

Abstract
Stress induces aggregation of RNA-binding proteins to form inclusions, termed stress granules (SGs). Recent evidence suggests that SG proteins also colocalize with neuropathological structures, but whether this occurs in Alzheimer's disease is unknown. We examined the relationship between SG proteins and neuropathology in brain tissue from P301L Tau transgenic mice, as well as in cases of Alzheimer's disease and FTDP-17. The pattern of SG pathology differs dramatically based on the RNA-binding protein examined. SGs positive for T-cell intracellular antigen-1 (TIA-1) or tristetraprolin (TTP) initially do not colocalize with tau pathology, but then merge with tau inclusions as disease severity increases. In contrast, G3BP (ras GAP-binding protein) identifies a novel type of molecular pathology that shows increasing accumulation in neurons with increasing disease severity, but often is not associated with classic markers of tau pathology. TIA-1 and TTP both bind phospho-tau, and TIA-1 overexpression induces formation of inclusions containing phospho-tau. These data suggest that SG formation might stimulate tau pathophysiology. Thus, study of RNA-binding proteins and SG biology highlights novel pathways interacting with the pathophysiology of AD, providing potentially new avenues for identifying diseased neurons and potentially novel mechanisms regulating tau biology.
Introduction
Cellular stress elicits a switch in protein translation from cap-dependent to cap-independent RNA translation, which inhibits synthesis of non-housekeeping proteins, and maintains synthesis of proteins that protect against stress, such as heat shock proteins (Kedersha and Anderson, 2007; Buchan and Parker, 2009; Liu-Yesucevitz et al., 2011). The translational stress switch is associated with cytoplasmic translocation of mRNA-binding proteins, and consolidation of these proteins with transcripts to form RNA protein complexes that are termed stress granules (SGs). The primary stress granule proteins include T-cell intracellular antigen 1 (TIA-1), RasGAP-associated endoribonuclease (G3BP), elongation initiation factor 3 (eIF3), and poly-A binding protein (PABP), but there are >500 RNA-binding proteins, many of which participate in the formation of SGs (Gilks et al., 2004; Anderson and Kedersha, 2008; Liu-Yesucevitz et al., 2011). Recent advances in molecular genetics highlight the potential importance of SG biology in disease by genetically associating multiple RNA-binding proteins linked to SGs with neurologic/neurodegenerative disease (Waelter et al., 2001; Liu-Yesucevitz et al., 2010). These proteins include Tar DNA-binding protein (TDP-43), Fused in Sarcoma protein (FUS), survival motor neuron protein (SMN), ataxin-2, senataxin (SETX), angiogenin, and fragile X mental retardation protein (FMRP), as well as other proteins (Lefebvre et al., 1995; Imbert et al., 1996; Grohmann et al., 2001; Chen et al., 2004; Greenway et al., 2006; Elden et al., 2010; Lagier-Tourenne et al., 2010; Corrado et al., 2011; Lee et al., 2011; Liu-Yesucevitz et al., 2011; Ross et al., 2011; Van Damme et al., 2011). The link between SG proteins and motor neuron disease raises the possibility that the stress-response of RNA-binding proteins and SG formation might also contribute to the pathophysiology of other neurodegenerative diseases, such as dementia.
Recent reports indicate that SGs are associated with the pathological inclusions in multiple neurodegenerative diseases, including Huntington's disease, Creutzfeldt-Jakob disease, amyotrophic lateral sclerosis (ALS), and frontotemporal dementia linked to ubiquitin (FTLD-U) (Waelter et al., 2001; Goggin et al., 2008; Liu-Yesucevitz et al., 2010). A potential relationship between SGs and tau is particularly interesting because tau can bind to RNA, and RNA is known to promote the assembly of tau to form paired helical filaments (PHFs) (Kampers et al., 1996). The high concentrations of RNA present in SGs raises the possibility that formation of SGs might impact on aggregation of tau to form inclusions in tauopathies.
In this study we investigate the biology of RNA-binding proteins and SGs in animal models of tauopathy and cases of Alzheimer's disease (AD). We report that phase transitions of RNA-binding proteins are evident early in the course of tauopathies, including cytoplasmic translocation and granule formation. SGs begin to merge with tau inclusions at the mid-stage of disease development in a process that appears to be associated with enlargement of tau inclusions. We demonstrate that TIA-1-positive SGs are strongly associated with tau pathology in AD. In contrast, SGs positive for G3BP show little colocalization with phospho-tau protein, and SGs positive for tristetraprolin (TTP) show colocalization with phospho-tau protein in cases with severe pathology.
Materials and Methods
JNPL3 mutant tau mice
The JNPL3 line of mice were generated as described by Lewis et al. (2000) and backcrossed to homozygous status on Swiss Webster background. This mouse line expresses the human P301L mutant 4R tau isoform driven by the mouse prion promoter. Mice exhibit motor deficits that correlate with an age- and gene-dose-dependent development of neurofibrillary tangles (NFT) and neuronal loss (Lewis et al., 2000). Mice from different age ranges were classified for severity of disease phenotype using a motor test (wire hang test and hindlimb clasping), which has been shown to correlate well with degree of pathology in hindbrain and spinal cord. Mice were classified as normal, mild, moderate, or severely affected based on motor tests noted above.
Male JNPL3 mice were killed by cervical dislocation and brains were dissected into hemispheres. One half was dissected and frozen, the other half was drop fixed in 4% paraformaldehyde overnight. Serial sections through the amygdala were taken from each mouse brain for IHC (N = 3 from each of the four groups). Swiss-Webster non-transgenic mice (N = 3) age-matched to the 11-month-old group (correlating with the oldest Tg mouse sample) were used for the control group.
Inducible rTg4510 tau mice
The rTg4510 line of tau transgenic mice were generated as described by Santacruz et al. (2005). Similar to the JNPL3 line, these mice express the human P301L mutant 4R tau isoform, but the transgene is conditionally expressed in the forebrain and hippocampus under control of the Ca2+-calmodulin kinase II promoter. Pretangles of tau appear in the brain at early stages (2.5 months), with silver staining of tau aggregates and Sarkosyl insoluble tau appearing at 4+ months. Severe pathology, mature NFTs, and neuronal loss are reported by 7+ months (Santacruz et al., 2005).
For this study, three male mice at 2.5, 5, and 8 months of age were used. Mice were killed by cervical dislocation and cortical brain tissue was dissected for biochemical analysis.
Postmortem human brain tissue
Brain tissue samples from human frontal cortex were obtained from the Boston University Alzheimer Disease Center, Mayo Clinic Jacksonville, the Loyola University Stritch School of Medicine, and Rush Medical Center. Paraffin-embedded postmortem human brain tissue was obtained from healthy (N = 3) controls, patients with the diagnosis of AD (N = 6), and five patients with FTDP-17 (Table 1). Demographic and clinical information for the human cases is described in Table 1. The sections were used for immunohistochemistry with staining for tau and SGs.
Table 1.
Human brain tissue samples
| Patient | Age | Gender | PMI | Diagnosis | Brodmann areas |
|---|---|---|---|---|---|
| 1 | 78 | M | Neurologic Nml | 8,9 | |
| 2 | 82 | F | 7 | Neurologic Nml | 8,9 |
| 3 | 83 | F | 13 | Neurologic Nml | 8,9 |
| 4 | 96 | M | 15 | AD | 8,9 |
| 5 | 88 | F | 15.5 | AD | 8,9 |
| 6 | 81 | M | 6.5 | AD | 8,9 |
| 7 | 79 | F | 12.5 | AD | 8,9 |
| 8 | 82 | F | 5.75 | AD | 8, 9 |
| 9 | 87 | M | 14 | AD | 8,9 |
| 10 | 65 | M | 12 | AD | 8,9 |
| 83 | 44 | F | 10 | FTDP-17 | Hippocampus |
| 253 | 51 | F | 12 | FTDP-17 | Hippocampus |
| 268 | 53 | F | 18 | FTDP-17 | Hippocampus |
| 300 | 53 | M | — | FTDP-17 | Hippocampus |
| 315 | 52 | M | 32 | FTDP-17 | Hippocampus |
PMI, postmortem interval (hours).
Biochemical fractionation
Tissue fractionation.
