

Abstract
This study investigates the effect of Mg2+ bound to the DNA major groove on DNA structure and dynamics. The analysis of a comprehensive dataset of B-DNA crystallographic structures shows that divalent cations are preferentially located in the DNA major groove where they interact with successive bases of (A/G)pG and the phosphate group of 5′-CpA or TpG. Based on this knowledge, molecular dynamics simulations were carried out on a DNA oligomer without or with Mg2+ close to an ApG step. These simulations showed that the hydrated Mg2+ forms a stable intra-strand cross-link between the two purines in solution. ApG generates an electrostatic potential in the major groove that is particularly attractive for cations; its intrinsic conformation is well-adapted to the formation of water-mediated hydrogen bonds with Mg2+. The binding of Mg2+ modulates the behavior of the 5′-neighboring step by increasing the BII (ε-ζ>0°) population of its phosphate group. Additional electrostatic interactions between the 5′-phosphate group and Mg2+ strengthen both the DNA-cation binding and the BII character of the 5′-step. Cation binding in the major groove may therefore locally influence the DNA conformational landscape, suggesting a possible avenue for better understanding how strong DNA distortions can be stabilized in protein-DNA complexes.
Introduction
Specific DNA base-cation interactions have instigated numerous studies (reviewed in [1]–[3]) because it is presumed that cations can modulate DNA structure in a sequence-dependent manner, thus affecting the readout and packaging of DNA. Most of these studies are devoted to divalent cations because, in contrast to monovalent alkali cations, divalent cations can be unequivocally located in X-ray structures when they are ordered in the lattice [1]–[4]. In addition, DNA-cation interactions seem preferentially involve divalent cations – at least Mg2+ – over monovalent cations [5].
Within crystals, hydrated divalent cations frequently mediate intermolecular contacts between adjacent DNA molecules. Mg2+, Ca2+, or Mn2+ cross-link DNA bases, especially guanines, to phosphates of neighboring helices [6]–[14]. Molecular dynamics simulations have shown that Mg2+ stabilizes groove-backbone interactions in right-handed DNA crossovers [15]. Cations mediating intermolecular interactions thus behave as adhesives between helices and may play a biological role in crowded DNA media.
Both NMR and crystallographic approaches have been used to localize cations in DNA grooves where more specific intramolecular interactions link cations and groove atoms.
Despite potential steric hindrance due to the minor groove width, the minor groove is able to accommodate divalent cations. NMR experiments shows that divalent cations interact with the 5′-ApA minor groove floor in tracts of ApApApA [16], [17]. Mg2+ and Ca2+ inter-strand coordination modes have been observed at pure A:T sites [7], [11], [18] and, in some cases, at mixed A:T/G:C sites [7], [9], [13]. The strong electronegative potential of A:T minor groove is likely a key factor for constituting favorable cation-binding sites.
In the major groove, cations may preferentially bind to purines, in which O6/N7 guanine and N7 adenine atoms offer excellent anchoring points for ions [19], [20]. Various experimental results have shown that divalent cation interactions require dinucleotides containing at least one guanine [7], [21]. NMR spectra of a series of oligomers in presence of either paramagnetic Mn2+ and Co2+ or diamagnetic Zn2+ have shown H8 resonance broadening or shift for guanines in various sequence contexts [21]–[24]. The most pronounced effects occur on guanines in GpG or GpA and, to a lesser extent, guanines in GpT and GpC. These interesting results can be better interpreted in light of high-resolution X-ray DNA structures, which provide a detailed description of cation intra- and inter-strand cross-links. Intra-stand interactions seem to be predominant, with Ca2+ and Mg2+ coordinating the N7 and O6 atoms of GpG or ApG [7], [9] and, in one case, of GpA [11]. Less frequently, inter-strand bridges have also been observed, with Mg2+ coordinating the guanines of GpC•GpC at one end of the Dickerson-Drew dodecamer [13], [25] or G and T in GpA•TpC in a different sequence obtained at low temperature [14]. Fourier-transform infrared investigations on calf thymus DNA demonstrate that the interaction between Mg2+ or Ca2+ and the N7 of purines was not an artifact due to the short lengths of the oligomers studied by NMR and crystallography [26]. From all of these results, one can conclude that divalent cations within the major groove are able to bridge a guanine to an opposite base but preferentially form intra-strand bridges within G-containing dinucleotides.
