

Abstract
Background
Major circulation pathologies are initiated by oxidative insult expansion from a few injured endothelial cells to distal sites; this possibly involves mechanisms that are important to understanding circulation physiology and designing therapeutic management of myocardial pathologies. We tested the hypothesis that a localized oxidative insult of endothelial cells (ECs) propagates through gap junction inter-cellular communication (GJIC).
Methodology/Principal Findings
Cultures comprising the bEnd.3 cell line, that have been established and recognized as suitable for examining communication among ECs, were used to study the propagation of a localized oxidative insult to remote cells. Spatially confined near infrared illumination of parental or genetically modified bEnd.3 cultures, pretreated with the photosensitizer WST11, generated O2•− and •OH radicals in the illuminated cells. Time-lapse fluorescence microscopy, utilizing various markers, and other methods, were used to monitor the response of non-illuminated bystander and remote cells. Functional GJIC among ECs was shown to be mandatory for oxidative insult propagation, comprising de-novo generation of reactive oxygen and nitrogen species (ROS and RNS, respectively), activation and nuclear translocation of c-Jun N-terminal kinase, followed by massive apoptosis in all bystander cells adjacent to the primarily injured ECs. The oxidative insult propagated through GJIC for many hours, over hundreds of microns from the primary photogeneration site. This wave is shown to be limited by intracellular ROS scavenging, chemical GJIC inhibition or genetic manipulation of connexin 43 (a key component of GJIC).
Conclusion/Significance
Localized oxidative insults propagate through GJIC between ECs, while stimulating de-novo generation of ROS and RNS in bystander cells, thereby driving the insult's expansion.
Introduction
ROS and RNS are highly potent chemical entities that play key roles in both normal and patho-physiological conditions. ROS are known as the spearheads of first line defense mechanisms against invading pathogens in the plant and animal kingdoms [1], [2]. Over the past two decades, numerous studies have shown that superoxide anion, hydrogen-peroxide and nitric oxide (O2•−, H2O2, and NO• respectively) are also important in regulating cell and tissue functions, including vascular cell growth [3], cell death [4], cell migration, vessel tone modulation, extra-cellular matrix modification [5], [6], and more. Evidently the level, lifetime, and biological context of ROS/RNS production, define their biological effect.
ROS and RNS are tightly linked to cardio-vascular functions under normal and patho-physiological circumstances [6]. Normally, their levels within the vascular lumen and tissues are well regulated both, enzymatically and non-enzymatically. However, under certain pathological conditions, immune and endothelial cells (ECs) produce large amounts of ROS and NO• [7], [8] instantaneously. These species may impair the delicate balance between ROS production and annihilation, inflicting deleterious effects that are often augmented by cross-talk between activated endothelial and immune cells. The subsequent vascular disorders, such as endothelial dysfunction and perfusion-arrest, underlie most of the cardio-vascular pathologies [7]. On the other hand ROS and RNS, generated by ionizing radiation or light activated sensitizers in the vascular lumen and/or ECs, function as the spearheads in therapeutic tumor ablation [9].
Pivotal to the cumulative damage of these pathologies and therapies, is the up and down propagation of the acute oxidative insult (OI) [10], [11], [12], [13], [14], [15], [16], termed “the bystander effect” [17]. Notably, the distal spreading of an OI presents a paradox; namely, the short life span of the involved radicals (microsecond and shorter times [18]) in the biological milieu does not allow for their migration (up to a few mm) from the primary site of insult to the observed boundaries of injury. Hence, any proposed mechanism should involve alternative elements of propagation [14] similar to, or different from those recently suggested for plant defense mechanisms [19], [20].
In recent years, several studies showed that gap junctions (GJs), composed of six transmembrane connexin (Cx) subunits arranged as cylindrical channels (∼1.5nm diameter) that connect adjacent cells, can facilitate the transfer of 1–3KDa molecules with some dependence on the cell type and physiological status [21], [22]. ECs express mostly Cx37, 40 and 43; the latter is considered an important component of GJIC in myocytes and ECs, and was previously detected in several endothelial cell lines in-vitro, as well as in locations of disturbed blood flow in-vivo [23], [24]. The involvement of this route of communication among neighboring cells was demonstrated in neuronal [25], [26] and myocardial ischemia–reperfusion injuries [22], as well as in liver injuries [27]. With regard to vascular therapeutic approaches, GJIC was shown to augment suicide gene-therapy [17] and is involved in the cancer cell bystander effects under ionizing radiation therapy [28]. However, to the best of our knowledge, no such study was conducted, thus far, with ECs and a locally generated oxidative insult.
In fact, most studies regarding ECs focused on their role in immune cell recruitment as a response to local injury. However, the function of ECs as vaso-regulators necessitates upstream and downstream signal transduction [29], as well as calcium ion mediated communication with the neighboring myocytes, in response to changes in oxygen and nitric oxide levels. GJIC, among ECs, has recently been recognized as being imperative in conducted vasodilatation [30], [31]. Hence, it is reasonable to hypothesize that GJIC plays an important role in mediating the OI propagation. Indeed, a recent study suggests that GJIC and H2O2 have a role in endothelial derived hyperpolarization in response to bradykinin [32], but the link between the two has not yet been clarified.
In this study, we propose novel means to experimentally induce confined OI (COI), in a small group of cultured ECs (bEnd.3 cell line), and to monitor the propagation of the insult to distal ECs within the monolayer. In brief, a computer steered laser beam is delivered through a photoactivation unit (spatial resolution ∼7µm), for 5 minutes, to a geometrically confined region within the EC monolayer culture, that has been pre-incubated with the bacteriochlorophyll based photosensitizer WST11 [33]. The photoactivated WST11 locally generates intracellular O2•− and •OH radicals [34]. ROS and RNS formation, apoptosis, calcium ion fluxes and stress related gene activation in bystander, non-illuminated cell populations is then followed, for up-to 24 hours post COI, by live cell time-lapse fluorescence microscopy (TLFM), as well as by other methods. Changes in the propagation distance and rate of the above events, in response to genetic manipulation of Cx43 [23] and chemical blockade of the endothelial GJs, suggest that GJIC provides a route for propagation of COI among ECs from the initial site of insult to remote cells, followed by cell death propagation.
Materials and Methods
Cell culture
PymT transformed mouse brain parental endothelial cell line (bEnd.3), and its clone bEnd.3-D2 [23] were kindly provided by Dr. Paolo Meda (University of Geneva Medical Center, Switzerland). Cells were cultured as monolayers in Dulbecco's modified Eagle's medium (DMEM) (Gibco/Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS), 2mmol/L glutamine, 0.06 mg/ml penicillin, and 0.1 mg/ml streptomycin (Biological Industries, Bet Haemek, Israel). In order to maintain bEnd.3-D2 clone purity, cells were grown in the presence of 200U hygromycin B/ml (Calbiochem, San Diego, CA).
H5V mouse heart endothelial cells [35] were cultured as monolayers in DMEM: F-12 (1∶1), containing 10% FCS, 1% non-essential amino acids mixture, 2mmol/L glutamine, 0.06 mg/ml penicillin, and 0.1 mg/ml streptomycin (Biological Industries). Cells were transfected with the Cyto-Hyper plasmid (Evrogen, Moscow, Russia) [36] and lipofectamine (Gibco/Invitrogen), according to manufacturer's guidelines to create clones of H5V-Hyper cells. Stable clones were selected by the addition of 2mg/ml neomycin G418 (Calbiochem) and fluorescence activated cell sorting (FACS) (Table S1).
All cell lines were cultured at 37°C in 5% CO2 humidified atmosphere.
Photosensitizer
WST11 [33] was provided by Steba Biotech Ltd. (Rehovot, Israel). Stock solution (2mmol/L) was prepared in PBS and further diluted to a final concentration of 20µmol/L in the culture medium. The Photosensitizer's concentration was determined spectroscopically in methanol at 748 nm, using ε = 1.2 105 mol−1 cm−1. Stock solution was stored in the dark at −20°C until used.
Imaging and photo-activation system
Our imaging system was constructed using an upright fluorescence Olympus BX61 microscope equipped with several auxiliary devices, all controlled by iQ Live Cell Imaging Software (Andor Technology, Belfast, N. Ireland). The light source for imaging was a Lambda DG-4 (Sutter Instruments, Novato, CA), equipped with a full spectrum xenon lamp. Excitation wavelength was controlled by the Lambda DG-4 filters, or by the microscope's motorized filter wheel. Emitted light from the sample was collected by a rear mounted Cascade 512B EM-CCD (Photometrics, Tucson, AZ), providing a 512X512 pixel matrix, 16µm per pixel. For WST11 photoactivation, a laser beam (755nm) was delivered using a Model 3900S Ti: Sapphire CW laser (Spectra-Physics, Irvine, CA), coupled to the system by a single mode fiber through an acousto-optic modulator to a fluorescence recovery after photobleaching and photo-activation (FRAPPA) unit (Andor Technology). The unit was aligned to the microscope's optical path, enabling computer steered high resolution temporal and spatial photoactivation. Olympus full water immersion objectives (UMPlanFlN/10X/0.3 N.A., and XLUMPlanFl X20/0.95 N.A.) and Zeiss (W Plan-Apochromat X63/1.0 N.A.) were used for imaging. An Olympus objective (PlanApon N 2X/0.08 N.A.), was used for photoactivation. All the filters used for imaging were purchased from Chroma (Bellows Falls, VT).
