mice fed NS or HS diet, with or without L-Arg supplementation. n= number of vessels (1 vessel studied per mouse).
Discussion
The major findings of this study are as follows: (1) Arterioles in HS mice have lower BH4 levels and higher O2- levels than those in NS mice. (2) ACh, which increases eNOS activity, further increases arteriolar O2- in HS mice only. (3) L-Arg supplementation prevents the reduction in BH4 in arterioles of HS mice, and these vessels do not have elevated O2- levels. (4) ACh-induced arteriolar dilation is smaller in HS mice than in NS mice, but not if HS mice are supplemented with L-Arg. Collectively, these findings suggest that eNOS uncoupling due to low BH4 bioavailability is responsible for O2- generation and reduced endothelium-dependent dilation in arterioles of mice fed a high salt diet.
Previous studies have documented that reduced BH4 bioavailability, leading to uncoupling of eNOS and O2- production, contributes importantly to vascular dysfunction in various other pathological states, including hypertension, diabetes, and chronic alcohol consumption [1,9,10,13,15,25,27,36]. In an earlier study on the mouse spinotrapezius muscle, we found that ingestion of a high salt diet leads to arteriolar O2- accumulation and that the NOS inhibitor L-NMMA prevents this O2- accumulation, implicating uncoupled eNOS as the source of this O2- [32]. This interpretation is further supported by our current finding that arteriolar wall BH4 levels in HS mice are almost 50% lower than those in NS mice (Figure 3), and that preserving normal BH4 levels in HS mice by dietary L-Arg supplementation is as effective as NOS inhibition in preventing arteriolar O2- accumulation (Figure 1).
In some cases, reduced BH4 bioavailability associated with vascular dysfunction is the result of insufficient de novo production due to reduced expression of GTPCH [25,27]. In other cases, it is due to accelerated BH4 oxidation by reactive oxygen species [15,16]. In the current study, we found no difference between NS and HS mice in GTPCH expression (Figure 5), suggesting that BH4 production may not have been reduced in HS mice. However, we did find that BH2, the stable oxidation product of BH4, is profoundly elevated in arterioles of HS mice (Figure 4), which indicates that the reduced BH4 levels are due at least in part to increased BH4 oxidation. The current study provides no direct evidence for the identity of the reactive molecule(s) responsible for accelerated BH4 oxidation in arterioles of HS mice. We and others have consistently documented the presence of elevated arteriolar O2- in both mice and rats fed high salt diets [17-19,32,43]. However, O2- is a relatively poor oxidizer of BH4 [16]. A more likely candidate is peroxynitrite (ONOO-), which is formed when O2- interacts with NO and is a strong oxidizer of BH4 [14,16].
Administration of exogenous L-Arg has previously been found to lessen or reverse endothelial dysfunction in some cardiovascular disease states [11,33,42], and our current findings are consistent with recent evidence that at least part of this effect is due to an increase in BH4 availability. In both normal and diabetic rats, dietary L-Arg supplementation leads to an increased plasma concentration of L-Arg, increased endothelial cell concentrations of L-Arg and BH4, and increased endothelial NO synthesis [13]. Studies on cultured endothelial cells indicate that increased cellular L-Arg increases intracellular BH4 levels by increasing the expression of GTPCH [41]. We were able to increase arteriolar wall L-Arg levels by the same amount in both dietary groups by providing supplemental L-Arg in the drinking water (Figure 2). However, this led to increased BH4 levels only in the arterioles of HS mice (Figure 3), and this occurred through a reduction in BH4 oxidation rather than an increase in GTPCH expression (Figures 4 and 5). The reason for the difference between our study and the previous study on endothelial cells is not clear, but it could be attributable to different methodologies (addition of L-Arg to endothelial cells in vitro vs. in vivo supplementation), differences in the duration of exposure to increased L-Arg levels (24-48 hours vs. 4 weeks in our study), or even species differences (bovine vs. murine). L-Arg is an effective scavenger of O2- [39], and this antioxidant property could be responsible for the reduced BH4 oxidation we found in HS mice. Increasing arteriolar wall L-Arg would lead to reduced O2-, and therefore less ONOO- formation. In this scenario, the lack of effect of increased L-Arg on arteriolar BH4 in NS mice would not be surprising because there is little if any O2- for the L-Arg to scavenge, and little ONOO- to begin with.