Many studies therefore concur and have established that divalent cations interact with DNA bases within the grooves. However, the effect of cations on DNA structure is still unknown and is not a trivial issue to address. The main problem is to separate cause from effect. Does cation binding induce structural adjustments in DNA, or do cations recognize an intrinsic structural property in the sequence that favors their binding? Further, because our knowledge is mainly based on crystallographic structures, observed DNA distortions are difficult to interpret, as they may be due to lattice influences, the presence of inter-molecular contacts and, more generally, biases intrinsic to the solid-state approach. Thus, the impact of divalent ions buried in minor grooves on groove dimensions remains controversial [7], [9], [13], [25]. There are nonetheless several indications that DNA curvature is sensitive to divalent cations in the major groove. In crystals, DNA locally bends when Ca2+ or Mg2+ bridge the two guanines in GpG, compressing the major groove [7], or when Mg2+ binds opposite guanines in GpC•GpC [13], [27]. Some experiments in solution also support the possibility that divalent counterions influence DNA curvature, but these experiments could not specify their locations. Mg2+, Ca2+, or Zn2+ increase the gel mobility anomaly of GGGCCC repeats [28]. Also, Zn2+- or Mg2+- induced bends have been detected in G:C rich regions embedded in d(A)n tracts by atomic force microscopy [29] and NMR measurements of diffusion coefficients [30]. However, the molecular basis of such possible effects of divalent counterions on DNA remains far from well-characterized and understood.
The present work focuses on divalent cations located in the DNA major groove. We first analyzed divalent cation interactions in high-resolution X-ray DNA structures, using recent deposits in the Protein Data Bank. Upon identification of one major interaction pattern, extensive molecular dynamics (MD) simulations were then used to examine in solution the stability of this identified DNA-Mg2+ interaction and its impact on DNA structure and dynamics. Poisson-Boltzman calculations helped to build a credible scenario of DNA-Mg2+ interactions. These investigations were done using the so-called Jun-Fos oligomer, which has been exhaustively studied by NMR and modeling [31]–[34]. Possible repercussions of these findings involve DNA-protein interactions, particularly nucleosome structures in which numerous cations coordinate DNA.
Results
Divalent cations in the DNA major groove: analysis of X-ray structures
The dataset of high resolution X-ray DNA structures (listed in Materials and Methods), including structures recently deposited in the PDB, provided for the examination a total of 23 major groove environments with divalent cations. In 20 cases, the DNA-cation interactions involve two successive purines of ApG or GpG steps (Ri–1piGi), confirming the predominance of intra-strand cross-links [7], [9]. Focusing our analysis on this frequent interaction pattern, we found that four Ca2+ and all of the 14 Mg2+ interact with the N7/O6 electronegative atoms of these bases via their first hydration shells (Figure 1). In two cases, Ca2+ directly coordinates the bases. These types of interaction have been previously described in detail (reviewed in [3]).
Figure 1. Interaction of Mg2+ in the DNA major groove.
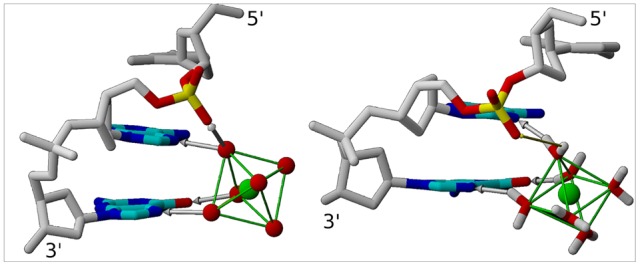
The 5′-CpApG-3′ fragment binds Mg2+ in both crystallographic (left) and simulated (right) structures. Mg2+ (green) forms water-mediated hydrogen bonds (gray arrows) with the N7 of both purines and the O6 of the guanine via its first hydration shell (left: oxygen atoms in red; right: explicit water molecules). The phosphate groups of CpA and ApG are in BII and BI conformation, respectively. Owing to the orientation of the O2P-O1P vector in the BII-CpA linkage, an additional hydrogen bond occurs between Mg2+ and O2P (left) in several X-ray structures. For the same reason, Mg2+ remains close to O2P in the simulated structures (right, thin yellow arrow).