Live cell Sample preparation for photoactivation and fluorescence imaging
Cell suspensions (20×104) were cultured for 48h before the experiment to 80–90% confluence in a 35mm Petri dish with, or without coverslips (Nunc, NY, USA). The Petri dish was mounted on the microscope's heated temperature controlled plate (37°C). In order to avoid pH changes during time-lapse fluorescence microscopy the culture medium was replaced by CO2 independent medium (Gibco/Invitrogen), supplemented with 10% FCS and 2mmol/L glutamine, 1h before the addition of WST11 (20µmol/l). Fluorescence indicators that were added prior to, during or after incubation were used according to experimental requirements as indicated in Table S1. This procedure is further designated as standard pre-incubation protocol.
Laser photo-activation and time-lapse fluorescence microscopy (TLFM) protocol
A 4×4mm field of a cell monolayer was randomly chosen using the X2 objective and a 300×300µm rectangle was selected for photoactivation by the FRAPPA unit. In experiments where physical cell/cell contact dependence was examined, linear scrapes were made (∼20µm wide) using a surgical blade causing the interruption of the monolayer mosaic. Pixel dwell time for photoactivation was set to 100msec and two repeats, providing total laser illumination time of 5min. Laser power and wavelength were set to 1.2mW (at the sample) and 755nm, respectively. Immediately after photoactivation, either TLFM was performed with the X10 or X20 water-immersion objectives, or the plate was placed in the culture incubator for later examination. Filter combination and camera parameters were determined according to experimental requirements, as indicated. Stacks and image recordings were analyzed off-line. Standard pre-incubation protocol, followed by laser photoactivation, is termed here and thereafter as confined oxidative insult (COI).
Fluorescent viability probes
Post COI cell death was tracked by two common membrane integrity probes, propidium iodide (PI) and calcein-acetoxy methyl ester green (CaAMg, Molecular Probes). Exposure to CaAMg probe was terminated by three successive rinses with pre-warmed probe free culture medium, while PI was present in the culture medium throughout TLFM experiment. For more details see Table S1.
Scrape load dye transfer (SLDT) experiment
To assess the extent of gap junctional intercellular communication (GJIC) in bEnd.3 cells, the scrape load dye transfer (SLDT) assay [37] was used. Briefly, the monolayers were incubated with carbenoxolone (CBX), a GJ uncoupler, or its' inactive analog, glycyrrhizic acid [38] (GZA, both 100µmol/L, Sigma, St. Louis, MO, USA), for 30 minutes in the culture incubator. The coverslips were then rinsed with PBS and the culture medium replaced by PBS containing 0.5mg/ml Lucifer yellow (LY) and 0.5mg/ml rhodamine–dextran conjugate (Sigma). Then, linear scrapes were gently made with a surgical blade and the monolayers were left in the dark for 5min to complete dye uptake and transfer. GJIC was measured by counting the number of cell rows to which LY migrated from the scratch. Rhodamine-dextran served as a control for the initial dye loading site.
ROS detection and ROS scavenging
After COI, 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was used to monitor intracellular ROS formation in the treated cultures, dihydroethidine (DHE) was used to specifically monitor superoxide in the treated cultures and the H5V-hyper clone (see “Cell culture” section) was used to monitor intra-cellular hydrogen-peroxide as detailed in Methods S1 and Table S1.
In order to determine the role of connexins in ROS propagation, monolayers were incubated with 100µmol/L CBX or GZA for 30 minutes prior to COI in the culture incubator. Three hours after COI, ROS propagation was analyzed with DCFH-DA.
To scavenge intracellular ROS, two antioxidants were used: N-acetyl-L-cysteine (NAC, final concentration 10mmol/L) and vitamin C (100µm/L) [39], both purchased from Sigma. The scavengers were added prior to, or after COI, according to experimental requirements.
Detection of apoptotic markers
Following COI, caspase activity was monitored with the CaspACE FITC-VAD-FMK assay kit (Promega), as previously described [40] and phosphatidylserine translocation to the outer membrane was monitored with the Annexin-V-FITC assay [41]. For more details see Methods S1 and Table S1.
Indirect immuno-fluorescence labeling of c-Jun N-terminal kinase (JNK), Cx43 and peroxynitrite
bEnd.3 monolayers were subjected to COI and placed back into the incubator for 3h. Next, the coverslips were washed three times with PBS, fixed with 4% paraformaldehyde for 20 minutes at 25°C, permeabilized for 5min with 0.1% tritonX-100 in PBS at 25°C, rinsed with PBS and blocked in 1% normal goat serum in PBS for 30min. For JNK: Monoclonal anti-diphosphorylated (activated) JNK (pJNK, Sigma J4750 [42]) and polyclonal rabbit anti-JNK (Sigma J4500, both diluted 1∶100 in blocking buffer) served as specific first antibodies and were incubated for one hour at 25°C. The coverslips were then rinsed in PBS and further incubated with secondary fluorescent Ab diluted 1∶200 (FITC conjugated donkey anti-mouse for pJNK and FITC conjugated goat anti-rabbit for gJNK, both purchased from Jackson Laboratories, Bar Harbor, ME), for one hour at 25°C, rinsed with PBS and counterstained with DAPI. Finally, the coverslips were mounted on glass slides, sealed with adhesive and kept refrigerated in the dark until examination by fluorescence microscopy as described in Table S1.
Connexin43 (Cx43) labeling
naïve untreated bEnd.3 cells fixation and staining procedures, were identical to those described above. Primary and secondary Ab comprised of monoclonal mouse anti-Cx43 (BD Cat. Number 610062) [43] diluted to 1∶100 and FITC conjugated donkey anti-mouse (Jackson Laboratories) diluted to 1∶200, respectively.
Peroxynitrite detection
bEnd.3 cells grown on cover slips were subjected to COI and placed back for 3h into the culture incubator. Next, coverslips were washed three times with PBS, placed for 1min in ethanol: acetic acid [95∶5] and incubated with anti-nitrotyrosine (10μg/ml, Millipore, Billerica, MA, Cat. Number 05–233), according to manufacturer's protocol [26]. Next, cells were incubated with secondary FITC conjugated donkey anti-mouse Ab (Jackson Laboratories) diluted to 1∶200 and counterstained with DAPI.
Calcuim ion (Ca+2) detection
Ca+2 levels in bEnd.3 cells was determined by the fluorescent indicator Fluo-4 (Molecular Probes) [44]. Briefly, bEnd.3 monolayers were incubated with Fluo-4 AM (5µmol/L) for one hour at 37°C in HBSS, supplemented with 20 mmol/L Hepes and 2.5 mmol/L probenicid, 0.1% (w/v) BSA and 0.042% (v/v) pluronic acid F-127 (all from Sigma). Cells were then washed twice with pre-warmed, probe-free CO2-independent medium containing PI and subjected to COI. The cells were imaged by TLFM every five minutes, with identical imaging parameters for CaAMg detailed in Table S1.
Data analysis
Image and stack analysis was performed by ImageJ freeware (NIH) [45] and CellProfiler [46]. Data analysis, statistical analysis and graphical representation were performed by MATLAB and Microsoft Excel. Student's t-test was used to determine statistical significance.
Results
Endothelial cell death propagation following confined oxidative insult (COI)
Endothelial bEnd.3 cell monolayers were subjected to COI in a 300×300µm2 rectangular field. The spatial precision of light applied through the FRAPPA unit, previously shown by others [47], is also shown in Fig. S1, indicating no stray light delivery out of the assigned area of illumination. Figure 1A presents frames selected at the indicated time points from a 24h time-lapse video, initiated at the end of the COI (Video S1). Cell death within the first 30 minutes after the end of illumination is strictly limited to the COI domain, as indicated by loss of CaAMg fluorescence (green) and increased PI staining (red). The circle of PI stained dead cells (PI+) expanded across the surrounding non-illuminated bystander cells at a rate of ∼20µm/h, increasing the dead cell area from ∼9 to 50×10 µm2 by 20h. Dark control (WST11, no illumination) and light control (laser illumination, no WST11) showed no cell death during the 20h period (Fig. 1B, C). To examine the possibility that cell death progression requires cell-cell contact, the continuity of the monolayer was disrupted by scratching a 20µm wide groove (using a surgical scalpel) prior to COI. As can be seen, cell death progression stopped at the scratch and did not cross it, forming a semicircular area of dead cells limited only to the side of the COI rectangle (Fig. 2A); this continued in all other directions confirming that cell death propagation is cell-contact dependent. The need for cell contact to enable the propagation of death appears to contrast recent reports [48], [49] suggesting that cell death propagation, following photodynamic insult, is mediated by diffusible cytotoxic agents released into the culture medium by the dying cells.
Figure 1. Propagation of cell death following COI.

A – bEnd.3 monolayers were subjected to COI. Cell death was monitored by changes in the fluorescence of membrane viability probes, CaAMg and PI, as followed by TLFM. Dead cells are defined as CaAMg negative (green), PI positive (red). Time zero represents termination of COI. B – Dark control (with WST11, no illumination). C – Light control (Illumination, of same rectangle, no WST11). Both control images were taken at 20h. All other details are as described in the Materials and Methods section.
Figure 2. Death propagation among bystander cells requires cell-cell contact and connexins.