In comparing the values for untreated NS vs. untreated HS mice, it is clear that high salt intake has no effect on endogenous arteriolar wall L-Arg levels (Figure 2). This is consistent with previous findings in Sprague-Dawley rats that ingestion of a high (8%) salt diet has no effect on plasma L-Arg levels [40], and that dietary salt over a wide range has no effect on systemic L-Arg synthesis or on the expression or activity of hepatic L-arginase, an enzyme that plays a central role in the regulation of plasma L-Arg levels [12].
The impact of high dietary salt intake on microvascular function has historically been studied almost exclusively in the rat [6]. We and others have found no contribution of NO to the endothelium-dependent dilation of mesenteric or skeletal muscle resistance vessels in rats fed high salt [4,5,35,43], presumably due to scavenging of NO by the O2- that accumulates in these vessels [17-19,43]. However, the rat is apparently different from the mouse in that this elevated O2- is due primarily to increased activity of NADP(H) oxidase and xanthine oxidase [18,43]. Many investigators now use the mouse to explore mechanisms of cardiovascular disease because of the availability of numerous murine models with targeted deletion or overexpression of genes relevant to cardiovascular control. Similar to our previous findings in the rat, our first study in mice fed high salt showed a correlation between increased vascular wall O2-, a loss of NO bioavailability, and blunted endothelium-dependent dilation in skeletal muscle arterioles [32]. The results of the present study confirm that increased arteriolar O2- is accompanied by a loss of NO in salt-fed mice; arteriolar responses to ACh, which were reduced 70-90 % by L-NAME in NS mice, were much smaller and insensitive to L-NAME in HS mice (Figure 7). The undiminished responses to ACh in HS mice supplemented with L-Arg are apparently due to preservation of the L-NAME-sensitive component of the dilation (Figures 6 and 7), in association with the maintenance of normal vascular wall BH4 levels and an absence of O2- accumulation. Dilation of arterioles to the NO donor SNP was identical in all experimental groups (Figure 8), indicating that guanylyl cyclase activity and cGMP-dependent pathways in smooth muscle are not influenced by dietary salt content or by levels of O2-, L-Arg or BH4 in the arteriolar wall. We therefore conclude that the effectiveness of L-NAME in reducing endothelium-dependent dilation of arterioles in HS mice given L-Arg is due to a preservation of NO bioavailability rather than an increase in arteriolar smooth muscle responsiveness to NO.
During application of ACh at the highest concentration, the residual dilation that persists after L-NAME treatment appears to be greater in NS mice given L-Arg than in untreated NS mice (Figure 7). Because L-Arg and L-NAME compete for the same binding site on eNOS, it is possible that eNOS was not fully inhibited under conditions in which arteriolar wall L-Arg levels were increased almost 60% by supplementation (Figure 2). However, L-NAME appears to have the same effect on dilations to ACh at lower concentrations in both groups, suggesting this may not be a consistent problem.
Because both the L-NAME used in this study and the L-NMMA used in our previous study [32] are nonspecific, they will inhibit the neuronal isoform of NOS (nNOS) as well as eNOS. Although nNOS expression and activity is normally low in mouse arterioles, it can be increased in certain pathological states [23,26]. Because low BH4 levels can also lead to uncoupling of nNOS [38], we can’t rule out the possibility that if nNOS is upregulated in salt-fed mice, this isoform could be the source of at least some of the O2- generated in arterioles of HS mice.