Regarding DNA structure, we examined the two crucial inter-base pair parameters of twist and roll, and the conformational state of the phosphate groups (BI and BII conformers, represented Figure 2), three descriptors that are correlated in crystals as well as in solution [32], [34]–[41]. From this structural point of view, Ri–1piGi interacting with ions are characterized by low twist (25.8±4.6° on average), positive roll (7.9±2.9° on average), and BI phosphate groups. Interestingly, the preceding steps, Cpi–1Ai–1 or Tpi–1Gi–1, also exhibit homogeneous structural features, with twist and roll of 49.3±4.7° and −9.8±2.8°, respectively; their phosphate linkages are in BII. These two contrasted profiles, low twist/positive roll/BI conformer and high twist/negative roll/BII conformer, illustrate the couplings imposed by the intrinsic mechanics of B-DNA [32], [34]–[41].
Figure 2. BI and BII phosphate groups.
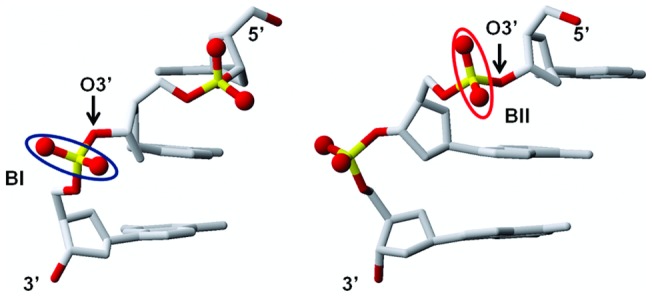
. The two panels represent two views of the same 5′-CpApG-3′ fragment and highlight the structural differences between BI (p in GpA, circled in blue in the left panel) and BII (p in CpA, circled in red in the right panel) phosphate groups. The orientation of the O1P-O2P vector is roughly perpendicular and parallel to the axis of the double helix in BI and BII, respectively. The O3′ atom points outside the helix in BI whereas it is turned towards the helix center in BII.
It is established that positive roll is required to allow divalent cation interaction with Ri–1piGi sites [7]. Thus, cations indirectly impose the BI backbone state, disqualifying the BII state, which is incompatible with positive roll. This is manifest on the phosphate groups of GpG, all in BI in the presence of cations, although they are particularly conducive to BII [42]. The observation of BII-pi–1 is more intriguing, especially for TpG that shows a non negligible but yet moderate BII propensity [42]. This feature can be better understood by considering the structural implications of the BI→BII transition and its relationship with cations. The BII backbone conformation differs from its canonical BI alternative in regard to the torsion angles ε and ζ, which are respectively trans/g- in BI (ε-ζ ∼ −90°) and g-/trans in BII (ε-ζ ∼ +90°) [37]. This ε/ζ crankshaft induces two major structural changes in the phosphate group (Figure 2): first, the orientation of the O1P-O2P vector is roughly perpendicular and parallel to the axis of the double helix in BI and BII, respectively; second, the O3′ atom that points outside the helix in BI is turned towards the center in BII. In particular, the phosphate group motion from BI to BII shortens the distance between O2P(pi–1) and N7(Ri–1) by roughly 1.8 Å. Therefore, any ion binding to N7(Ri–1) will be closer to O2P(pi–1) when pi–1 is in BII. In nine occurrences of our dataset, hydrogen bonds were observed between O2P(pi–1) and one water molecule belonging to the cation hydration layer (Figure 1). In the absence of visible or stable hydrogen bonds (11 cases), the short distances (5.8±0.2 Å on average) separating O2P and O3′ from cations implies the existence of significant electrostatic interactions.
This analysis of crystallographic data confirms the previous observation that divalent cations in the major groove preferentially cross-link dinucleotide steps containing at least one guanine. These steps are characterized by low twist, positive roll and a BI backbone state. However, this analysis also suggests that cations influence the structure and dynamics of the 5′-step by interacting with its phosphate group. This was further explored using theoretical modeling approaches.
Overview of the simulations
Molecular dynamics (MD) simulations in explicit solvent were done on the Jun-Fos oligomer. In addition to benefiting from an experimental support [31]–[34], this 14 bp oligomer contains a motif, CpApG, which appears to be recurrently contacted by cations in our X-ray dataset.