A – Cell contact in bEnd.3 monolayer was impaired by surgical scalpel scratch (white arrows, scratch extends far beyond the field of view), and the culture was subjected to COI (60×60 µm, top left image, yellow rectangle) in one side of the scratch. Cell death (monitored as described in Fig. 1) propagates only in that side. Lower panel – an additional COI site was imposed at t = 150 min in the same side of the scratch (white rectangle). B – bEnd.3 cells were incubated with GZA (inactive analog, upper panel) or the GJ uncoupler CBX (middle panel). After 30min Lucifer yellow (LY, pseudo yellow) was added and SLDT was carried out (n = 12). White arrow marks the linear scratch. Rhodamine-dextran (red, lower panel) served as initial loading site control. C – bEnd.3 monolayers immunostained for Cx43, (pseudo-colored green) seen on cell membranes (arrow heads) and around the nuclei (counter stained with DAPI, pseudo-colored blue). Control experiments showed no non-specific binding of the secondary antibody (n = 3). D – The graph illustrates time dependent registration of PI+ cells beyond the COI rectangle in GZA treated (solid line), CBX treated (dashed line) and bEnd.3-D2 cells (dotted line) following COI. Values are displayed as means ± SD. * P<0.01 between the GZA and CBX/ bEnd.3-D2 experiments from t = >1.5h onwards (n = 4). All other details are described in Materials and Methods section.
Gap junction intercellular communication (GJIC) among bEnd.3 endothelial cells is essential for the oxidative insult propagation – the role of connexin (Cx) 43
We hypothesized that GJIC connecting adjacent endothelial cells enables death propagation from primarily photoactivated bEnd.3 cells via non-illuminated bystander cells to remote sites. Connexin (Cx) 43 is a key component of GJIC in myocites and was previously reported to be upregulated in bEnd.3 ECs within areas of disturbed blood flow [23], [24] and other stress conditions, such as ionizing radiation [50]. To test the possible involvement of GJIC in the bEnd.3 endothelial cell death propagation, we first examined its' functionality, and then checked the effect of inhibiting Cx43 function on the impact of COI, as described below.
Cx43 expression and function of GJIC of bEnd.3 cells
The scrape load dye transfer (SLDT) assay is a common and reliable method for analyzing and quantifying GJIC function [37]. Two fluorescent dyes were used: Lucifer yellow (LY), a low molecular weight dye (457Da) that can be transmitted by GJIC, and a rhodamine-dextran conjugate (70 KDa) that cannot pass through GJ due to its large size, which serves as an initial loading site indicator. A monolayer of bEnd.3 cells was injured by a linear scratch, as described above; both dyes were added to the culture medium and allowed to enter the scratched cells via their injured membrane. Figure 2B shows selected images of LY distribution among bEnd.3 cells after performing the SLDT procedure in the presence of carbenoxolone (CBX), a connexin inhibitor, or its inactive analog, glycyrrhizic acid (GZA) [38]. LY migrated through at least three rows of bystander cells in the presence of GZA (average propagation distance: 3.41±1.47 rows, Fig. 2B, upper panel). In contrast, CBX inhibited LY migration beyond the scratch (average propagation distance: 1.10±0.31 cell rows P<0.01, Fig. 2B lower panel). Rhodamine-Dextran, however, could not be detected beyond the injured cells on the scratch boundary, underscoring expression and functionality of GJIC in parental bEnd.3 endothelial cells. This conclusion was further substantiated by immunofluorescence staining of bEnd.3 cells (Fig. 2C), which revealed the presence of Cx43 in the peri-nuclear region (due to turnover, Cx43 has a half-life of 1–2h) and cell membranes, in agreement with previous reports [23].
GJIC blockers inhibit death propagation among bEnd.3 cells
To test if GJIC controls death propagation in bEnd.3 endothelial cultures, COI was performed in the presence of CBX or GZA, to consider any nonspecific effects of the glycyrrhetinic acid. The fluorescence of the viability markers (PI and CaAMg) was monitored by TLFM and used to count numbers of dead bystander cells beyond the COI rectangle. Monolayers treated with GZA showed extensive bystander death propagation, however treatment with CBX significantly inhibited this effect (Fig. 2D).
In the bEnd.3-D2 clone, the expression of the Cx43-βGal protein elicits a dominant-negative fivefold inhibition effect in GJIC function [23], as determined by the dye coupling assays which were reproduced in our lab by the scrape load dye transfer assay (not shown). Therefore, any GJIC process mediated by Cx43 should be strongly attenuated in the mutant strain [23]. Figure 2D (dotted line) graphically shows that the dominant negative inhibition of GJIC strongly attenuated bystander cell death, to the extent seen upon CBX inhibition. In summary, the above results imply that GJIC and, Cx43 in particular, play a critical role in mediating bystander cell death propagation after COI.
Intra-cellular ROS and RNS generation in bystander cells following COI
ROS and RNS have been recognized in the last few years as important mediators of acute oxidative insult and promoters of cell death and survival [51]. Hence, for further elucidation of the underlying features of bystander cell death we monitored the temporal evolution and propagation of these species in the treated cell cultures.
bEnd.3 monolayers were pre-incubated with both WST11 and 2′,7′-dichlorofluorescin diacetate (DCFH-DA), a cell-permeable non-fluorescent probe (Table S1). Once in the cell, DCFH-DA undergoes de-esterification to its membrane impermeable 2′,7′-dichlorofluorescein form and remains intracellular. Oxidation modifies DCFH to its fluorescent form 2′,7′-dichlorofluorescein (DCF), which enables detection of H2O2, •OH and peroxynitrite (ONOO−) generation within cells [52]. Figure 3A shows a representative image from a set of experiments in which endothelial monolayers were pre-incubated with DCFH-DA, rinsed with fresh culture medium containing PI, subjected to COI and placed back into the culture incubator. Three hours later, the cells were imaged by fluorescence microscopy to detect changes in DCF fluorescence and cell death. Dead bystander cells (PI+), at the upper left corner, are at a distance of 50–100µm from the rim of the COI. Remarkably, live cells positioned in close proximity to the dead cell area (0–100µm) demonstrate high DCF fluorescence, indicative of intra-cellular ROS. The DCF fluorescence intensity gradually declined, reaching background levels at a distance of 500–700µm (Fig. 3B). Control cells, exposed only to laser illumination (no WST11) and imaging light to detect DCF fluorescence, show low fluorescence values, indicating minimal probe oxidation by light. Since the lifetimes of the reactive species in the biological milieu are very short (µsec to msec) [18], the fluorescence, due to intracellular ROS observed hours after COI completion, suggests continuous de-novo ROS generation in the bystander cells, far from the illuminated region, long after illumination. This conclusion was further confirmed by the negligible effect of extra-cellularly added superoxide dismutase, or catalase (Fig. 4) on the intensity and time profile of the propagation signal.
Figure 3. De-novo ROS generation in endothelial bystander cells and the effect of ROS scavengers and GJIC inhibition.

bEnd.3 monolayers were incubated with DCFH-DA, rinsed with fresh culture medium and subjected to COI. After illumination the plates were placed in the incubator for 3h and then imaged for PI (pseudo-colored red) and DCF (ROS, pseudo-colored green) fluorescence. A – An overlay of two pseudo-colored captures, the white arches represent distance increments of 100µm from the COI (white rectangle), representative of n = 3. PI+ bystander cells on the top left corner are at 50–100µm from the rim of the COI. Adjacent to them on the first arch (0–100µm) are DCF positive, PI negative bystander cells. B – A plot of DCF fluorescence intensity (mean ± SD of five cells in three separate experiments). LC – light control, cells exposed to laser illumination, without WST11 incubation, imaged for DCF fluorescence 3h later. C – bEnd.3 monolayers were subjected to COI under the following conditions: Control: encompassing standard COI; Vit. C Pre-COI: encompassing incubation with 100µmol/L vitamin C for 1h, rinse and then subjected to COI; Vit. C Post-COI: encompassing the addition of 100µmol/L vitamin C immediately after COI; NAC: incubation with 10mmol/L NAC prior to COI. 3h after COI, cell death was determined by PI fluorescence. Notably, in all cases COI resulted in a complete cell death within the primary illuminated square. Values represent averaged percent of bystander cell death (mean ± SD of at least n = 3 separate experiments in each group) relative to the control. The control values are statistically significant higher than all treated groups (P<0.01). D, E – bEnd.3 monolayers were incubated with GZA (D) or CBX (E), respectively, subjected to COI and probed with DCFH-DA. Dead bystander cells (red) at a distance of 60–80µm from the rim of COI, are accompanied by PI−, DCF+ (representative of n = 3 experiments) in (D) but not in (E). The insert in (E) illustrates the mean DCF fluorescence of CBX compared to GZA treated cells (right and left colum respectively, n = 10 cells) at equal distances from the dead bystander cells. *- P<0.01 between the treatments. # - P<0.05. F – bEnd.3 monolayers were scratched by a surgical scalpel (arrow), underwent COI (near the upper right corner), and placed in the incubator for 10h. Intra-cellular ROS propagation (DCF+) is blocked at the scratch. Insert – right and left columns representing mean DCF fluorescence intensities in the photoactivation side and beyond the scratch, respectively). * P<0.01.
Figure 4. Superoxide anion (O2 •−) and H2O2 detection in bystander cells following localized oxidative insult.