Cortical tissue was extracted from rTg4510 and non-transgenic mice and homogenized in chilled RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm sodium fluoride, 1 mm Na3VO, 1 mm PMSF, and 1 μg/ml protease inhibitors). The homogenate was centrifuged at 20,000 × g for 20 min at 4°C to remove cellular debris. The supernatant containing the total tau fraction was retained and assayed for total protein concentration.
Triton-X fractions.
Lysates from the preparation above were combined 1:1 with RIPA buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% NP-40, 0.1% SDS, 0.5 mm sodium deoxycholate) with 1× Halt protease inhibitor cocktail (Thermo Scientific), 1× phosphatase inhibitor cocktail (PhosSTOP, Roche), and 2% Triton X-100, and mixed. Lysates were first centrifuged for 1 h at 100,000 × g 4°C, and the supernatants were collected as RIPA buffer soluble proteins. To prevent contamination caused by carrying over, the pellets were resonicated and recentrifuged at 100,000 × g for 30 min at 4°C. RIPA buffer-insoluble pellets were dissolved in urea buffer (7 m urea, 2 m thiourea, 4% CHAPS, 30 mm Tris-HCl, pH 8.5) and sonicated. Soluble and insoluble proteins were analyzed by Western blot.
Sarkosyl insoluble and soluble tau fractions.
To produce a fraction enriched for tau, 10 μl of a 10% solution of Sarkosyl detergent was added to 100 μg of supernatant. Additional RIPA was used to produce a final volume of 100 μl. This sample was rotated at room temperature for 1 h before a 1 h centrifugation at 100,000 × g spin at room temperature. The supernatant was collected in a separate tube and the Sarkosyl pellet was resuspended in 100 μl of sample buffer containing 100 mm DTT. The heat-soluble tau-enriched fractions were generated by boiling the supernatant for 3 min at 95°C and centrifuging for 20 min at 20,000 × g.
Coimmunoprecipitation.
Equal amounts of lysate were precleared by rec-Protein G-Sepharose 4B Conjugate beads (Invitrogen) for 1 h at 4°C. Samples were spun down and lysates were removed, 0.5 μl of PHF-1 antibody or 1 μl of Tau-5 antibody (Abcam) was added to each cell lysate (300 μg), and the samples were incubated overnight at 4°C on a rotating wheel. Protein G rec-Protein G-Sepharose 4B Conjugate beads (50 μl) were added to the samples, then the samples were incubated for an additional 1 h at 4°C. The beads were spun down and washed three times in coimmunoprecipitation buffer. The beads were boiled at 95°C for 5 min in SDS sample buffer. Proteins were then analyzed by Western blot.
Immunohistochemistry: free-floating
Floating transgenic mouse brain tissue sections were washed 2 × 5 min in PBS and heated to 85°C in tris-sodium citrate buffer for 30 min. The slices were washed with 0.2% Triton X-100 for 15 min and blocked for 1 h with 5% donkey serum in PBS. Incubation was performed with primary antibodies for tau and SGs overnight in 5% donkey serum/PBS. Primary antibodies used were used as follows: for tau: MC-1, PHF-1, and Alz-50 (1:1000, generously provided by Peter Davies, Albert Einstein College of Medicine, Bronx, NY). For stress granules: TIA-1 (1:300, Santa Cruz Biotechnology, sc-1751), eIF3η (1:300, Santa Cruz Biotechnology, sc-16377), TTP (1:300, Santa Cruz Biotechnology, sc-14030), TDP-43 (1:1000, Proteintech, 12892–1-AP), FUS (1:300, Proteintech, 11570-1-AP), and G3BP (1:200, Becton Dickinson, 611126). The sections were then washed 3 × 10 min TBS-Triton X-100 (0.05%) and incubated in secondary antibodies for 2 h. Secondary antibodies were as follows: Dylight 488-conjugated donkey anti-host IgG and Dylight 594-conjugated donkey anti-host IgG (1:600, Jackson ImmunoResearch). The sections were washed 4 × 10 min in TBS/0.02% Tween; the third wash included DAPI (1 μg/ml). Background fluorescence was then quenched by incubating for 3 min in 1% sudan black followed by five quick dips in PBS and 4 × 5 min PBS washes. Mounting was performed using Prolong-Gold anti-fade reagent (Invitrogen).
Immunohistochemistry: paraffin-embedded
Tissue sections were deparaffinized and rehydrated, then treated with 0.1 m glycine for 30 min. The sections were then treated as described for the free-floating sections. Incubation was performed with primary antibodies for tau as follows: CP-13, PHF-1, mouse monoclonal Tau-5 (1:1000, Abcam, ab3931–500); and for SG as described above.
Immunocytochemistry: SH-SY5Y
Coverslips were washed 3 × 5 min in PBS and washed with 0.1% Triton X-100 for 15 min. Coverslips were washed 3 × 3 min in PBS and blocked for 1 h with 10% donkey serum in PBS. Incubation was performed with primary antibodies for tau and TIA-1 overnight in 5% donkey serum/PBS. Primary antibodies used were as follows, for tau: CP-13, PHF-1 (1:100, generously provided by Peter Davies); for stress granules: TIA-1 (1:300, Santa Cruz Biotechnology, sc-1751). Subsequent treatment was as for immunohistochemistry.
SH-SY5Y transfection protocol
Cells were seeded on 18 mm poly-d-lysine coated coverslips in 12-well plates (200,000/coverslip) 1 d before transfection in antibiotic-free media (1:1 DMEM/F-12, 10% FBS). Cells were transfected at 1:2 ratio DNA:Lipofectamine (Invitrogen), delivering 2 μg of total DNA per well. Lipofectamine in OptiMEM (Invitrogen) is incubated for 5 min before the addition of the DNA/OptiMEM. The transfection mixture is then allowed to sit for 20 min at room temperature before being added to the cells. Of media, 500 μl is removed and 500 μl of transfection mixture was added dropwise to each well. Cells were incubated for 24 h, after which the addition of 15 μm arsenite was added to half of the plates and allowed to incubate for 24 h. At 48 h after transfection, the cells were fixed in 4% paraformaldehyde and stored in PBS.
Confocal microscopy
Microscopy was performed using a Carl Zeiss LSM 510 META confocal laser-scanning microscope carrying lasers at 405, 488, 543, and 633 nm. Images were captured using a 63× oil objective. LSM proprietary software was used for digital image analysis. Images were combined into figures using Adobe Photoshop software. Quantitative and area analysis was done using an Olympus BX-60 (Olympus) equipped with epifluorescence optics, digital camera (AxioCam MRm; Zeiss), and analyzed with Zeiss Axiovision software. To analyze the potential interaction between tau inclusions and stress granules, quantification of colocalization was done using the Carl Zeiss LSM software, and R-values for the degree of colocalization were recorded. SG density was quantified using Fiji software by calculating the number of puncta >1 μm2 per 63× frame controlled for the number of cells.
Statistical analysis
All statistical calculations were done using one-way ANOVA followed by Tukey's post hoc test. Statistical calculations were performed using Graphpad Prism software. Error bars in the figures are shown as SEM.
Results
Stress granules increase in size and number with disease severity
To investigate the evolution of SG biology during the course of disease, we examined the distribution of markers for SGs and phospho-tau in JNPL3 and rTg4510 mice at varying stages of disease in the amygdala and cortex, respectively. SG inclusions increased in number, size, and distribution with the severity of the disease (Fig. 1A). We examined TIA-1 and eIF-3η, which are RNA-binding proteins that are considered to be primary SG proteins. TIA-1 is nuclear under basal conditions, but translocates to the cytoplasm with stress to form punctate granules (Fig. 1A) (Liu-Yesucevitz et al., 2011). The nuclear TIA-1 is not apparent because we used Sudan Black to quench low-level fluorescence (such as in nuclei) and highlight consolidated cytoplasmic TIA-1 reactivity, such as occurs in SGs, because this type of fluorescence tends to be very bright. Nuclear TIA-1 was readily apparent when tissues were examined without prestaining with Sudan Black (Fig. 1B).
Figure 1.