Four simulations of the Jun-Fos oligomer were considered (Table 1, detailed in Materials and Methods). The RMSD values calculated between canonical B-DNA and the simulated structures were very stable during all simulations. The structural characteristics of B-DNA such as χ values and sugar conformation were realistic. Convergence of the MD simulations with respect to the DNA structure was checked by comparing S1–S3. Statistics for the DNA descriptors give very similar results when extracted from any of these three trajectories. For instance, the average twist values of A22pG23 -which binds Mg2+- are 28.7±4.4°, 28.7±4.2° and 29.2±3.9° in S1, S2 and S3, respectively. Also, the phosphate group equilibrium, which is crucial for our study, is well reproduced through the trajectories, with between 36 and 40% of BII conformers on C21pA22 in S1–S3.
Table 1. MD simulations performed on the Jun-Fos oligomer.
| Name | Ions | Restraints on Mg2+ | ttotal (ns) | tint (ns) | ff for Mg2+ |
| S0 | Na+ | N.A. | 50 | N.A. | N.A |
| S1 | Na+ Mg2+ | No | 50 | 20 | Amber default |
| S2 | Na+ Mg2+ | No | 25 | 18.5 | [15] |
| S3 | Na+ Mg2+ | Yes | 75 | 75 | Amber default |
The simulations were performed with solvent containing Na+ or Na+ and Mg2+. Mg2+ was either free of restraints or restrained to be close (<5.5 Å) to A22 and G23. ttotal is the total duration of the trajectories. For S1–S3, tint is the duration of the interaction between Mg2+ in the major groove of A22 and G23. Two force field parameters for Mg2+ (ff for Mg2+) were used. N.A.: not applicable.
Mg2+ binding in the DNA major groove: effect on DNA structure and dynamics
We first present the results for S1 and S2, in which Mg2+ is free to escape from its initial position, close to A22 and G23. Mg2+/DNA interactions, monitored by the distances between Mg2+ and the electronegative atoms of A22 and G23 (example from S1: Figure 3), are observed during 20 and 18.5 ns in S1 and S2, respectively. These similar results show that the cation force field parameters do not significantly impact the Mg2+ behavior, since S1 and S2 use different parameters for the cation, either from Amber (S2) or from Varnai and Timsit [15]. During the time where Mg2+ interact with the oligomer, the low root mean square fluctuation (RMSF) values of Mg2+ (1.55 Å2 on average) reflect the existence of a well-defined attractive cation pocket. In the simulated structures, three water molecules of the Mg2+ hexahydrate cluster form hydrogen bonds with N7(A22, G23) and O6(G23), as observed in the crystallographic structures (Figure 1). The residence times of Mg2+ in the major groove indicates that B-DNA is likely not a Mg2+ binder as strong as RNA [43], [44], for instance the major groove of G•U wobble pairs [45]. However, the observation of ∼20 ns DNA-Mg2+ interactions ensures that the X-ray pattern is not an artifact, and that B-DNA in solution can transiently capture Mg2+.
Figure 3. Distances characterizing the interaction between Mg2+ and DNA during the MD simulations.

The distances between Mg2+ and the electronegative atoms N7(A22 and G23), O6(G23) and O2P(p22) belonging to the p22A22p23G23 fragment were extracted from S1. Mg2+ interacts with A22 and G23 via its first hydration shell for roughly 20 ns, as shown by the values of the distances d(Mg2+ – N7(A22)), d(Mg2+ – N7(G23)), and d(Mg2+ – O6(G23)). O2P(p22) is transitorily close to Mg2+. Short Mg2+ – O2P(A22) distances are associated with p22 in BII (BI in blue, BII in red). Identical interaction patterns are observed in S2 and S3.
The Mg2+/DNA interactions extracted from S1–S3 present identical characteristics. Thus, the snapshots of the trajectories S0–S3 (Table 1) were sorted in two groups. The first one contains the structures in which Mg2+ does not interact with the DNA major groove, for a total duration of 86.5 ns. The second group includes the snapshots characterized by water mediated hydrogen bonds between Mg2+, A22 and G23, which cover 113.5 ns.