A – Time dependent DHE fluorescence increase (superoxide generation) in bEnd.3 cells at four selected distances from the COI boundary, within the monolayers (n = 10 cells, ± SD). B, C – H5V-Hyper monolayers supplemented with PI and 100UI/ml catalase, were subjected to COI and imaged. B – An overlay of Hyper protein fluorescence (H2O2 elevation, intense green, 520nm emission, chevrons) and PI+ (red, at a distant of 30–40µm from the white rectangle COI rim, arrowheads) emissions acquired 3h after the COI. C – Time dependent fluorescence intensity of Hyper-low (solid line), Hyper-high (dashed line) and PI (dashed dotted line) levels, in a single bystander H5V-Hyper cell adjacent to PI positive cells that were normalized to maximum. The Hyper low fluorescence decreased by 20%, while the Hyper-high emission elevated within 90 minutes by ∼25%.
The observed ROS propagation from the COI domain could be either the cause, or the result of a bystander cell death propagation mechanism. To resolve this dilemma, we treated bEnd.3 cell monolayers with intracellular antioxidants. We anticipated that in the first case intracellular ROS quenching would result in increased cell survival, whereas in the second case such quenching would have no effect on the cell death. We chose two common cell permeable ROS scavengers, N-acetyl-L-cysteine (NAC) and vitamin C [39] to answer this question. bEnd.3 monolayers were pre-incubated with vitamin C or NAC for 1h and then rinsed free of the extra-cellular ROS scavengers. The monolayers were then subjected to COI. Subsequently, the plates were returned to the culture incubator for 3h and the extent of cell death was determined with PI and CaAMg viability probes. In both treated groups, a significant reduction in the number of dead bystander cells was observed, as compared to untreated controls (Fig. 3C). The mean bystander cell death in these groups were reduced by 73% (vitamin C) and 43% (NAC), P<0.01. In order to verify that the effect does not involve quenching of the primary COI insult, we also added vitamin C after completion of COI. This way, any decline in bystander cell death could be exclusively attributed to scavenging secondary ROS formation. The mean bystander cell death level in this group was reduced by 48% as compared to the control (Fig. 3C, P<0.01). These results strongly suggest that the observed ROS are part of the driving force of bystander death propagation, rather than passive participants in the process. Moreover, the significant response to NAC, which exerts its effect mainly by enhancing intra-cellular pools of reduced glutathione [39], depicts the involvement of intracellular ROS. The difference in cell death levels between pre and post illumination vitamin C introduction, appears therefore, to reflect primarily the contribution of the COI generated ROS.
Apparently GJIC is important for both ROS generation and injury propagation. Figure 3D, E shows that in cell cultures incubated with CBX, de-novo ROS generation was at least two folds lower (p<0.01, n = 3 including 10 cells/experiment) than in GZA treated cultures (Fig. 3E, insert). Moreover, scratching the cell monolayer prior to COI (Fig. 3F) also reduced DCF fluorescence by a factor of 2 beyond the scratch (Fig. 3F, insert). In summary, these results suggest that cell to cell contact and, more specifically, GJ coupling is required for the propagation of oxidative stress followed by death of bystander endothelial cells.
De-novo generation of superoxide in bEnd.3 bystander cells
The most likely de-novo produced ROS in response to the COI, are O2 •−, H2O2 (the latter as a product of superoxide dismutation), and •OH, that can be formed by the Fenton reaction, or decomposition of peroxynitrite [8]. Potential generators of O2 •− in cells are complex I within the mitochondria, xanthine oxidase and NADPH oxidase [53]. Several patho-physiological states were shown to activate these enzymes and, thereby, induce pathological O2 •− formation, followed by cell apoptosis [7]. Thus, we hypothesized that O2 •− is likely to be one of the ROS identified above; It was submitted for a more specific examination, using dihydroethidine (DHE), which was, converted to fluorescent 2-hydroxyethidium (and other compounds) upon oxidation by O2 •− [54], [55]. Figure 4A shows the spatial and temporal evolution of O2 •− in bystander cells post COI, at increasing distances from the activated rectangle on a time scale of hours. Cells closer to the COI rim produced O2 •− faster, to higher levels and at earlier times than more distant ones. Remote cells showed very low basal O2 •− formation. The O2 •− values directly correlate to the cell distance from the COI priming site. As noted above, the observed evolution and decay times of the O2 •− signal are orders of magnitude longer than the literature lifetimes of superoxide radicals [18]; they, therefore, cannot reflect the lag of oxygen radical diffusion from the site of primary insult. These dynamics, instead, suggest de-novo generation of O2 •− in bystander cells following the propagation of some stress signal from the COI region. Incubation with superoxide dismutase (100IU/ml) during and after COI of bEnd.3 cell cultures had no effect on the O2 •− signal formation, nor on the cell death propagation rates, reinforcing the notion that superoxide is formed intracellularly.
De-novo generation of hydrogen peroxide (H2O2) in bEnd.3 bystander cells
The importance of H2O2 has also been recognized in both physiological and pathophysiological situations [7]. Although its life time is longer than that of a superoxide radical, its high oxidation activity renders its lifetime shorter in the biological milieu and minimizes possible migration from the photoactivation domain. Thus, H2O2 molecules can only be monitored in bystander cells a few hours post photoactivation, if generated within these cells shortly before, or during detection. To explore the contribution of H2O2 to the above DCF monitored ROS signal in bystander cells (Fig. 3), we followed fluorescence of the Hyper-H5V clone (see Materials and Methods section) after the COI. Throughout these experiments we included 100 IU/ml catalase in the culture medium to block extra-cellular generation of H2O2. Figure 4B shows Hyper-H5V fluorescence at 520nm, due to the H2O2 generation in live cells (chevrons), which precedes cell death (PI+, arrowheads). To quantify the HyPer signal, the ratiometric changes in the 520 nm fluorescence, were recorded upon excitation at 420 (hyper-low) and 490 nm (hyper-high), as illustrated in Fig. 4C. This plot shows TLFM of a representative bystander cell at ∼ 70µm from the rim of the COI, during 3–5 hours post photoactivation. The 520nm emission decreases overtime upon excitation at 420nm (Hyper-high), and mirrors the increase in emission upon excitation at 490nm (HyPer-high). The increase of the PI signal (signifying cell death) lags by 30 minutes after the H2O2 signal.
De-novo generation of Peroxynitrite (ONOO−)
ECs generate nitric oxide (NO•) following a plethora of physiological and pathophysiological situations [8], [56]. In most pathological situations, the NO• secretion coincides with the evolution of O2 •−, resulting in the generation of deleterious peroxynitrite [53], known to induce endothelial dysfunction. The nitrosylation of tyrosine residues in proteins is the common footprint for the presence peroxynitrite in-vivo [26]. Hence, we set out to detect the evolution of peroxynitrite in bystander cells following COI by immune-staining for nitrosylated proteins. Heavy staining for nitro-tyrosine was observed in viable bystander cells adjacent to dead bystander ones three hours post COI (Fig. 5A). No nitro-tyrosine staining was observed in controls (Fig. 5B, C). These observations show that death propagation is preceded by nitrosative stress propagation, exemplified by peroxynitrite, which is most probably due to the concomitant generation of NO• and O2 •− in the bystander cells.
Figure 5. Peroxynitrite generation in bEnd.3 bystander cells following COI.

A – Monolayers were subjected to COI, placed back in the incubator for three hours and then subjected to anti-nitrotyrosine antibodies first, and then to fluorescing secondary antibodies (pseudo-colored green). Arrowheads mark dead bystander cells (PI+); blue depicts DAPI nuclear stain. B – Control, as in (A) but with secondary antibodies only, right side arrows mark dead bystander cells. C – Control, untreated monolayers subjected to primary and secondary antibodies. All images represent at least three similar experiments.
Mode of bystander cell death
Essentially, nitrosative or oxidative insults may result in necrotic and/or apoptosis cell death. Necrosis is characterized by initial impairment of the cell membrane integrity, enabling nuclei staining by PI. In apoptosis, the phosphatidylserine is translocated to the outer membrane, enabling Annexin-V-FITC staining; however, the membrane remains initially non-permeable to PI [57]. Thus (Annexin-V-FITC)+/PI− suggests apoptosis. An additional critical marker is cysteine-aspartic proteases (caspase) activation. Figure 6 presents bEnd.3 cells stained for activated caspases (using CaspAceTM, Fig. 6A), phosphatidylserine (using Annexin-V-FITC, Fig. 6B) and cell permeabilization/death (using PI) after COI. Both CaspAce and Annexin-V-FITC staining precede PI staining by 3–4 cell rows.
Figure 6. Apoptotic cell death in bystander cells following COI.