TIA-1-positive SGs increase with disease state. A, Immunohistochemistry for SGs (TIA-1, red) show increased abundance with disease state, and that colocalization with phospho-tau (PHF-1, S396,404, green) increases with disease state. Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence and highlight the fluorescence associated with pathology, which is brighter. Arrows point to colocalization. The bottom set of panels (High Mag) show higher magnification images of the images directly above (severe). Scale bar, 10 μm. B, TIA-1 immunofluorescence in amygdala from a non-transgenic mouse (SW Ctrl) or young JNPL3 mouse (Nml; age, 2 months). The endogenous nuclear TIA-1 fluorescence evident in sections not treated with Sudan Black is not evident in sections treated with Sudan Black. C, Colocalization of eIF3-positive (red) granules with PHF-1-positive (green) tau in brain tissue from JNPL3 mice. Arrows point to colocalization. The bottom set of panels (High Mag) show higher magnification images of the images directly above (severe). Nuclei are identified with DAPI (blue). D, Demonstration of antibody specificity by immunoabsorption. Sections of amygdala from a JNPL3 mouse with severe disease were probed with antibodies to PHF-1 and eTIA-1. Pre-adsorbing the primary antibody with the epitope peptides abolished all reactivity for the corresponding antibody. E, Density of SGs increase greatly during the moderate and severe disease state. Density was determined using Fiji software to quantify SG puncta per 63× frame with a threshold set to >1 μm2 (controlled for cell number). F, The extent of colocalization between SGs with phospho-tau increase with inclusion size. Colocalization is largely absent in Tg Normal and SW Control mice and is not reported here. G, Colocalization of Tau with TIA-1 increases greatly in JNPL3 mice with moderate and severe disease. Inclusions that react with PHF-1-positive Tau and TIA-1 show high levels of colocalization within each inclusion. H, Within each lesion, the TIA-1 and PHF-1 signal were highly localized. Scale bar, 10 μm.
eIF-3 is constitutively cytoplasmic, but participates in the formation of primary SGs. TIA-1 and eIF-3 were both evident in SGs at each stage of disease, and SGs containing these two proteins showed similar patterns of evolution during the course of disease, increasing in abundance with disease stage (TIA-1 shown in Fig. 1A; eIF-3 for severe only shown in Fig. 1C to demonstrate colocalization). Specificity of the TIA-1 and eIF3 reactivity was demonstrated by immunoadsorption, which eliminated all fluorescence (Fig. 1D, TIA-1 shown), consistent with our prior study using these antibodies (Liu-Yesucevitz et al., 2010). Statistically significant increases in SG density were evident in the transition from mild to moderate and moderate to severe disease (Fig. 1E, p < 0.001). However, even early stage mice showed a trend (p < 0.1) toward increased numbers of SGs compared with non-transgenic mice, which indicates the presence of stress early in the disease course, either due to overexpression of tau or the presence of pathological tau that is not yet apparent by immunohistochemistry. Similar results were observed for rTg4510 mice (under conditions of tau induction) of varying disease stages (Fig. 2).
Figure 2.

SG pathology in P301L tau transgenic mice. The patterns of evolution of reactivity for the RNA-binding proteins were very similar between the JNPL3 and rTg4510 mouse models, except that the latter develops more severe pathology at end-stage disease. Few SGs are evident in 2-month-old mice with no evident disease and no evident phospho-tau (PHF-1) reactivity. In moderate disease (5 months), TIA-1 (red) reactivity is present but frequently does not colocalize with tau pathology (PHF-1, green). In severe disease (8 months), TIA-1 reactivity strongly colocalizes with tau pathology. In contrast, G3BP (red) reactivity frequently does not colocalize with tau pathology. Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence. Scale bar, 10 μm.
Interestingly, the size distribution of SGs shifted toward larger SGs with increasing disease severity (Fig. 1F, Table 2). JNPL3 tau mice at an early, or predisease stage showed SGs that were overwhelmingly <5 μm and generally ∼1–2 μm in diameter, while mice at a moderate stage of disease had large inclusions (Fig. 1A,F). The SGs that were present in early stage mice showed a distribution that included perinuclear reactivity, as well as reactivity that was distal from nuclei and presumably in processes (Fig. 1A). The size of SG inclusion showed a distinct transition in mice with moderate disease (Fig. 1F). The frequency of SGs exhibiting a diameter that was >5 μm increased over tenfold (Fig. 1F). Mice at the severely affected stage showed SG reactivity that was broadly distributed and generally >5 μm (Fig. 1A,C,F).
Table 2.
JNPL3 tau mouse, SW control mouse, human control, and human AD brain quantification of tau and SG inclusions and colocalization of tau and SG pathology (±SD)
| Tissue | Density of SG/ 63× field | Average area of SG inclusions (TIA1+) (μm2) | % Colocalization of Tau (PHF1+) with SG | Average degree of colocalization |
|---|---|---|---|---|
| Mouse | ||||
| SW control | 0.9 ± 0.7 | 2.7 ± 0.7 | n/a | n/a |
| Tg normal | 3.0 ± 0.8 | 2.7 ± 0.9 | n/a | n/a |
| Tg mild | 3.7 ± 0.7 | 2.8 ± 1.1 | 11.1 ± 11.7 | 0.692 ± 0.079 |
| Tg moderate | 8.7 ± 0.9 | 5.0 ± 2.3 | 45.4 ± 8.8 | 0.778 ± 0.084 |
| Tg severe | 22.2 ± 2.5 | 8.4 ± 3.8 | 69.1 ± 8.7 | 0.824 ± 0.098 |
| Human | ||||
| Control | 0.9 ± 0.5 | 4.3 ± 1.8 | 1.1 ± 0.6 | 0.387 ± 0.172 |
| Alzheimer's | 29.1 ± 3.2 | 9.5 ± 3.0 | 64.2 ± 6.6 | 0.840 ± 0.069 |
The term Tg Normal refers to the age of 2 months, when no tau pathology is present.
The anatomic distribution of SGs also paralleled the distribution of phospho-tau protein. SGs in the JNPL3 mouse tissue were found only in areas where there was tau pathology, such as the brain stem and amygdala. SG inclusions were not observed in cortex, where only sparse tau pathology is observed in the JNPL3 mouse strain (data not shown). Similarly, SGs in the rTg4510 mouse tissue were found only in the cortex and hippocampus, where tau pathology occurs and not in the brain stem or amygdala (Fig. 2).
Stress granules merge with tau pathology at moderate and severe stages
Next, we investigated the relationship between SG inclusions and hyperphosphorylated or conformationally altered tau reactivity across disease state. We examined three different antibodies that are associated with tau pathology, PHF-1, MC-1, and Alz-50, and compared colocalization with TIA-1, which is an accepted marker of SGs (Kedersha et al., 2000; Anderson and Kedersha, 2008). Results were largely consistent for each tau marker (Fig. 1G). Colocalization of SGs and pathological tau greatly increased with disease severity (Fig. 1G). In mice at an early stage, with mild pathology, SG and tau inclusions existed largely in distinct physical locations; only 10% of tau inclusions colocalized with SG (Fig. 1A,G). As disease progressed the percentage of colocalized inclusions increased. Approximately 50% of tau inclusions colocalized with TIA-1-positive inclusions in mice with moderate pathology (Fig. 1A,G). Colocalization between tau and TIA-1 inclusions increased to ∼70% in mice with severe pathology (Fig. 1A,G).
Inclusions exhibiting both tau and TIA-1 reactivity were examined to determine the degree of colocalization (R-value). The degree of colocalization provides insight into the relationship between tau and the SG proteins within individual inclusions. The degree of tau/TIA-1 colocalization within inclusions was found to increase with disease stage, from mild to moderate to severe, with R-values ranging from 0.643 to 0.751 to 0.819 respectively (Fig. 1A,H, p < 0.001). Similar results were obtained for each of the three antibodies used, PHF-1, MC-1, and Alz-50 (Figs. 1H).
The frequency of large inclusions identified by either phospho-tau or SG markers increased with disease severity. To investigate whether the structure of large and small inclusions might differ, we stratified Tau/TIA-1 colocalization by inclusion size. Colocalization between inclusions containing phospho-tau and TIA-1 steadily increased with inclusion size (Fig. 1A,F). The correlation between inclusion size and percentage of colocalization was apparent at every stage of disease, although the abundance of large tau inclusions was low in mice at early stages of the disease (Fig. 1A). Large tau inclusions strongly colocalized with the TIA-1 SG marker in all disease states, but the rare large inclusion in mildly affected mice showed a lower percentage of colocalization than large inclusions in moderately or severely affected mice.