Independently of the presence of Mg2+, A22pG23 is characterized by low twist (28.8±4° on average) and positive roll (4.6±4° on average). p23 is confined in BI, with less than 4% of BII. Indeed, ApG is not conducive to BII [42]. These ApG structural features are ideally adapted to the octahedral structure of hydrated Mg2+ (Figure 4). The distance between the two N7 atoms of A22 and G23 is 3.5±0.2 Å on average, allowing the bridging of two water molecules belonging to the first solvation shell of Mg2+. The other conformational profile encountered in B-DNA, i.e. high twist/negative roll/BII phosphate, pushes the two N7 atoms away (N7–N7 distance of 4.2±0.2 Å on average) and is thus less convenient for binding Mg2+. Our simulations confirm that bridging two successive purines by Mg2+ necessitates low twist and positive roll [7] and, indirectly, BI phosphate group.
Figure 4. ApG step with and without Mg2+.
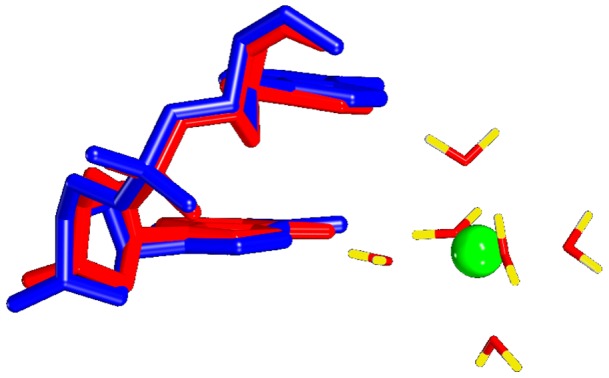
Superimposition of the average structures of A22pG23 either free of Mg2+ (blue) or binding Mg2+(red). In both cases, this step shows the same structural characteristics (low twist, positive roll and BI phosphate group). ApG is thus ideally adapted to the octahedral structure of hydrated Mg2+.
In contrast with p23, p22 undergoes BI↔BII transitions. C21p22A22p23G23 with p23 in BI and p22 either in BI or BII differ by ∼1 Å of RMSD. BI/BII populations of p22 are modulated by Mg2+ (Figure 5). In absence of Mg2+, p22 presents on average 24% of BII conformers ((ε-ζ) >0°). This percentage reaches 39% when hydrated Mg2+ coordinates A22 and G23. Thus, Mg2+ interacting with A22 and G23 increases the proportion of the p22 BII population. This latter result and the observations made on the crystallographic structures prompted a careful examination of the spatial relationship between Mg2+ and the atoms belonging to p22. The distances between O2P/O3′ (p22) and N7(A22) depend on the conformational state of p22 (Figure 6) owing to the structural changes during the BI↔BII transition, as mentioned above (Figure 2). When Mg2+ interacts with A22, both d(O2P(p22)-Mg2+) and d(O3′ (p22)-Mg2+(A22)) are shortened for stable BII conformers ((ε-ζ) >75°) (Figure 6). These distances are compatible with significant electrostatic interactions between Mg2+, O3′ (p22) and O2P(p22) but remain generally too great to allow the formation of additional hydrogen bonds. In particular, Mg2+-O2P water-mediated hydrogen bonds are observed in nearly the half of the crystallographic interactions whereas they were only marginal in our simulations (3.5% of the DNA-Mg2+ interactions). Actually, the Mg2+ locations in X-ray and simulated structures differ slightly (Figure 1). Mg2+ remains coplanar with the guanine during the trajectories, a position that increases the distance between the cation and O2P compared to the X-ray structures. Without excluding a possible force field bias, we cannot reasonably expect that Mg2+ locations are exactly identical in the solid state (i.e. from the crystallographic dataset) and in solution (i.e. from simulations).
Figure 5. (ε-ζ) distribution without and with Mg2+ in the DNA major groove.