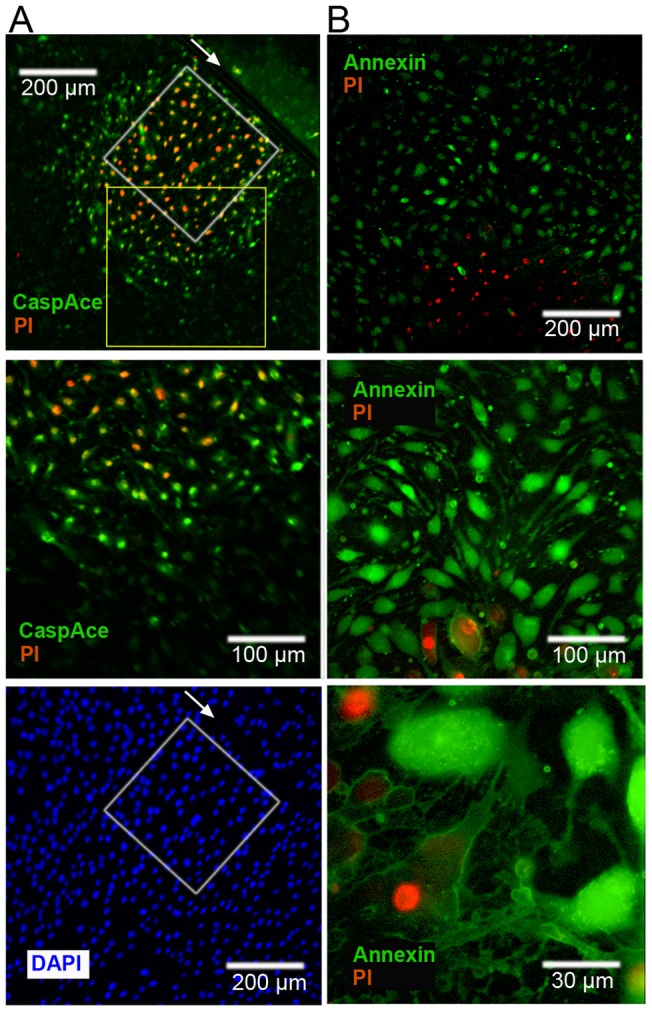
A – bEnd.3 monolayers were linearly scratched by a surgical scalpel (arrow), subjected to COI (white rectangle, left of scratch upper panel) and placed back in the incubator for 3h. Then, CaspACE FITC-VAD-FMK, PI and DAPI were added to the cells. Upper and lower panels are snapshots of the same field of view. Middle panel is a higher magnification snapshot of the yellow rectangle in upper panel. Cell death (PI+) is preceded by CaspACE staining at 100–200µm beyond the COI, left of the scratch only. B – bEnd.3 monolayers were placed in the incubator after COI for 3h, and then subjected to Annexin-V-FITC (for phosphatidylserine depolarization) and PI. Phosphatidylserine depolarization ((Annexin-V)+ cells (green)) clearly precedes cell death and permeabilization (PI+). Panels show different magnifications. Images are a representative of n = 3 experiments.
c-Jun N-terminal kinase (JNK) activation in bystander cells
The stress-activated kinase JNK is a member of the mitogen-activated protein kinases superfamily and, upon activation, can control proliferation, differentiation and apoptosis. Intra-cellular ROS and RNS generation readily activate JNK, eventually leading to apoptotic cell death [58], [59]. Hence, we hypothesized that the apoptotic death propagation observed in the bystander ECs should be accompanied by JNK activation. To test this hypothesis, we fixed bEnd.3 monolayer cells 3h post COI and stained them by immunofluorescence for activated (phosphorylated) JNK (pJNK), or general JNK (gJNK) [42]. Figure 7 presents gJNK immunofluorescence of control, untreated cultures and displays gJNK images of bystander cells three hours post COI. Arrowheads mark dead (PI+) bystander cells. Untreated cells exhibit diffuse gJNK stain with no nuclear localization. Bystander cells, on the other hand, show nuclear gJNK localization, indicating their activation [60]. This nuclear translocation is observed to a distance of two cell rows from the PI+ bystander cells. To substantiate this observation, we immunostained pJNK under the same experimental conditions. PI− pJNK+ bystander cells encompass two cell rows (100µm) from the PI+ cells, with negligible nonspecific binding and no pJNK stain beyond that point (Fig. 7). We conclude that JNK is phosphorylated and translocated into the nucleus, in response to the oxidative stress propagation. The chain of events that can lead to ROS induced JNK activation was detailed by others and involves several signaling pathways [59].
Figure 7. JNK activation and nuclear translocation in bystander cells.
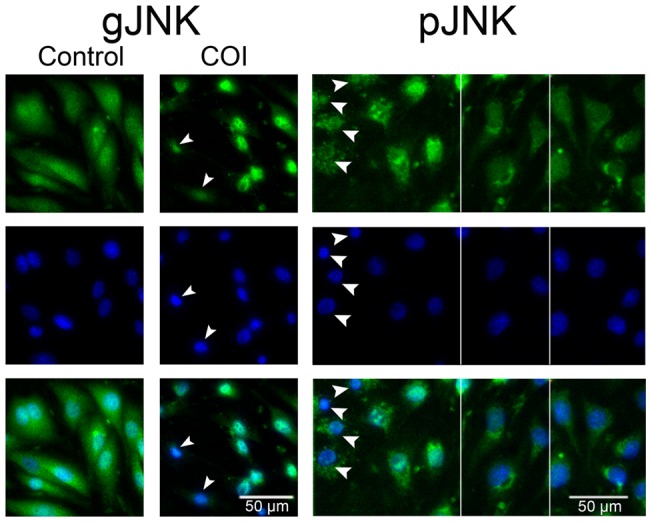
bEnd.3 monolayers, were subjected to COI or left untreated and returned into the incubator for 3 more h, then fixed with 4% PFA and stained for gJNK or pJNK (pseudo-colored green) as indicated. After immune-staining, the cell nuclei were counter stained by DAPI (pseudo-colored blue) and imaged by fluorescence microscope. gJNK: Untreated cells exhibit JNK distribution throughout the cell. In treated monolayers, bystander cells adjacent to PI+ cells (marked by arrowheads, 60–80µm away from rim of the COI) show distinct JNK nuclear localization. pJNK: a collage comprised of three snapshots separated by vertical white lines. Bystander cells adjacent to PI+ cells show pJNK in the nucleus (left side) while distant cells have no pJNK staining (right side). Lowest panels are a merge of the upper ones.
Calcium ion involvement in bystander cell death propagation
Several secondary messengers may mediate bystander cell death propagation by means of GJIC, including inositol trisphosphate, cAMP and calcium ions (Ca2+) [61], [62]. Recently, Ca2+ transients were shown during cell death propagation in a rat C6 Glioma Cytochrome-C model [40]. Changes in cytosolic Ca2+ may be due to transfer from adjacent cells through GJIC, or their release from intra-cellular storages by inositol trisphosphate. In a preliminary attempt to monitor such changes we used the Fluo-4 probe (which exhibits increased fluorescence intensity upon calcium binding in situ), as detailed in the Materials and Methods section. Figure 8 illustrates the normalized time dependent fluo-4 fluorescence intensity of five individual cells lying approximately 30µm from the rim of the photoactivation region. The bystander cells show sharp transients representing 20–30% elevation in Ca2+ levels within 5–10 minutes post COI. Approximately two hours later these cells became PI positive (not shown).
Figure 8. Cytosolic Ca2+ ions elevation in bystander cells.
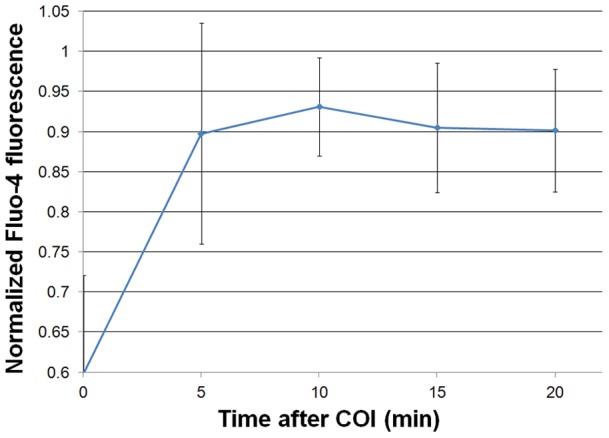
bEnd.3 monolayers were incubated with Fluo-4, for Ca2+ staining, subjected to COI and then followed by TLFM at 5min intervals. The plot illustrates the maximal normalized Fluo-4 emission intensity in n = 5 bystander cells close to the rim of the COI rectangle.
Discussion
Understanding the chain of events leading from a locally confined oxidative insult, to a widespread tissue injury is a major challenge in basic and translational research of vascular patho-physiology. Among other means, ECs use GJIC to communicate with each other and with the myocytes surrounding them, as well as performing their physiological functions [63]. It is reasonable to assume that this communication plays a role in promoting, or possibly controlling, damage propagation from the point of injury to remote sites. Nevertheless, limited research efforts have been invested in exploring the initial response and progression modality of localized oxidative insults in the endothelium, while numerous studies focused on the myocyte response [64], [65].
In the present study, we used confluent endothelial cell cultures as a model for a living endothelium, to study its response to a spatially confined oxidative insult (COI) and the OI's spread therein. We used bEnd.3 cells, derived from primary mouse brain endothelial cells, transduced with a PymT expressing retrovirus [66]. These cells retain key features of differentiated endothelium, such as: specific protein expression (CD31, VEGF receptor 2), internalization of acetylated LDL and adhesion molecule expression, upon stimulation with pro-inflammatory cytokines [67]. The relevance of this cell line to living endothelium was further established in our lab, by demonstrating the ability of bEnd.3 cells to form 3D capillary tubes on basement membrane matrix (Fig. S2); this requires unique expression and arrangement of multiple adhesion and adherence molecules [68]. On this basis, bEnd.3 cells appear to represent an authentic model for examination of gap junction communication among endothelial cells, as also noted by others [24]. The use of these cells in our experimental setup allowed us to produce a well-defined COI and to follow the response of the entire cell population by multiple probes and tests, a task extremely challenging for in-vivo or ex-vivo studies. Nevertheless, endothelial cells in-vivo, interact with their surrounding vascular tissue and blood cells, and are thereby exposed to changing shear stress, as well as hemodynamic forces. As a result, they may express different connexin repertoires. Therefore, confined oxidative insults and damage propagation in-vivo, may also be affected by other factors, which were not addressed in our in-vitro model, described in the present study.