The impact of the salt-dependent reduction in NO bioavailability on the regulation of muscle blood flow in the mouse is unknown. Nitric oxide contributes importantly to the arteriolar dilation and hyperemia that accompanies muscle contraction in the rat, hamster and mouse [21,22,28,30], and we have previously reported that arteriolar dilation during high frequency contraction of the spinotrapezius muscle is reduced in rats on the same high salt diet regimen as the mice in this study [21]. It is reasonable to expect that active hyperemia (and other forms of local blood flow control that involve NO) would be reduced in mice fed a high salt diet, but additional studies will be required to assess the precise physiological influence of dietary salt on blood flow regulation in the mouse.
Acknowledgments
This investigation was supported by National Institutes of Health grants HL-44012 (M.A. B.), ES-018274 (T.R.N.) and ES-015022 (T.R. N.), and by American Heart Association Grants 0755264B (M.A.B.), 2250298 (M.A. B.) and 0755024Y (G.W.). The authors would like to acknowledge the expert technical assistance of Kimberly Wix in this study.
References
- 1.Bagi Z, Koller A. Lack of nitric oxide mediation of flow-dependent arteriolar dilation in Type 1 diabetes is restored by sepiapterin. J Vasc Res. 2003;40:47–57. doi: 10.1159/000068938. [DOI] [PubMed] [Google Scholar]
- 2.Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, Yokoyama M, Kawashima S, Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–71. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 3.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 4.Boegehold MA. Effect of dietary salt on arteriolar nitric oxide in striated muscle of normotensive rats. Am J Physiol Heart Circ Physiol. 1993;264:H1810–H1816. doi: 10.1152/ajpheart.1993.264.6.H1810. [DOI] [PubMed] [Google Scholar]
- 5.Boegehold MA. Flow-dependent arteriolar dilation in normotensive rats fed low- or high-salt diets. Am J Physiol Heart Circ Physiol. 1995;269:H1407–H1414. doi: 10.1152/ajpheart.1995.269.4.H1407. [DOI] [PubMed] [Google Scholar]
- 6.Boegehold MA. Microvascular structure and function in salt-sensitive hypertension. Microcirculation. 2002;9:225–241. doi: 10.1038/sj.mn.7800139. [DOI] [PubMed] [Google Scholar]
- 7.Boegehold MA, Bohlen HG. Arteriolar diameter and tissue oxygen tension during muscle contraction in hypertensive rats. Hypertension. 1988;12:184–191. doi: 10.1161/01.hyp.12.2.184. [DOI] [PubMed] [Google Scholar]
- 8.Channon KM. Tetrahydrobiopterin. Regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med. 2004;14:323–327. doi: 10.1016/j.tcm.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Cosentino F, Patton S, d’Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, Malinski T, Lüscher TF. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest. 1998;101:1530–1537. doi: 10.1172/JCI650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong H-J, Hsiao G, Cheng T-H, Yen M-H. Supplementation with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension. 2001;38:1044–1048. doi: 10.1161/hy1101.095331. [DOI] [PubMed] [Google Scholar]
- 11.Kitazono T, Faraci FM, Heistad DD. L-arginine restores dilator responses of the basilar artery to acetylcholine during chronic hypertension. Hypertension. 1996;27:893–896. doi: 10.1161/01.hyp.27.4.893. [DOI] [PubMed] [Google Scholar]
- 12.Kitiyakara C, Chabrashvili T, Jose P, Welch WJ, Wilcox CS. Effects of dietary salt on plasma arginine. Am J Physiol Reg Int Comp Physiol. 2001;280:R1069–R1075. doi: 10.1152/ajpregu.2001.280.4.R1069. [DOI] [PubMed] [Google Scholar]