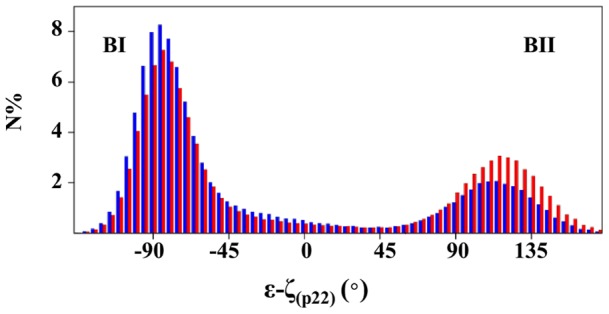
N is the percentage of snapshots within each (ε-ζ)p22 interval. The distribution of (ε-ζ)p22 values was calculated on two groups of structures extracted from S0–S3. The first group (blue) corresponds to snapshots in which Mg2+ does not interact with the DNA major groove. In the second group (red), Mg2+ binds to the N7 and O6 atoms of A22/G23 through three water-mediated hydrogen bonds. The BII population of the 5′ phosphate group, p22, increases.
Figure 6. Effect of p22 conformational state on Mg2+-p22A22 distances.
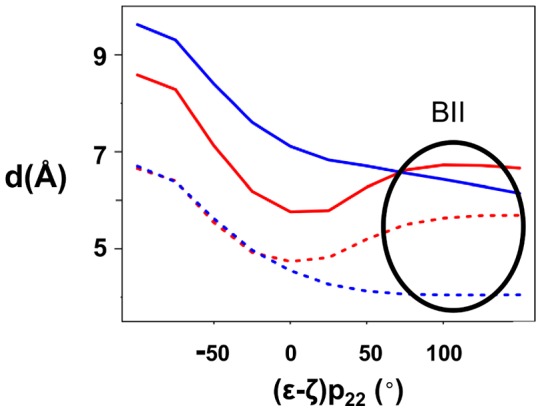
The distances between the OP2 and O3′ atoms of p22 and A22 (d(O2P(p22)-N7(A22)), indicated by a dashed red line; (d(O3′ (p22)-N7(A22) by a dashed blue line) depend on the conformational state of p22, represented here by (ε-ζ) p22 values. When Mg2+ interacts with A22 and G23, d(Mg2+-O2P(p22)) (red) and d(Mg2+-O3′ (p22)) (blue) are parallel to d(O2P(p22)-N7(A22)) and d(O3′ (p22)-N7(A22), respectively. The standard deviations for distances are 1.2 Å. The distances were examined on the two groups of structures defined in Figure 5 caption.
The twist and roll of C21pA22 cover a large range of values, positive/negative rolls and low/high twists being associated with BI and BII conformers, respectively, according to the intrinsic B-DNA mechanics. The evolution of twist and roll as a function of (ε-ζ), shown Figure 7, is common to all the simulations. A clear transition between low and high twist values occurs around (ε-ζ) = −30°. Roll values change more slowly and are more gradual. Mg2+-mediated increase in the pure BII population of p22 leads to an increase in the proportion of the high twist/negative roll conformation in C21p22A22 without affecting the structural couplings characteristic of the double helix.
Figure 7. Changes in inter-base pair parameters as a function of (ε-ζ) values.
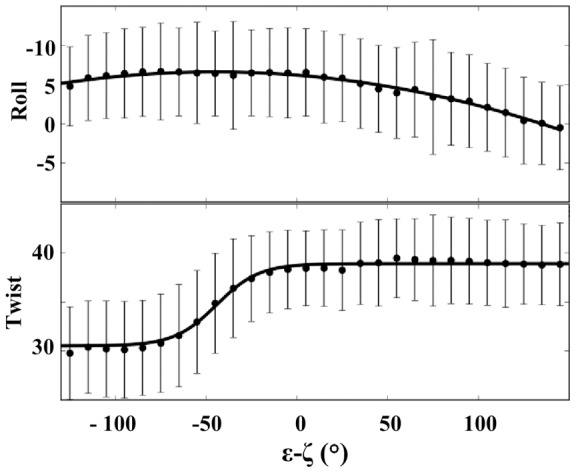
The C21p22A22 average values of roll (°) and twist (°) and their standard deviations (bars) are plotted as a function of the (ε-ζ) values of p22, considering S0–S3. Mg2+-mediated enhancement of the BII population of p22 leads to an increase in the proportion of conformations characterized by negative rolls and high twists.
In sum, the interaction between Mg2+ and the DNA major groove involves the C21p22A22p23G23 fragment. The intrinsic structural properties of A22p23G23 match the octahedral configuration of the hydrated cation. Mg2+ modulates p22 behavior, favoring BII conformers. In turn, the conformational landscape explored by C21p22A22 is modified in terms of roll and twist. The proximity between Mg2+ and both O2P(p22) and O3′ (p22) indicates that p22 actively participates in the electrostatic component of the interaction, which is investigated in the next section.