Our study shows, for the first time, that GJIC among ECs can also serve as a major route for the expansion of a localized oxidative insult. A major step towards achieving this goal was the construction of a system that enables the generation of a confined primary oxidative insult (acute local production of O2•- and •OH) to a well-defined single cell, or a small cell population, with high spatial resolution (∼7µm). This enabled, for the first time, the follow up of oxidative stress propagation from a primary insult site, in a small group of ECs, to remote sites (Figs. 1,2). By monitoring the appearance of different ROS and RNS, using several fluorescing probes, in the presence of extracellular ROS scavengers, we could differentiate between intra and extracellular reactive species, which have been recognized as important in such insults and the resulting damage propagation. It was then clearly demonstrated that a primary localized burst of oxygen radicals was followed by intercellular cues, and then mobilized through gap junctions that trigger the propagation of the oxidative assault. The ECs subjected directly to the COI appeared to undergo necrosis (rapid nuclei exposure to PI that is not preceded by phosphatidylserine translocation, Figs. 1,2). However, the bystander cells undergo apoptotic cell death following the ROS (Figs. 3 and 4) and the RNS (Fig. 5) generation, as reflected by both, phosphatidylserine translocation to the external cell membrane, and the activation of caspase-3 (Fig. 6). The two markers were examined three hours after photoactivation and were observed at a distance of 4–5 cell rows from the primary insult rim, preceding the PI staining (cell death). This observation is reinforced by the positive staining for phosphorylated and nucleus translocated JNK, a stress activated kinase [42], [58], that can lead to cell apoptosis by several mechanisms as detailed elsewhere [59]. We wish to point out that the use of several unrelated chemicals and genetically encoded ROS and RNS probes (Fig. 3, 4 and 5) significantly minimize the possibility of an experimental artifact as a cause for these results. Since bystander cell death was significantly inhibited by ROS scavengers, even when added after COI (Fig. 3), it appears that ROS play a central role in the mechanism of cell death propagation. This observation reflects the dual nature of ROS in the vascular system, acting as death inducing agents, as well as secondary messengers. Moreover, this observation may have an impact on future clinical interventions for certain vascular disorders.
The spatial spreading of newly generated ROS in the cell monolayer is restricted by their short half-life time and therefore the diffusion limits. Consequently, it is extremely unlikely that radicals formed in the primary COI will cause cell death by diffusion through GJIC, until reaching the remote death sites. However, within the current state of knowledge we cannot exclude the possibility that ROS with longer life times, such as H2O2, traverse through GJIC as secondary messengers and cause ROS generation and bystander cell death at larger distances. The identity of other potential messengers usually requires radioactive labeling of suspect compounds for their follow-up [61]. The major difficulty stems from the need to distinguish whether a molecule observed in a receiver cell is transmitted by GJIC from a donor cell or is the down-stream result of a different secondary messenger. While such investigation is beyond the scope of this study, our model can serve as a convenient platform to address it in the future.
The reported area of bystander cell death propagation in our study is markedly larger than previously reported in non-endothelial systems. Decrock and colleagues have shown the spread of apoptosis via Cx43 hemichannels after Cytochrome-C electroporation into C6 glioma cells [40]. Moreover, these authors reported apoptosis for 10–20% of the bystander cells in an area of 0–200µm from the electroporation site by six hours post cytchrome-C loading and 4–5% in the area of 200–370µm. The “apoptotic wave” shown here is far-reaching and encompasses all cells to a distance of ∼350µm from the primary square of insult. This propagation is more specific than the previously discussed release of extracellular signals (e.g. by hydrogen peroxide) that function as death signals to adjacent cells [48]. The de-novo generated peroxynitrite (the product of NO• and O2 •− interaction, Fig. 5), probably following the overwhelming activation of endothelial NADPH oxidase and eNOS [56] through the intercellular communication, may also suggest new therapeutic targets. These radicals are known to initiate apoptosis when overwhelmingly produced, as was indeed shown by our present study (Figs. 3, 4 and 5). Since GJIC also functions between ECs and myocytes [63], they may provide an interesting route to initiate damage propagation within the major components of the myovascular system, a process that might be fatal. Such a process can be mediated by Ca2+ mobilization from one cell to the other (endothelial bystander cells showed Ca2+ elevation, Fig. 8). Finally, the phosphatidylserine exposure on route of the apoptotic wave propagation is an “eat me signal” that would recruit phagocytes and, thereby, start an inflammatory process which will augment the primary oxidative damage [69].
Conclusions
The presented findings are highly relevant to the clinical arena in several aspects:
(1) The Cx43 component that was found here to be critical for the insult and death propagation, was previously found to be up-regulated in regions of disturbed blood flow and shear-stress [24]. (2) GJIC appears to mediate other forms of damage expansion, such as the propagation of neuronal injury during ischemia-reperfusion injury [26]. (3) Studies of non-endothelial systems have suggested that the bystander effect, e.g. following deuteroporphyrin photo-activation, can be mediated by extracellular propagation of ROS [48]. However ROS, in particular O2 •− and •OH radicals, have very short lifetimes in the physiological milieu [18]; they therefore cannot migrate for more than a few microns from the site of their generation before reacting with the bio-milieu, unless undergoing a propagation mechanism, as reported for some plant systems [20]. Hence, it is extremely unlikely for this propagation to be in the form of ROS or RNS transit via GJIC.
The hereby-reported propagation of “de-novo” ROS generation utilizing GJIC enables long distance progression of an oxidative insult that augments the damage of localized vascular injury. (4) Apoptosis is an energy dependent mechanism and, therefore, expected to take place mostly in re-oxygenated, rather than ischemic tissues in-vivo [70]. In several animal models, massive apoptosis of cardio-myocytes is observed after blood is restored to ischemic tissue regions, thus enhancing the initial damage [71]. As noted above, the exposure of phosphatidylserine on the outer leaflet of the plasma membrane is the most universally seen alteration on the surface of apoptotic cells [72], [73]. Such exposure on ECs in-vivo would lead to rapid thrombus formation [74], [75] and blockade of blood flow into the acutely insulted domain. However, a wide spread of ischemic damage caused by such a response may result in organ failure, as observed in several vascular pathologies. Hence, a balance that can be assisted by therapeutic intervention, aiming at limiting the propagation of an oxidative insult, apoptotic death, and immune response, must be maintained, and may present an additional therapeutic target. Fortunately, the rate of insult propagation (in the order of hours, Figs. 1 and 2) should allow for effective intervention.
Supporting Information
Illumination region accuracy control. Agar gel sections (3×3mm2) were immersed in a solution of 2mmol/L of WST11 for 1h and then left to dry overnight at 25°C. The dry gel sections were placed on a glass slide and mounted onto the microscope. WST11 fluorescence was recorded with a near infra-red filter set (Ex. 740/30nm, Em. >780nm, beam-splitter >770nm). A random field of view was chosen with the X2 objective (PlanApo 2X/0.08N.A) and a 300×300µm2 rectangle was assigned for laser photo-activation with the FRAPPA unit (2mW laser power, 100e3 pixel dwell time, 2 repeats, 755nm). Immediately after, the objective was replaced with the X10 (UMPlanFlN/10X/0.3 N.A 0.3) for imaging. The illuminated rectangle was recorded and averaged (panel A). B – A quantitative display of the cross section shown in A (white horizontal rectangle). Images are representative of n = 10 similar experiments.
(JPG)
{kind=link}
bEnd.3 cell line tube formation assay. Basement membrane matrix (MatrigelTM, BD) was added to wells in a 24 well plate and gel was allowed to polymerize for 30min at 37°C. Next, a cell suspension (500µl, 40e3 cells, 5% FCS) was added to each well and the plate was placed in a humidified 37°C incubator. 24h later, tube formation was examined and imaged with a light microscope, as presented above.
(JPG)
{kind=link}
Fluorescent probes used and their application details.
(DOCX)
Supporting materials about methods.
(DOCX)
Time lapse fluorescence microscopy (TLFM) of endothelial cell death propagation, following confined oxidative insult (COI). bEnd.3 cell monolayers in 3cm Petri-dishes were incubated with 20 µmol/L WST11 and viability probes (CaAMg and PI) as detailed in the materials and methods section and Table S1. Cells were subjected to COI, represented by the white 300×300µm2 rectangle. TLFM was launched immediately after illumination. The movie is an overlay of pseudo-colored snaps (green – CaAMg fluorescence, red – PI fluorescence). Time intervals are depicted on movie. Scale = 100µm.
(AVI)
Acknowledgments
The authors would like to thank Mr. Asher Auerbach for his technical advice and assistance with the construction and maintenance of the imaging system. We would also like to thank Mr. Ronen Levi for his ongoing logistical support and Ms. Martie Spiegel for reviewing the manuscript. This work is in partial fulfillment of the PhD thesis of I.F. at the Feinberg Graduate School, The Weizmann Institute of Science. Y.S. is the incumbent of the Tillie and Charles Lubin Professorial Chair in Biochemical Endocrinology. A.S. is the incumbent of the Robert and Yaddele Sklare Professorial Chair in Biochemistry.