Small SG inclusions (including those surrounding large inclusions) did not colocalize with phospho-tau inclusions even in areas with high SG density and even in mice with severe pathology (Fig. 1A,D). A corollary of this observation is that many cells without visible phospho-tau display abundant SG pathology in mice with moderate and severe pathology (Fig. 1A,D), which highlights the presence of a significant stress response in the absence of visible phospho-tau.
Microglia and astrocytes form SGs
We hypothesized that the presence of SGs in cells lacking visible pathological tau could be due to the occurrence of SGs in non-neuronal cells. This hypothesis prompted us to determine how the distribution of SGs correlated with cell type. JNPL3 mouse brain tissue sections were labeled with markers for neurons (NeuN), reactive astrocytes (GFAP), and microglia (Iba1), as well as with TIA-1. SGs identified with TIA-1 were evident in NeuN-positive cells at every stage of disease (Fig. 3A). Double labeling with NeuN also emphasizes the changes in TIA-1 distribution with disease state. NeuN is a nuclear protein, while TIA-1 can be nuclear or cytoplasmic. TIA-1 reactivity in non-transgenic and asymptomatic JNPL3 mice was largely evident in nuclei, where it directly colocalized with NeuN. In contrast, TIA-1 reactivity in JNPL3 mice with more overt pathology was evident in both cytoplasmic (frequently as granules) and nuclear domains, which is consistent with the cytoplasmic translocation and SG formation that is known to occur with TIA-1 in response to stress. SG markers were also present in astrocytes, and appeared to steadily increase with disease stage, although total SG load remained far below that of neurons (Fig. 3B). SGs were evident in microglia (identified by Iba-1 reactivity), but appeared to peak at the mild stage of disease, and then decrease in microglial localization with increasing severity of disease (Fig. 3C). Microglial SGs were most prominent in mice with mild pathology, and less abundant in mice with moderate pathology. In brain tissue from cases with severe disease, cytoplasmic TIA-1 granules overlapped with Iba-1 reactivity rarely, and overlap that did present was not in the same focal plane, suggesting that the proteins were not colocalized (Fig. 3C). These data suggest that the relationship of SGs to the disease process might differ between neurons and inflammatory cells, such as microglia.
Figure 3.

Correlation between TIA-1-positive SG expression and cell type. A, Colabeling TIA-1 (red) to identify SGs and NeuN (green) to identify neurons indicates that SG formation in neurons increases with disease state. Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence. B, Colabeling identifies SGs (TIA-1, red) and astrocytes (GFAP, green) and shows modestly the presence of cytoplasmic SGs in astrocytes at each stage of the disease process. C, Colabeling SGs (TIA-1, red) and microglia (Iba-1, green) indicates that SGs are present in microglia mainly at the mild stage of disease. White arrows in the merged panels point to sites of colocalization. Scale bar, 10 μm.
G3BP and TTP identify RNA granules that often are not associated with PHF-1
The composition of the RNA protein complexes that comprise RNA granules vary with the complex and the cellular conditions. For instance, while TIA-1 is associated with translational silencing, G3BP and TTP are associated with degradation of some mRNA and translational stimulation of other mRNA, although they both have many different functions (Irvine et al., 2004; Zekri et al., 2005; Ronkina et al., 2010; Liu-Yesucevitz et al., 2011). We examined the expression of four other SG-associated proteins to explore specific SG profiles in neurodegenerative disease. The proteins examined were G3BP, TTP, FMRP, SMN, and Hu antigen R (HuR) (Anderson and Kedersha, 2008). Disease-related differences were observed for G3BP and TTP. Three other secondary SG protein components, FMRP, HuR, and SMN, did not show any change in expression pattern with disease state (data not shown). Analysis of the JNPL3 mouse amygdala showed a steady increase in the amount of punctate cytoplasmic G3BP reactivity with disease severity, paralleling the trend for TIA-1 (Fig. 4A). The number of all granules and number of large granules increased with disease state, suggesting formation of large RNA granules. However, unlike TIA-1, G3BP-positive granules showed only weak colocalization with PHF-1 or TIA-1 (Fig. 4A, PHF-1 shown). Interestingly, large G3BP granules were strongly evident at the onset of disease (mild), but not in healthy mice, suggesting that G3BP might be a sensitive marker of disease onset (Fig. 4A). The specificity of the G3BP reactivity was demonstrated by immunoadsorption, which eliminated all fluorescence (Fig. 4B). Inclusions containing TTP also increased with disease state (Fig. 4D). TTP-positive inclusions showed a pattern that varied distinctly with disease state. Up through the moderate stage of disease, TTP showed little if any colocalization with PHF-1-positive inclusions, even when apparently occurring within the same cell (Fig. 4D). However, in severe disease, TTP was found to strongly colocalize with phospho-tau protein (Fig. 4D). Specificity of the TTP reactivity was demonstrated by immunoadsorption, which eliminated all fluorescence (Fig. 4C). The pattern of G3BP reactivity in rTg4510 mice was similar to that for JNPL3 mice (Fig. 4E). The presence of inclusions composed of RNA-binding proteins that accumulate with disease but that are often not colocalized with PHF-1 reactivity contrasts with the pattern seen for TIA-1 and suggests that the strong colocalization of TIA-1 with PHF-1-positive tau reflects a selective association between pathological tau protein and particular RNA-binding proteins. This highlights a potentially important biological process, and also provides an internal control indicating that colocalization between pathological tau and TIA-1 is specific and does not reflect a non-specific association between aggregated proteins. Finally, the presence of G3BP-positive inclusions in JNPL3 and rTg4510 mouse brains identifies a novel type of inclusion occurring in the brain of an animal with tauopathy whose localization appears to be largely independent of the classical NFT.
Figure 4.
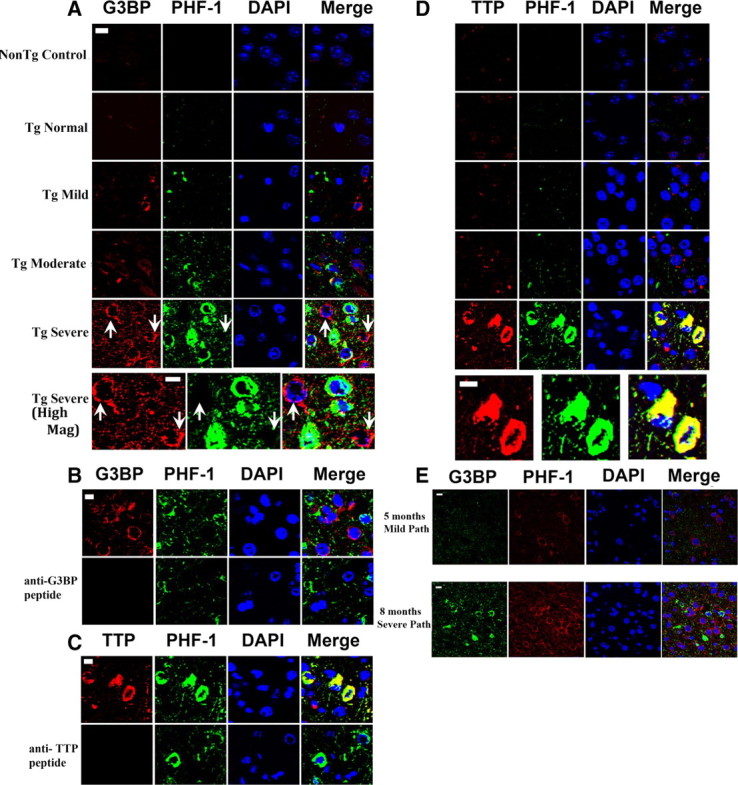
G3BP occurs in cells lacking PHF-1 reactivity. A, The cytoplasmic reactivity of G3BP (red) occurs early in the disease and increases with disease state but does not colocalize with phospho-tau protein (PHF-1, green), even at the severe stage of disease. Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence. The bottom set of panels (High Mag) show higher magnification images of the images directly above (severe). Scale bar, 10 μm. B, C, Demonstration of antibody specificity by immunoabsorption. Sections of amygdala from a JNPL3 mouse with severe disease were probed with antibodies to PHF-1 and either G3BP (B) or TTP (C). Pre-adsorbing the primary antibody with the epitope peptides abolished all reactivity for the corresponding antibody. D, The cytoplasmic reactivity of TTP (red) increases with disease state and does not colocalize with phospho-tau protein (green), until the stage of severe disease. The bottom set of panels (High Mag) show higher magnification images of the images directly above (severe). E, The pattern of evolution of reactivity for G3BP was very similar between the JNPL3 and rTg4510 mouse models, except that the latter develops more severe pathology at end-stage disease. Few SGs were evident in 5-month-old mice with no evident disease and no evident phospho-tau (PHF-1) reactivity. In severe disease (8 months), G3BP (red) reactivity frequently does not colocalize with tau pathology. Nuclei are identified with DAPI (blue). Scale bar, 10 μm.