- 13.Kohli R, Meininger CJ, Haynes TE, Yan W, Self JT, Wu G. Dietary L-arginine supplementation enhances endothelial nitric oxide synthesis in streptozotocin-induced diabetic rats. J Nutr. 2004;134:600–608. doi: 10.1093/jn/134.3.600. [DOI] [PubMed] [Google Scholar]
- 14.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid and thiols. Implications for uncoupling endothelial nitric oxide synthase. J Biol Chem. 2003;278:22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 15.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 17.Lenda DM, Sauls BA, Boegehold MA. Reactive oxygen species may contribute to reduced endothelium-dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol. 2000;279:H7–H14. doi: 10.1152/ajpheart.2000.279.1.H7. [DOI] [PubMed] [Google Scholar]
- 18.Lenda DM, Boegehold MA. Effect of a high salt diet on oxidant enzyme activity in skeletal muscle microcirculation. Am J Physiol Heart Circ Physiol. 2002;282:H395–H402. doi: 10.1152/ajpheart.0354.2001. [DOI] [PubMed] [Google Scholar]
- 19.Lenda DM, Boegehold MA. Effect of a high salt diet on microvascular antioxidant enzymes. J Vasc Res. 2002;39:41–50. doi: 10.1159/000048992. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Fredricks KT, Roman RJ, Lombard JH. Response of resistance arteries to reduced PO2 and vasodilators during hypertension and elevated salt intake. Am J Physiol Heart Circ Physiol. 1997;273:H869–H877. doi: 10.1152/ajpheart.1997.273.2.H869. [DOI] [PubMed] [Google Scholar]
- 21.Marvar PJ, Nurkiewicz TR, Boegehold MA. Reduced arteriolar responses to skeletal muscle contraction after ingestion of a high salt diet. J Vasc Res. 2005;42:226–236. doi: 10.1159/000085461. [DOI] [PubMed] [Google Scholar]
- 22.Maxwell AJ, Schauble E, Bernstein D, Cooke JP. Limb blood flow during exercise is dependent on nitric oxide. Circulation. 1998;98:369–374. doi: 10.1161/01.cir.98.4.369. [DOI] [PubMed] [Google Scholar]
- 23.McKinnon RL, Lidington D, Bolon M, Ouellette Y, Kidder GM, Tyml K. Reduced arteriolar conducted vasoconstriction in septic mouse cremaster muscle is mediated by nNOS-derived NO. Cardiovasc Res. 2006;69:236–244. doi: 10.1016/j.cardiores.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Meininger CJ, Wu G. Regulation of endothelial cell proliferation by nitric oxide. Methods Enzymol. 2002;352:280–295. doi: 10.1016/s0076-6879(02)52026-7. [DOI] [PubMed] [Google Scholar]
- 25.Meininger CJ, Marinos RS, Hatakeyama K, Martinez-Zaguilan R, Rojas JD, Kelly KA, Wu G. Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem J. 2000;349:353–356. doi: 10.1042/0264-6021:3490353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng W, Ayata C, Waeber C, Huang PL, Moskowitz MA. Neuronal NOS-cGMP-dependent ACh-induced relaxation in pial arterioles of endothelial NOS knockout mice. Am J Physiol. 1998;274:H411–H415. doi: 10.1152/ajpheart.1998.274.2.H411. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell BM, Dorrance AM, Webb RC. GTP cyclohydrolase 1 downregulation contributes to glucocorticoid hypertension in rats. Hypertension. 2003;41:669–674. doi: 10.1161/01.HYP.0000051889.62249.5D. [DOI] [PubMed] [Google Scholar]
- 28.Murrant CL, Sarelius IH. Multiple dilator pathways in skeletal muscle contraction-induced arteriolar dilations. Am J Physiol Reg Int Comp Physiol. 2002;282:R969–R978. doi: 10.1152/ajpregu.00405.2001. [DOI] [PubMed] [Google Scholar]