Footnotes
Competing Interests: A.S. and Y.S. are the inventors of WST11; patent owned by the Weizmann Institute of Science, Rehovot, Israel, licensed to Steba Biotech, France (Scherz A, Brandis A, Mazor O, Salomon Y, Scheer H, (2011) Water-soluble anionic bacteriochlorophyll derivatives and their uses, US patent 7,947,672, reference #33). A.S. and Y.S. serve as consultants to Steba Biotech. This does not alter adherence to all the PLoS ONE policies on sharing data and materials.
Funding: This research is funded by grant # BCTR0707388 from ‘The Susan Komen Foundation for the Cure’, a non-profit organization and the “Women's Health Research Center”, Weizmann Institute of Science. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Bogdan C, R llinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Current opinion in immunology. 2000;12:64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- 2.Miller G, Schlauch K, Tam R, Cortes D, Torres MA, et al. The plant NADPH oxidase RBOHD mediates rapid systemic signaling in response to diverse stimuli. Science's STKE. 2009;2:ra45. doi: 10.1126/scisignal.2000448. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Gines J, Lopez-Ongil S, Gonzalez-Rubio M, Gonzalez-Santiago L, Rodriguez-Puyol M, et al. Reactive oxygen species induce proliferation of bovine aortic endothelial cells. Journal of cardiovascular pharmacology. 2000;35:109. doi: 10.1097/00005344-200001000-00014. [DOI] [PubMed] [Google Scholar]
- 4.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 5.Irani K. Oxidant signaling in vascular cell growth, death, and survival: a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circulation research. 2000;87:179. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 6.Touyz R, Schiffrin E. Reactive oxygen species in vascular biology: implications in hypertension. Histochemistry and cell biology. 2004;122:339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 7.Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends in cardiovascular medicine. 2007;17:48. doi: 10.1016/j.tcm.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovascular research. 1999;43:652. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 9.Gross S, Gilead A, Scherz A, Neeman M, Salomon Y. Monitoring photodynamic therapy of solid tumors online by BOLD-contrast MRI. Nature Medicine. 2003;9:1327–1331. doi: 10.1038/nm940. [DOI] [PubMed] [Google Scholar]
- 10.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences. 1997;94:514. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:29. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 12.Morre DM, Lenaz G, Morre DJ. Surface oxidase and oxidative stress propagation in aging. Journal of Experimental Biology. 2000;203:1513. doi: 10.1242/jeb.203.10.1513. [DOI] [PubMed] [Google Scholar]
- 13.Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radical Biology and Medicine. 2000;28:1456–1462. doi: 10.1016/s0891-5849(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 14.Cai H. NAD (P) H oxidase–dependent self-propagation of hydrogen peroxide and vascular disease. Circulation research. 2005;96:818–822. doi: 10.1161/01.RES.0000163631.07205.fb. [DOI] [PubMed] [Google Scholar]
- 15.Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, et al. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. Journal of Molecular and Cellular Cardiology. 2010;49:565–575. doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Dorado D, Ruiz-Meana M. Propagation of Cell Death During Myocardial Reperfusion. News in physiological sciences: an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society. 2000;15:326. doi: 10.1152/physiologyonline.2000.15.6.326. [DOI] [PubMed] [Google Scholar]
- 17.Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: role of gap-junctional intercellular communication. Cancer Res. 2000;60:3989–3999. [PubMed] [Google Scholar]
- 18.Kavdia M. A computational model for free radicals transport in the microcirculation. Antioxidants & redox signaling. 2006;8:1103–1111. doi: 10.1089/ars.2006.8.1103. [DOI] [PubMed] [Google Scholar]
- 19.Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell. 2005;8:89–91. doi: 10.1016/j.ccr.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Wong HL, Shimamoto K. Sending ROS on a bullet train. Science's STKE. 2009;2:pe60. doi: 10.1126/scisignal.290pe60. [DOI] [PubMed] [Google Scholar]
- 21.Higgins GC, Beart PM, Shin YS, Chen MJ, Cheung NS, et al. Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. Journal of Alzheimer's Disease. 2010;20:453–473. doi: 10.3233/JAD-2010-100321. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovascular research. 2004;61:386. doi: 10.1016/j.cardiores.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 23.Kwak BR, Pepper MS, Gros DB, Meda P. Inhibition of endothelial wound repair by dominant negative connexin inhibitors. Mol Biol Cell. 2001;12:831–845. doi: 10.1091/mbc.12.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwak BR, Silacci P, Stergiopulos N, Hayoz D, Meda P. Shear stress and cyclic circumferential stretch, but not pressure, alter connexin43 expression in endothelial cells. Cell Commun Adhes. 2005;12:261–270. doi: 10.1080/15419060500514119. [DOI] [PubMed] [Google Scholar]
- 25.Lin J, Weigel H, Cotrina M, Liu S, Bueno E, et al. Gap-junction-mediated propagation and amplification of cell injury. Nature neuroscience. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- 26.Forman L, Liu P, Nagele R, Yin K, Wong PYK. Augmentation of nitric oxide, superoxide, and peroxynitrite production during cerebral ischemia and reperfusion in the rat. Neurochemical research. 1998;23:141–148. doi: 10.1023/a:1022468522564. [DOI] [PubMed] [Google Scholar]
- 27.De Maio A, Vega VL, Contreras JE. Gap junctions, homeostasis, and injury. Journal of Cellular Physiology. 2002;191:269–282. doi: 10.1002/jcp.10108. [DOI] [PubMed] [Google Scholar]
- 28.Azzam EI, de Toledo SM, Little JB. Oxidative metabolism, gap junctions and the ionizing radiation-induced bystander effect. Oncogene. 2003;22:7050–7057. doi: 10.1038/sj.onc.1206961. [DOI] [PubMed] [Google Scholar]
- 29.Pries AR, Höpfner M, Le Noble F, Dewhirst MW, Secomb TW. The shunt problem: control of functional shunting in normal and tumour vasculature. Nature Reviews Cancer. 2010;10:587–593. doi: 10.1038/nrc2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bagher P, Segal S. Regulation of blood flow in the microcirculation: Role of conducted vasodilation. Acta Physiologica. 2011;202:271–284. doi: 10.1111/j.1748-1716.2010.02244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.W lfle SE, Schmidt VJ, Hoyer J, K hler R, De Wit C. Prominent role of KCa3. 1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovascular research. 2009;82:476. doi: 10.1093/cvr/cvp060. [DOI] [PubMed] [Google Scholar]
- 32.Hammond S, Mathewson AM, Baker PN, Mayhew TM, Dunn WR. Gap junctions and hydrogen peroxide are involved in endothelium-derived hyperpolarising responses to bradykinin in omental arteries and veins isolated from pregnant women. European Journal of Pharmacology. 2011;668:225–232. doi: 10.1016/j.ejphar.2011.06.050. [DOI] [PubMed] [Google Scholar]
- 33.Brandis A, Mazor O, Neumark E, Rosenbach Belkin V, Salomon Y, et al. Novel Water soluble Bacteriochlorophyll Derivatives for Vascular targeted Photodynamic Therapy: Synthesis, Solubility, Phototoxicity and the Effect of Serum Proteins. Photochemistry and Photobiology. 2005;81:983–992. doi: 10.1562/2004-12-01-RA-389. [DOI] [PubMed] [Google Scholar]
- 34.Ashur I, Goldschmidt R, Pinkas I, Salomon Y, Szewczyk G, et al. Photocatalytic generation of oxygen radicals by the water-soluble bacteriochlorophyll derivative WST11, noncovalently bound to serum albumin. The Journal of Physical Chemistry A. 2009;113:8027–8037. doi: 10.1021/jp900580e. [DOI] [PubMed] [Google Scholar]
- 35.Garlanda C, Parravicini C, Sironi M, De Rossi M, Wainstok de Calmanovici R, et al. Progressive growth in immunodeficient mice and host cell recruitment by mouse endothelial cells transformed by polyoma middle-sized T antigen: implications for the pathogenesis of opportunistic vascular tumors. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:7291. doi: 10.1073/pnas.91.15.7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belousov V, Fradkov A, Lukyanov K, Staroverov D, Shakhbazov K, et al. Bacterial redox sensors. Nature Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 37.Trosko J, Chang C, Wilson M, Upham B, Hayashi T, et al. Gap Junctions and the Regulation of Cellular Functions of Stem Cells during Development and Differentiation. Methods. 2000;20:245–264. doi: 10.1006/meth.1999.0941. [DOI] [PubMed] [Google Scholar]
- 38.Rozental R, Srinivas M, Spray D. How to close a gap junction channel. Methods in molecular biology. 2001;154:447–476. doi: 10.1385/1-59259-043-8:447. [DOI] [PubMed] [Google Scholar]
- 39.Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, et al. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. Journal of Thrombosis and Haemostasis. 2007;5:1302–1308. doi: 10.1111/j.1538-7836.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- 40.Decrock E, De Vuyst E, Vinken M, Van Moorhem M, Vranckx K, et al. Connexin 43 hemichannels contribute to the propagation of apoptotic cell death in a rat C6 glioma cell model. Cell Death & Differentiation. 2008;16:151–163. doi: 10.1038/cdd.2008.138. [DOI] [PubMed] [Google Scholar]