TDP-43 and FUS exhibit cytoplasmic translocation early in the disease process
Some frontotemporal lobar dementias are not associated with tau inclusions but are associated with TDP-43 inclusions (FTLD-U). We recently reported that the TDP-43 inclusions present in brains of subjects with FTLD-U are associated with other markers of SGs, such as TIA-1 (Liu-Yesucevitz et al., 2010). However, TDP-43 inclusions are not reported in cases of FTDP-17 associated with tauopathy, such as subjects with the P301L tau mutation. The presence of SGs/RNA granules associated with TIA-1, eIF3, G3BP, and TTP raised a question of whether TDP-43 inclusions might also form in the brains of JNPL3 mice. We double labeled sections for TDP-43 and TIA-1 or NeuN to determine colocalization of TDP-43 with SGs and neuronal localization (Fig. 5A,B). TDP-43 was largely nuclear, and showed a modest increase in cytoplasmic inclusion formation with disease severity (Fig. 5A). Nuclear TDP-43 reactivity tended to decrease with disease severity (Fig. 5A). TDP-43 expression was largely localized to neurons (Fig. 5B). Interestingly, the cytoplasmic TDP-43 granules tended to occur only in brain tissue from animals with moderate or severe pathology (Fig. 5A), and frequently did not colocalize with TIA-1 or PHF-1 inclusions (Fig. 5A).
Figure 5.

TDP-43 and FUS form cytoplasmic inclusions in transgenic mice with moderate to severe disease. A, Immunocytochemical reactivity of TDP-43 (green), TIA-1 (red), and nuclei (DAPI, blue) in brains of mice of varying disease states. B, Immunocytochemical reactivity of TDP-43 (green), neurons (NeuN, red), and nuclei (DAPI, blue) in brains of mice of moderate disease state. Sections were treated with Sudan Black to quench endogenous fluorescence. C, Immunocytochemical reactivity of FUS (red), PHF-1 (green), and nuclei (DAPI, blue) in brains of mice of varying disease states. D, Immunocytochemical reactivity of FUS (green), TIA-1 (red), and nuclei (DAPI, blue) in brains of mice of moderate disease state. Scale bar, 10 μm.
Studies of amyotrophic lateral sclerosis have identified mutations in FUS, which is another RNA-binding protein with properties similar to that of TDP-43 (Kwiatkowski et al., 2009; Vance et al., 2009). Studies in humans show that FUS also forms inclusions in human brain, but that FUS inclusions mainly occur in brains lacking TDP-43 inclusions (Blair et al., 2010). This background led us to investigate the expression of FUS in the JNPL3 mice. Expression of FUS was largely nuclear but demonstrated some formation of cytoplasmic granules with increasing disease severity (Fig. 5C). Nuclear FUS expression was strong throughout the disease process and exhibited a modest increase with disease severity (Fig. 5C). FUS expression was localized to neurons, and did not colocalize with PHF-1 reactivity or with TIA-1 reactivity (Fig. 5D). The pattern of FUS reactivity also differed from that of TDP-43 in that nuclear expression remained strong throughout the disease process, and FUS formed larger cytoplasmic granules than TDP-43 did (Fig. 5A,C). These data suggest that TDP-43 and FUS react to the disease process in mice with tauopathy, but exhibit changes that are more modest than that for TIA-1, G3BP, or TTP.
SG markers colocalize with tau in postmortem human brain tissue
Next, we investigated whether tau inclusions in brains (frontal cortex) of subjects with AD also colocalized with SG markers (Fig. 6A). Differentiating SG reactivity from lipofuscin can be challenging, so to overcome this problem we use Sudan Black to quench lipofuscin fluorescence (Liu-Yesucevitz et al., 2010). Use of Sudan Black highlights consolidated cytoplasmic TIA-1 reactivity, such as occurs in SGs, because this type of fluorescence tends to be very bright. However, weaker fluorescence, such as the endogenous nuclear TIA-1 reactivity, tends to be quenched; hence nuclear TIA-1 is difficult to observe in tissues treated with Sudan Black (Fig. 6B). We investigated a range of anti-tau antibodies, including antibodies detecting phosphorylated tau epitopes (PHF-1, CP-13), antibodies detecting pathological conformational tau epitopes (MC-1, Alz-50), and antibody detecting total tau (Tau-5). In each case, reactivity with the tau epitopes (Fig. 6A, PHF-1, CP-13, and Tau-5) showed a strong degree of colocalization with TIA-1, a classic SG marker (Fig. 6A, Table 2). As was the case with the JNPL3 mice, larger tau inclusions were more likely to be colocalized with SG markers than the small tau inclusions. When examining the brain sections from the cognitively normal control samples, some sparse tau reactivity was found throughout the frontal cortex as has been previously noted (Jaworski et al., 2009; Glodzik et al., 2011). SGs were also present in older normal control brain tissues, but were small to medium in size, sparse, and not associated with tau pathology.
Figure 6.

Colocalization of TIA-1 with tau pathology in the Alzheimer brain. A, Frontal cortex from a subject with AD was probed with antibodies to tau (phospho-tau, CP-13, PHF-1; total tau, Tau-5, green) and TIA-1 (red). Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence. Large inclusions containing TIA-1 colocalize with each of the markers of tau protein that were tested. Figures from AD case 5 shown. B, TIA-1 immunofluorescence in Alzheimer cortex (frontal) and Control cortex (occipital) treated with ± Sudan Black. The endogenous nuclear TIA-1 fluorescence is not evident in sections treated with Sudan Black. Scale bar, 10 μm.
SG markers in human AD brain show similar molecular pathology to JNPL3 tissue
As with the JNPL3 mouse tissue, we note modest FUS and TDP-43 translocation and inclusion formation in the cytoplasm of the human AD brain (Fig. 7, frontal cortex). FUS reactivity colocalized with TIA-1 moderately; however, TDP-43 colocalized with TIA-1 inclusions infrequently (Fig. 7). Strong G3BP and TTP signals were also evident in the human AD tissue (Fig. 7A–C). The G3BP signal did not colocalize with the SG marker, TIA-1 (Fig. 7A,C). We also examined the relationship to phospho-tau. FUS, TDP-43, and G3BP did not show colocalization with PHF-1 reactivity, which is similar to the results obtained with the JNPL3 mouse (Fig. 7). TTP also showed strong reactivity, and the reactivity partially colocalized with tau inclusions, which correlates with results observed in the most severe stage of pathology in the JNPL3 mouse. Similar results were also observed in five cases of FTDP-17 (Fig. 7D).
Figure 7.

Expression of FUS, G3BP, TTP, and TDP-43 in the Alzheimer brain. A, Frontal cortex from a subject with AD was probed with antibodies to FUS, G3BP, and TDP-43 (green) and double labeled with anti-TIA-1 antibody (red). Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence. FUS showed some colocalization with TIA-1, while neither G3BP nor TDP-43 showed significant colocalization with TIA-1. Control represents a health control cortex with rabbit serum substituted for primary antibody; similar results were observed using AD cortex. B, Frontal cortex from a subject with AD probed with antibodies to FUS, G3BP, TDP-43, and TTP (green) and double labeled with PHF-1 antibody (red). Nuclei are identified with DAPI (blue). FUS, G3BP, and TDP-43 did not show significant colocalization with PHF-1, while TTP did partially colocalize with PHF-1. Control represents FTDP-17 cortex with rabbit serum substituted for primary antibody. Figures from AD cases 5 and 10 shown. C, Merged images from B shown at higher magnification. Red, RNA-binding protein; green, PHF-1; blue, DAPI. D, Frontal cortex from different subjects with FTDP-17 (numerical identifications shown) probed with antibodies to TTP, TIA-1, and G3BP (green), and double labeled with PHF-1 antibody (red). Nuclei are identified with DAPI (blue). Only the green and merged images are shown. Scale bar, 10 μm.