- 29.Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol. 2005;25:1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- 30.Musch TI, McAllister RM, Symons JD, Stebbins CL, Hirai T, Hageman KS, Poole DC. Effects of nitric oxide synthase inhibition on vascular conductance during high speed treadmill exercise in rats. Exp Physiol. 2001;86:749–757. doi: 10.1111/j.1469-445x.2001.tb00040.x. [DOI] [PubMed] [Google Scholar]
- 31.Nurkiewicz TR, Boegehold MA. High dietary salt alters arteriolar myogenic responsiveness in normotensive and hypertensive rats. Am J Physiol Heart Circ Physiol. 1998;275:H2095–H2104. doi: 10.1152/ajpheart.1998.275.6.H2095. [DOI] [PubMed] [Google Scholar]
- 32.Nurkiewicz TR, Boegehold MA. High salt intake reduces endothelium-dependent dilation of mouse arterioles via superoxide anion generated from nitric oxide synthase. Am J Physiol Reg Int Comp Physiol. 2007;292:R1550–R1556. doi: 10.1152/ajpregu.00703.2006. [DOI] [PubMed] [Google Scholar]
- 33.Pieper GM, Siebeneich W, Dondlinger LA. Short-term oral administration of L-arginine reverses defective endothelium-dependent relaxation and cGMP generation in diabetes. Eur J Pharmacol. 1996;317:317–320. doi: 10.1016/s0014-2999(96)00831-x. [DOI] [PubMed] [Google Scholar]
- 34.Shi W, Meininger CJ, Haynes TE, Hatakeyama K, Wu G. Regulation of tetrahydrobiopterin synthesis and bioavailability in endothelial cells. Cell Biochem Biophys. 2004;41:415–434. doi: 10.1385/CBB:41:3:415. [DOI] [PubMed] [Google Scholar]
- 35.Sofola OA, Knill A, Hainsworth R, Drinkhill M. Change in endothelial function in mesenteric arteries of Sprague-Dawley rats fed a high salt diet. J Physiol. 2002;543:255–260. doi: 10.1113/jphysiol.2002.022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun H, Patel KP, Mayhan WG. Tetrahydrobiopterin, a cofactor of NOS, improves endothelial dysfunction during chronic alcohol consumption. Am J Physiol Heart Circ Physiol. 2001;281:H1863–H1869. doi: 10.1152/ajpheart.2001.281.5.H1863. [DOI] [PubMed] [Google Scholar]
- 37.Vásquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vásquez-Vivar J, Hogg N, Martásek P, Karoui H, Pritchard KA, Jr, Kalyanaraman B. Tetrahydrobiopterin-dependent inhibition of superoxide generation from neuronal nitric oxide synthase. J Biol Chem. 1999;274:26736–26742. doi: 10.1074/jbc.274.38.26736. [DOI] [PubMed] [Google Scholar]
- 39.Wascher TC, Posch K, Wallner S, Hermetter A, Kostner GM, Graier WF. Vascular effects of L-arginine: anything beyond a substrate for the NO-synthase? Biochem Biophys Res Commun. 1997;234:35–38. doi: 10.1006/bbrc.1997.9994. [DOI] [PubMed] [Google Scholar]
- 40.Wood PA, Hamm DA, Chen PY, Sanders PW. Studies of arginine metabolism and salt sensitivity in the Dahl/Rapp rat models of hypertension. Mol Gen Metab. 1998;64:80–83. doi: 10.1006/mgme.1998.2696. [DOI] [PubMed] [Google Scholar]
- 41.Wu G, Kelly KA, Hatakeyama K, Meininger CJ. Regulation of endothelial tetrahydrobiopterin synthesis by L-arginine. In: Blau N, Heilbronn TB, editors. Pterins, Folates and Neurotransmitters in Molecular Medicine. SPS Verlagsgesellschaft mbh; Germany: 2004. [Google Scholar]
- 42.Zhou MS, Kosaka H, Tian RX, Abe Y, Chen QH, Yoneyama H, Yamamoto A, Zhang L. L-arginine improves endothelial function in renal artery of hypertensive Dahl rats. J Hypertens. 2001;19:421–429. doi: 10.1097/00004872-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Zhu J, Huang T, Lombard JH. Effect of high-salt diet on vascular relaxation and oxidative stress in mesenteric resistance arteries. J Vasc Res. 2007;44:382–390. doi: 10.1159/000102955. [DOI] [PubMed] [Google Scholar]