- 41.Gatti R, Belletti S, Orlandini G, Bussolati O, Dall'Asta V, et al. Comparison of annexin V and calcein-AM as early vital markers of apoptosis in adherent cells by confocal laser microscopy. J Histochem Cytochem. 1998;46:895–900. doi: 10.1177/002215549804600804. [DOI] [PubMed] [Google Scholar]
- 42.Shaul Y, Seger R. Use of inhibitors in the study of MAPK signaling. Methods In Molecular Biology-Clifton Then TOTOWA- 2004;250:113–126. doi: 10.1385/1-59259-671-1:113. [DOI] [PubMed] [Google Scholar]
- 43.Hunter A, Barker R, Zhu C, Gourdie R. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Molecular biology of the cell. 2005;16:5686. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mulders ACM, Hendriks-Balk MC, Mathy MJ, Michel MC, Alewijnse AE, et al. Sphingosine Kinase–Dependent Activation of Endothelial Nitric Oxide Synthase by Angiotensin II. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2043–2048. doi: 10.1161/01.ATV.0000237569.95046.b9. [DOI] [PubMed] [Google Scholar]
- 45.Abr moff MD, Magalh es PJ, Ram SJ. Image processing with ImageJ. Biophotonics international. 2004;11:36–42. [Google Scholar]
- 46.Carpenter A, Jones T, Lamprecht M, Clarke C, Kang I, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayer M, Depken M, Bois JS, Julicher F, Grill SW. Anisotropies in cortical tension reveal the physical basis of polarizing cortical flows. Nature. 2010;467:617–621. doi: 10.1038/nature09376. [DOI] [PubMed] [Google Scholar]
- 48.Chakraborty A, Held KD, Prise KM, Liber HL, Redmond RW. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat Res. 2009;172:74–81. doi: 10.1667/RR1669.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubio N, Fleury S, Redmond R. Spatial and temporal dynamics of in vitro photodynamic cell killing: extracellular hydrogen peroxide mediates neighbouring cell death. Photochemical & Photobiological Sciences. 2009;8:457–464. doi: 10.1039/b815343d. [DOI] [PubMed] [Google Scholar]
- 50.Banaz-Yasar F, Tischka R, Iliakis G, Winterhager E, Gellhaus A. Cell line specific modulation of connexin43 expression after exposure to ionizing radiation. Cell Communication and Adhesion. 2005;12:249–259. doi: 10.1080/15419060500514101. [DOI] [PubMed] [Google Scholar]
- 51.Decrock E, Vinken M, De Vuyst E, Krysko D, D'Herde K, et al. Connexin-related signaling in cell death: to live or let die&quest. Cell Death & Differentiation. 2009;16:524–536. doi: 10.1038/cdd.2008.196. [DOI] [PubMed] [Google Scholar]
- 52.Myhre O, Andersen J, Aarnes H, Fonnum F. Evaluation of the probes 2′, 7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochemical pharmacology. 2003;65:1575–1582. doi: 10.1016/s0006-2952(03)00083-2. [DOI] [PubMed] [Google Scholar]
- 53.Cai H, Harrison D. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation research. 2000;87:840. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 54.Posen Y, Kalchenko V, Seger R, Brandis A, Scherz A, et al. Manipulation of redox signaling in mammalian cells enabled by controlled photogeneration of reactive oxygen species. Journal of Cell Science. 2005;118:1957–1969. doi: 10.1242/jcs.02323. [DOI] [PubMed] [Google Scholar]
- 55.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free Radical Biology and Medicine. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 56.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological reviews. 2007;87:315. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mao X, Seidlitz E, Truant R, Hitt M, Ghosh H. Re-expression of TSLC1 in a non-small-cell lung cancer cell line induces apoptosis and inhibits tumor growth. Oncogene. 2004;23:5632–5642. doi: 10.1038/sj.onc.1207756. [DOI] [PubMed] [Google Scholar]
- 58.Karin M, Chang L, Honda S, Maeda S, Hirata H, et al. Reactive Oxygen Species Promote TNF¿-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 59.Shen H, Liu Z. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radical Biology and Medicine. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 60.Parola M, Robino G, Marra F, Pinzani M, Bellomo G, et al. HNE interacts directly with JNK isoforms in human hepatic stellate cells. Journal of Clinical Investigation. 1998;102:1942. doi: 10.1172/JCI1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldberg GS, Lampe PD, Sheedy D, Stewart CC, Nicholson BJ, et al. Direct isolation and analysis of endogenous transjunctional ADP from Cx43 transfected C6 glioma cells. Exp Cell Res. 1998;239:82–92. doi: 10.1006/excr.1997.3872. [DOI] [PubMed] [Google Scholar]
- 62.Saez JC, Connor JA, Spray DC, Bennett M. Hepatocyte gap junctions are permeable to the second messenger, inositol 1, 4, 5-trisphosphate, and to calcium ions. Proceedings of the National Academy of Sciences. 1989;86:2708. doi: 10.1073/pnas.86.8.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxidants & redox signaling. 2009;11:251–266. doi: 10.1089/ars.2008.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. Journal of Clinical Investigation. 1997;100:1813. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pimentel DR, Amin JK, Xiao L, Miller T, Viereck J, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circulation research. 2001;89:453. doi: 10.1161/hh1701.096615. [DOI] [PubMed] [Google Scholar]
- 66.Montesano R, Pepper M, M hle-Steinlein U, Risau W, Wagner E, et al. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62:435. doi: 10.1016/0092-8674(90)90009-4. [DOI] [PubMed] [Google Scholar]
- 67.Pepper MS, Tacchini-Cottier F, Sabapathy TK, Montesano R, Wagner EF. Endothelial cells transformed by polyoma virus middle T oncogene: a model for hemangiomas and other vascular tumors. Tumour angiogenesis Oxford University Press, Oxford, United Kingdom. 1997. pp. 309–331.
- 68.Matsumura T, Wolff K, Petzelbauer P. Endothelial cell tube formation depends on cadherin 5 and CD31 interactions with filamentous actin. The Journal of Immunology. 1997;158:3408–3416. [PubMed] [Google Scholar]
- 69.Opal SM. Phylogenetic and functional relationships between coagulation and the innate immune response. Critical care medicine. 2000;28:S77. doi: 10.1097/00003246-200009001-00017. [DOI] [PubMed] [Google Scholar]
- 70.McLaughlin R, Kelly C, Kay E, Bouchier-Hayes D. The role of apoptotic cell death in cardiovascular disease. Irish Journal of Medical Science. 2001;170:132–140. doi: 10.1007/BF03168827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gottlieb RA, Burleson K, Kloner RA, Babior B, Engler R. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. Journal of Clinical Investigation. 1994;94:1621. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. The Journal of Experimental Medicine. 2010;207:1807. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature-London- 2000. pp. 784–788. [DOI] [PubMed]
- 74.Meems H, Meijer AB, Cullinan DB, Mertens K, Gilbert GE. Factor VIII C1 domain residues Lys 2092 and Phe 2093 contribute to membrane binding and cofactor activity. Blood. 2009;114:3938. doi: 10.1182/blood-2009-01-197707. [DOI] [PubMed] [Google Scholar]
- 75.Zitvogel L, Kepp O, Kroemer G. Decoding Cell Death Signals in Inflammation and Immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Illumination region accuracy control. Agar gel sections (3×3mm2) were immersed in a solution of 2mmol/L of WST11 for 1h and then left to dry overnight at 25°C. The dry gel sections were placed on a glass slide and mounted onto the microscope. WST11 fluorescence was recorded with a near infra-red filter set (Ex. 740/30nm, Em. >780nm, beam-splitter >770nm). A random field of view was chosen with the X2 objective (PlanApo 2X/0.08N.A) and a 300×300µm2 rectangle was assigned for laser photo-activation with the FRAPPA unit (2mW laser power, 100e3 pixel dwell time, 2 repeats, 755nm). Immediately after, the objective was replaced with the X10 (UMPlanFlN/10X/0.3 N.A 0.3) for imaging. The illuminated rectangle was recorded and averaged (panel A). B – A quantitative display of the cross section shown in A (white horizontal rectangle). Images are representative of n = 10 similar experiments.
(JPG)
bEnd.3 cell line tube formation assay. Basement membrane matrix (MatrigelTM, BD) was added to wells in a 24 well plate and gel was allowed to polymerize for 30min at 37°C. Next, a cell suspension (500µl, 40e3 cells, 5% FCS) was added to each well and the plate was placed in a humidified 37°C incubator. 24h later, tube formation was examined and imaged with a light microscope, as presented above.
(JPG)
Fluorescent probes used and their application details.
(DOCX)
Supporting materials about methods.
(DOCX)
Time lapse fluorescence microscopy (TLFM) of endothelial cell death propagation, following confined oxidative insult (COI). bEnd.3 cell monolayers in 3cm Petri-dishes were incubated with 20 µmol/L WST11 and viability probes (CaAMg and PI) as detailed in the materials and methods section and Table S1. Cells were subjected to COI, represented by the white 300×300µm2 rectangle. TLFM was launched immediately after illumination. The movie is an overlay of pseudo-colored snaps (green – CaAMg fluorescence, red – PI fluorescence). Time intervals are depicted on movie. Scale = 100µm.
(AVI)