The presence of SGs in multiple cell types in the JNPL3 mice led us to investigate the cell type distribution of TIA-1 in the brains of subjects with AD. The cell type markers NeuN, Iba1, and GFAP were used as markers for neurons, microglia, and astrocytes, respectively. In all the frontal cortex samples analyzed, TIA-1-positive SG formation was evident in all cell types with the predominance of SGs found in neurons (Fig. 8). Marked microgliosis and astrocytosis were evident from the morphology of reactivity, and TIA-1-positive SGs were observed in both cell types, perhaps indicating three divergent roles for TIA-1-positive SGs in human AD tissue (Fig. 8).
Figure 8.

TIA-1-positive SGs are evident in neurons, microglia, and astrocytes in the Alzheimer brain. A, Frontal cortex tissue from a subject with AD was incubated with antibodies (green) to NeuN, Iba-1, and GFAP to identify neurons, microglia, and astrocytes, respectively. TIA-1-positive (red) reactivity was present in all three cell types. Nuclei are identified with DAPI (blue). Sections were treated with Sudan Black to quench endogenous fluorescence. B, A similar study performed in frontal cortex from a healthy, aged control. Figures from AD cases 5 and 10 and neurologically normal control case 1 are shown. Scale bar, 10 μm.
TIA-1 and TTP protein levels increase with disease progression, with soluble TIA-1 protein complexes interacting with tau protein
Formation of SGs and other RNA granules is driven by reversible aggregation mediated by hydrophobic domains of the RNA-binding proteins that frequently have homology to prion proteins. Because the progression of tauopathies is marked by development of highly insoluble tau fibrils, we were curious about whether RNA-binding proteins involved in the pathophysiology of tauopathies might also show progressive insolubility. Whole brain lysates were obtained from inducible rTg4510 Tau transgenic mice at ages ranging from 3–8 months, which represented mild to severe stages of disease. These samples were biochemically fractionated to obtain Triton-X soluble and insoluble fractions, as well as a heat stable (soluble tau) fraction that was produced separately. The samples were immunoblotted for TIA-1, TTP, G3BP, TDP-43, and FUS.
Results obtained from the whole brain lysates indicated that the levels of TIA-1, TTP, and G3BP increased over the course of the disease; however, changes in G3BP were only significant in the transition from moderate to severe disease (Fig. 9A). The small increase in G3BP observed in whole brain lysates contrasts with the large increase in G3BP observed on immunocytochemistry and might reflect the enhanced ability to image RNA granules due to consolidation of the fluorescence in response to granule formation. In contrast, levels of TDP-43 and FUS remained relatively constant throughout the disease course. As expected, levels of PHF-1 increased with disease state as is known in this mouse model (Fig. 9A). Next, we examined the soluble tau (heat stable) fraction to see whether any of the RNA-binding proteins partition into that fraction with tau protein. TIA-1 partitioned with soluble tau, exhibiting levels that increased with disease severity (Fig. 9C,D). Both TIA-1 and TTP exhibited increased levels in the soluble tau fraction in mice with severe pathology, but variable levels in mice with mild or moderate pathology (Fig. 9C,D). Next, we examined the Triton insoluble fractions. TDP-43, FUS, and phosho-tau (identified with the PHF-1 antibody) showed strong reactivity in the Triton insoluble fractions, but TIA-1, TTP, and G3BP were not evident (Fig. 9E). The absence of TIA-1 is consistent with our results published previously (Liu-Yesucevitz et al., 2010). We also examined the Sarkosyl insoluble (aggregated tau) fraction. None of the RNA-binding protein markers were evident in the Sarkosyl insoluble fraction (data not shown).
Figure 9.

Disease severity modulates levels and biochemical behavior of RNA-binding proteins. A, Immunoblots showing levels of TIA-1, TTP, G3BP, FUS, TDP-43, Tau5, and PHF-1 in whole brain lysates from rTg4510 mice at differing levels of disease severity. B, Quantification of immunoblots of whole brain lysates from rTg4510 mice (N = 3). C, Immunoblots showing levels of TIA-1, TTP, G3BP, FUS, and TDP-43 in heat stable fractions (soluble tau) from rTg4510 mice at differing levels of disease severity. D, Quantification of immunoblots of heat stable fractions (soluble tau) from rTg4510 mice (N = 3). E, Immunoblots showing levels of TIA-1, TTP, G3BP, FUS, TDP-43, and PHF-1 tau, in Triton-X insoluble lysates from rTg4510 mice at differing levels of disease severity. F, Immunoprecipitation of total tau with the Tau-5 antibody, followed by immunoblotting with Tau-5, TIA-1, TTP, or G3BP. Note: No immunoreactivity was observed at the molecular weight of holo-G3BP (47 KDa), however, a band reactive with the anti-G3BP antibody was apparent at 35 KDa. This band is labeled as 35 KDa G3 band. G, Quantification of immunoblots from the Tau-5 immunoprecipitations. H, Immunoprecipitation of phosphorylated tau with the PHF-1 antibody, followed by immunoblotting with Tau-5, TIA-1, TTP, or G3BP. I, Quantification of immunoblots from the PHF-1 immunoprecipitations.
Next, we investigated whether SG proteins, such as TIA-1, directly associate with tau. Total and phosphorylated tau were immunoprecipitated with the antibodies Tau-5 (total tau) or PHF-1 (phospho-tau), and the resulting immunoprecipitates were immunoblotted with antibodies to TIA-1, TTP, or G3BP (Fig. 9F,H). TIA-1 and TTP associate with tau, and also exhibit increasing association with phospho-tau with increasing severity of disease (Fig. 9F–I). G3BP was not detectable in PHF-1 (or tau-5) immunoprecipitates, but an immunoreactive band at 35 KD that reacted with the anti-G3BP antibody was readily apparent in the tau-5 immunoprecipitates, but only weakly apparent in the PHF-1 immunoprecipitates (Fig. 9F–I). These data present evidence suggesting a direct interaction between tau and some RNA-binding proteins.
Overexpressing TIA-1 increases formation of PHF-1/CP13- positive inclusions
Binding of TIA-1 to tau suggests that the two proteins might interact under physiological or pathophysiological conditions. To investigate this putative interaction, SH-SY5Y cells were transfected with TIA-1 ± WT or P301L Tau. The following day the cells were treated ± arsenite (15 μm, a mild treatment) to induce formation of stress granules, and fixed after 24 h (Liu-Yesucevitz et al., 2010). The cells were double labeled with antibodies to PHF-1 and TIA-1 (Fig. 10). Similar experiments were also performed using CP13 for the immunocytochemistry; the results observed with CP13 labeling were the same as for PHF-1 labeling (data not shown). Phospho-tau inclusions (PHF-1 positive) were not observed under basal conditions, even when tau or TIA-1 were overexpressed (Fig. 10A,C). However, TIA-1-positive inclusions were apparent in cells overexpressing TIA-1, which is similar to what we, and others, have observed previously (Fig. 10A,B) (Liu-Yesucevitz et al., 2010). Interestingly, coexpressing TIA-1 with tau (WT or P301L) yielded more TIA-1-positive inclusions (Fig. 10A,B). Treating the cells with arsenite (15 μm, 24 h) greatly increased the number of inclusions positive for tau and TIA-1 (Fig. 10A–C). Overexpressing tau with TIA-1 greatly increased the formation of PHF-1-positive inclusions compared with cells overexpressing tau alone, TIA-1 alone, or neither (Fig. 10A,B). Some of the tau inclusions exhibited double labeling for TIA-1, however, the fraction of tau inclusions showing robust colocalization with TIA-1 was less than unity (Fig. 10A–C). Expressing tau also increased the number of TIA-1-positive inclusions, despite no striking differences evident in the levels of PHF-1 (or CP13) positive tau (Fig. 10A); no difference was observed in tau or SG inclusion formation between WT and P301L tau (Fig. 10B). These data suggest that the TIA-1 stimulates tau inclusion formation under stressful conditions, and that tau expression facilitates SG formation.
Figure 10.

Coexpressing TIA-1 and tau induces inclusion formation in the context of exogenous stress. A, SH-SY5Y cells were transfected with TIA-1 ± tau (WT or P301L) and grown under basal conditions (left panels labeled “No Arsenite”) or with arsenite (“+ Arsenite”, 15 μm, 24 h). Arrows point to examples of inclusions that formed. The white box delineates areas that are shown at higher magnification in the right-hand column. Scale bar, 3 μm. B, Quantification of the number of TIA-1-positive inclusions with a size >1 μm. C, Quantification of the number of PHF-1 tau-positive inclusions with a size >1 μm.
Discussion
Recent studies suggest that stress granules are a form of pathology associated with neurodegenerative diseases. Studies of Huntington's disease, Creutzfeldt-Jakob disease, and our recent study of ALS and frontotemporal dementia (FTLD-U) all exhibit inclusions composed of the pathological protein aggregate that are colocalized with TIA-1, a marker of SGs (Waelter et al., 2001; Goggin et al., 2008; Liu-Yesucevitz et al., 2010). The current study examines the evolution of SG pathology in two transgenic mouse models of tauopathy, in human cases with AD, and in human cases of FTDP-17. It is important to note that the tau-driven transgenic mice used in this study model the human disease FTDP-17, whereas in AD both β-amyloid peptide and tau protein play a role in pathogenesis. Future studies will examine the relative roles of tau and Aβ in SG induction. Our results indicate that TIA-1-positive SGs are abundant in cases of AD and FTDP-17, and they increase with disease severity in the JNPL3 and Tg4510 mutant tau mouse models. Colocalization of tau with TIA-1-positive SGs varies with disease severity. Pathological tau and SGs occur in separate compartments when inclusions are small (1–3 μm), but colocalization increases dramatically with disease severity and SG size. The absence of colocalization in mice with little or mild pathology partially reflects the prevalence of SGs at this stage in inflammatory cells, including microglia and astrocytes. However, SGs are also present in neurons at the mild stage, which suggests that SGs and pathological tau (as defined by the epitopes of PHF-1, MC-1, and Alz-50) form at physically distinct sites. As the disease process progresses, SGs become larger, more strongly localized to neurons and more strongly colocalized with pathological tau. Thus, our study uncovers a complex evolution of SG pathology that varies by cell type and degree of colocalization with pathological tau depending on disease stage.
The colocalization of SGs with tau pathology might reflect a functional interaction between SGs and tau. Microtubule-associated proteins are known to regulate SG formation (Chernov et al., 2009; Tsai et al., 2009). Expansions of polyglutamine repeats in ataxin-2, a RNA-binding protein, are associated with supranuclear palsy (Ross et al., 2011). Our own data demonstrate that SG proteins, such as TIA-1 and TTP, bind phosphorylated tau, and that binding increases with disease severity. Using the mouse models, we observed that TIA-1 and TTP inclusions appear at locations that are physically distinct from the initial inclusions of phosphorylated tau. However, the transfection experiments suggest that TIA-1 can stimulate formation of tau inclusions under conditions of stress, and that some of these inclusions colocalize with TIA-1. The reason for the difference between the in vivo and cellular experiments is unclear at this juncture, but might reflect the acute nature of the transfection experiments or stresses associated with transient transfection. The inclusions that formed in transfected cells did not appear to be filamentous, which suggests that they do not contain mature, aggregated tau, such as might occur in NFTs. However, this putative interaction raises the possibility that SG proteins might contribute to generating tau pathology, perhaps by creating local environments with high concentrations of phosho-tau; conversely, tau pathology might stimulate formation of SGs through direct and indirect mechanisms.
The contrasting trajectory of SG evolution in neurons, astrocytes, and microglia provides insight into the disparate responses of these three cell types to disease progression. Neurons exhibit steady increases in SGs throughout the disease course. In contrast, microglia exhibit a bell-shaped SG evolution, with levels of microglial SGs peaking early in the disease course and decreasing with disease severity. The reduction in SGs among inflammatory cells might reflect the evolution of inflammation with disease state. SG proteins, such as TTP, have been shown to associate with transcripts for inflammatory proteins, such as cyclooxygenase-2 (COX-2) and tumor necrosis factor-α (TNF-α), and they appear to function as translational silencers of the proinflammatory cascade (Piecyk et al., 2000; Khera et al., 2010). Extending this scenario to the brain raises the possibility that SGs function in inflammatory cells as part of a mechanism to dampen the inflammatory response. As the disease progresses, protein aggregates and degenerating neurons might overcome the immunosuppressive defenses, inhibit SG formation in inflammatory cells, and activate the inflammatory response. In contrast, neuronal SGs are thought to promote the stress response, by shifting protein synthesis toward uncapped mRNA, which includes protective proteins such as heat shock proteins (Thomas et al., 2011). Thus, increasing neuropathology could lead to a widespread SG response in neurons but a dampened SG response in inflammatory cells.
The differential patterns of expression of particular SG proteins are a particularly novel finding in our study. This is the first study to compare the evolution of RNA granule formation by different RNA-binding proteins over the course of disease. Our study highlights a close relationship between TIA-1-positive granules and tau pathology. In contrast, TTP and G3BP also show strong disease-linked increases in cytoplasmic levels and granule formation with increasing disease severity, but the inclusions often occur separately from classic markers of pathological tau. TDP-43 and FUS show modest increases, and FMRP, SMN, and HuR exhibit no increase in levels or granule formation with disease severity.
Cytoplasmic granules composed of TTP and G3BP are particularly striking because they become more abundant and widely distributed as disease severity progresses, yet G3BP does not colocalize with tau inclusions at all, and TTP colocalizes only late in the disease process. The presence of RNA granules that accumulate in tauopathies, but are not colocalized with tau pathology, points to novel types of molecular neuropathology that is not associated with classic markers of disease pathology. This contrasting localization of TIA-1/Tau, G3BP, and TTP pathologies might also reflect independent mechanisms controlling the process of aggregation. The tendency of TIA, G3BP, and TTP to aggregate contrasts with the behavior of FMRP, SMN and HuR, yet all of these proteins have similar structures and a similar tendency to stimulate formation of SGs in cell culture, and they can also be observed in other types of granules in healthy neurons (Liu-Yesucevitz et al., 2010). The differential responses in tauopathies highlight potentially different signaling pathways regulating each type of protein.
The RNA-binding proteins addressed in this study (TIA-1, G3BP, TTP, FUS, and TDP-43) are essential players in RNA metabolism, with the mRNPs (messenger ribonucleoprotein particles) forming a dynamic regulatory system for all aspects of the life of an mRNA, including nuclear processing, transport, translation, and decay (Anderson and Kedersha, 2008). These proteins are key in the coordination of gene expression of many proteins, and their disruption could impair cell function and interfere with appropriate distribution and translation of mRNA in response to signaling (Irvine et al., 2004; Anderson and Kedersha, 2008; Ronkina et al., 2010). These proteins are able to act either as activators, repressors, or both with substrate selectivity. Of particular interest to the scope of this paper are the function of TIA-1 and TTP in the repression of inflammatory transcripts COX-2 and TNF-α, thus dampening the cellular inflammatory response (discussed above) (Piecyk et al., 2000; Pietrzik et al., 2002; Cok et al., 2003). In contrast, G3BP is able to act as either an activator or repressor in a transcript-dependent manner, and has been shown to be involved in cell proliferation, cytoskeletal reorganization, cell adhesion, and protein trafficking (Irvine et al., 2004; Zekri et al., 2005; McDonald et al., 2011). The knock-out of G3BP gene is embryonically lethal, indicating the integral role G3BP plays in development and basic cell functions (Irvine et al., 2004). The relationship of G3BP to protein turnover could be particularly important to disease processes. G3BP is able to function as regulator of the ubiquitin-specific protease, USP-10, a component of the deubiquitinating complex (Soncini et al., 2001). The interaction of G3BP with USP-10 rescues Sec23 and β-COP transcripts from degradation, maintaining the activity of anterograde and retrograde protein secretion pathways (Cohen et al., 2003a,b). Thus, G3BP might form cytoplasmic granules in neurons as part of a stress response that promotes survival. G3BP was also found to bind tau mRNA and stabilize the transcript, increasing its translation, which promotes microtubule stability, neurite outgrowth, and cytoskeletal remodeling (Atlas et al., 2004). The presence of cytoplasmic G3BP granules in neurons that are PHF-1-negative raises the possibility that G3BP might function to protect against hyperphosphorylated tau (specifically) or pathological tau (more generally) and related dysfunction of the microtubule network.
This study of SG evolution in tauopathies highlights previously uncharacterized molecular pathologies in the brains of patients with AD and in animal models of FTDP-17. SG biology offers multiple potentially important avenues of further study. The process of protein aggregation leading to SG formation can be rapidly reversed through signal transduction mechanisms, such as pathways regulated by eIF2α (Kedersha and Anderson, 2002). Such signal transduction pathways highlight potentially novel targets for therapeutic development in AD. The differential expression of RNA-binding proteins in tauopathies also suggests that these proteins might represent novel biomarkers whose expression, aggregation, or posttranslational modification might reflect distinct elements of the disease process. It would appear that SG markers could be used to identify injured neurons independent of classical pathological markers, such as antibodies (e.g., PHF-1) or chemical probes (e.g., thioflavin-S) recognizing pathological tau. Finally, SG biology highlights a potentially important interplay between the evolution of inflammatory and neurodegenerative biology in tauopathies. Future studies will begin to delineate a unique role for each of these SG proteins in the pathophysiology of neurodegenerative disease.
Footnotes
This work was supported by grant awards to B.W. (NIH Grants NS066108, NS073679, ES15567, and NS060872) and a fellowship award to T.V. (NIH AG042213). We thank the brain banks of the Boston University Alzheimer Disease Center, Loyola University Stritch School of Medicine, and Alzheimer Disease Research Center of the Washington University in St. Louis for generously providing human brain specimens. We would also like to specifically thank Dennis Dickson (Mayo Clinic Jacksonville) for his assistance in providing FTDP-17 cases.
References
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Atlas R, Behar L, Elliott E, Ginzburg I. The insulin-like growth factor mRNA binding-protein IMP-1 and the Ras-regulatory protein G3BP associate with tau mRNA and HuD protein in differentiated P19 neuronal cells. J Neurochem. 2004;89:613–626. doi: 10.1111/j.1471-4159.2004.02371.x. [DOI] [PubMed] [Google Scholar]
- Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J, Blumbergs PC, Vucic S, Kiernan MC, Nicholson GA. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2010;81:639–645. doi: 10.1136/jnnp.2009.194399. [DOI] [PubMed] [Google Scholar]
- Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernov KG, Barbet A, Hamon L, Ovchinnikov LP, Curmi PA, Pastré D. Role of microtubules in stress granule assembly: microtubule dynamical instability favors the formation of micrometric stress granules in cells. J Biol Chem. 2009;284:36569–36580. doi: 10.1074/jbc.M109.042879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Dargemont C. Deubiquitination, a new player in Golgi to endoplasmic reticulum retrograde transport. J Biol Chem. 2003a;278:51989–51992. doi: 10.1074/jbc.C300451200. [DOI] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R, Dargemont C. Ubp3 requires a cofactor, Bre5, to specifically deubiquitinate the COPII protein, Sec23. Nat Cell Biol. 2003b;5:661–667. doi: 10.1038/ncb1003. [DOI] [PubMed] [Google Scholar]
- Cok SJ, Acton SJ, Morrison AR. The proximal region of the 3′-untranslated region of cyclooxygenase-2 is recognized by a multimeric protein complex containing HuR, TIA-1, TIAR, and the heterogeneous nuclear ribonucleoprotein U. J Biol Chem. 2003;278:36157–36162. doi: 10.1074/jbc.M302547200. [DOI] [PubMed] [Google Scholar]
- Corrado L, Mazzini L, Oggioni GD, Luciano B, Godi M, Brusco A, D'Alfonso S. ATXN-2 CAG repeat expansions are interrupted in ALS patients. Hum Genet. 2011;130:575–580. doi: 10.1007/s00439-011-1000-2. [DOI] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glodzik L, de Santi S, Tsui WH, Mosconi L, Zinkowski R, Pirraglia E, Wang HY, Li Y, Rich KE, Zetterberg H, Blennow K, Mehta P, de Leon MJ. Phosphorylated tau 231, memory decline and medial temporal atrophy in normal elders. Neurobiol Aging. 2011;32:2131–2141. doi: 10.1016/j.neurobiolaging.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggin K, Beaudoin S, Grenier C, Brown AA, Roucou X. Prion protein aggresomes are poly(A)+ ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim Biophys Acta. 2008;1783:479–491. doi: 10.1016/j.bbamcr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Jr, Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, Bertini E, Leonhardt-Horti H, Muntoni F, Ouvrier R, Pfeufer A, Rossi R, Van Maldergem L, Wilmshurst JM, Wienker TF, Sendtner M, Rudnik-Schöneborn S, Zerres K, Hübner C. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N, Dürr A, Didierjean O, Stevanin G, Agid Y, Brice A. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14:285–291. doi: 10.1038/ng1196-285. [DOI] [PubMed] [Google Scholar]
- Irvine K, Stirling R, Hume D, Kennedy D. Rasputin, more promiscuous than ever: a review of G3BP. Int J Dev Biol. 2004;48:1065–1077. doi: 10.1387/ijdb.041893ki. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Psujek M, Bartosik-Psujek H. Total-tau and phospho-tau(181Thr) in cerebrospinal fluid of neurologically intact population increase with age. Folia Biol (Praha) 2009;55:126–131. doi: 10.14712/fb2009055040126. [DOI] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans. 2002;30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera TK, Dick AD, Nicholson LB. Mechanisms of TNFalpha regulation in uveitis: focus on RNA-binding proteins. Prog Retin Eye Res. 2010;29:610–621. doi: 10.1016/j.preteyeres.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, de Carvalho M, Meyer T, Tysnes OB, Auburger G, Gispert S, Bonini NM, Andersen PM, Gitler AD. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet. 2011;20:1697–1700. doi: 10.1093/hmg/ddr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. 2011;20:1400–1410. doi: 10.1093/hmg/ddr021. [DOI] [PubMed] [Google Scholar]
- Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000;19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 2002;21:5691–5700. doi: 10.1093/emboj/cdf568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronkina N, Menon MB, Schwermann J, Tiedje C, Hitti E, Kotlyarov A, Gaestel M. MAPKAP kinases MK2 and MK3 in inflammation: complex regulation of TNF biosynthesis via expression and phosphorylation of tristetraprolin. Biochem Pharmacol. 2010;80:1915–1920. doi: 10.1016/j.bcp.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Ross OA, Rutherford NJ, Baker M, Soto-Ortolaza AI, Carrasquillo MM, DeJesus-Hernandez M, Adamson J, Li M, Volkening K, Finger E, Seeley WW, Hatanpaa KJ, Lomen-Hoerth C, Kertesz A, Bigio EH, Lippa C, Woodruff BK, Knopman DS, White CL, 3rd, Van Gerpen JA, et al. Ataxin-2 repeat-length variation and neurodegeneration. Hum Mol Genet. 2011;20:3207–3212. doi: 10.1093/hmg/ddr227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini C, Berdo I, Draetta G. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001;20:3869–3879. doi: 10.1038/sj.onc.1204553. [DOI] [PubMed] [Google Scholar]
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: the good, the bad and the ugly. Cell Signal. 2011;23:324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, Tsui YC, Wei LN. Dynein motor contributes to stress granule dynamics in primary neurons. Neuroscience. 2009;159:647–656. doi: 10.1016/j.neuroscience.2008.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76:2066–2072. doi: 10.1212/WNL.0b013e31821f445b. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zekri L, Chebli K, Tourrière H, Nielsen FC, Hansen TV, Rami A, Tazi J. Control of fetal growth and neonatal survival by the RasGAP-associated endoribonuclease G3BP. Mol Cell Biol. 2005;25:8703–8716. doi: 10.1128/MCB.25.19.8703-8716.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]