Electrostatic relationships between Mg2+ and DNA
Poisson-Boltzmann calculations were undertaken on a series of snapshots extracted from S1 to investigate the effects of the dinucleotidic sequence and structure on the DNA-Mg2+ interaction.
First, the electrostatic attractiveness of the DNA major groove for cations was examined along the 6 bp fragment T20pC21pA22pG23pA24pA25. We focused on a series of 0.5 Å sided cubes, calculating and averaging the electrostatic potentials of 9 points per cube (8 vertices and the center). These cubes were positioned close to each dinucleotide Ni–1piNi so that they reproduced the average location of Mg2+ at A22pG23 in the simulations.
The electrostatic potential values of each Ni–1piNi major groove region, sorted according the conformational state of pi–1, are summarized Table 2. The proximity between the electronegative atoms of pi–1 and Ni–1 induced by pi–1 in BII tends to reinforce the electronegative character of the major groove. However, the electrostatic potentials are primarily subordinated to the dinucleotide sequence. Owing to their electronegative atoms in the major groove, G-containing dinucleotides, especially ApG, induce stronger negative potentials than ApA, CpA or TpC.
Table 2. Electrostatic potential values at cation locations in the DNA major groove.
| pi–1Ni–1piNi | Ep (kT/e) for pi–1 in BI | Ep (kT/e) for pi–1 in BII |
| p20T20p21C21 | −2.4 (0.6) | −2.2 (0.2) |
| p21C21p22A22 | −2.7 (0.5) | −3.2 (0.6) |
| p22A22p23G23 | −3.8 (0.7) | −4.3 (0.6) |
| p23G23p24A24 | −3.3 (0.7) | −3.5 (0.5) |
| p24A24p25A25 | −2.9 (0.4) | −3.2 (0.5) |
Poisson-Boltzmann electrostatic potentials (Ep) were calculated for 0.5 Å sided cubes centered on the average position of Mg2+ observed in MD structures (Figure 1). These squares were shifted along the five Ni–1piNi dinucleotide steps composing the T20p21C21p22A22p23G23p24A24p25A25 fragment. The potentials are sorted according to the conformational state of pi–1. A total of 100 snapshots extracted from S1 were examined. Standard deviations are given in parentheses.
Additional Poisson-Boltzmann calculations were done to better define the region and the atoms the most exposed to the electrostatic effect of Mg2+ during its interaction with DNA. Electrostatic potentials were calculated on DNA atomic positions, upon either removing (Epref) or keeping Mg2+ (EpMg2+). We consider here the difference between these two values, ΔEp = Epref – EpMg2+, and examined the different DNA components, i.e. the sugars, bases and phosphate groups (P, O1P, O2P, O3′ and O5′), along the sequence. As expected, important modifications in electrostatic potentials occur in the region surrounding the bases that bind the cation, A22 and G23. The results are summarized in Table 3. Mg2+ induces strong changes on the electrostatic potential of the electronegative atoms of A22 and G23. In addition, the A22 neighbor, A24, and the partners of A24 and A22, T5 and T7, also feel the presence of the cation. The electrostatic potentials of neither the sugar nor the phosphate groups are affected by Mg2+, apart from the noticeable case of p22. The electrostatic changes on p22, especially on O2P and O3′, are particularly marked when p22 adopts the BII conformation. According to the MD simulations, this electrostatic effect is not sufficient to completely trap p22 in BII. However, it explains the shift of the BI↔BII equilibrium towards BII. Therefore, N7(A22), N7(G23), O2P(p22) and O3′ (p22) are major players in the interaction of Mg2+ within the DNA major groove.
Table 3. Effect of Mg2+ on DNA atomic electrostatic potentials around the cation binding site.
| DNA atom or group of atom | ΔEp (kT/e) for p22 in BI | ΔEp (kT/e) for p22 in BII |
| O4(T7) | −1.3 (0.3) | −1.5 (0.3) |
| O4(T5) | −2.1 (0.2) | −2.1 (0.3) |
| N7(A22) | −3.1 (0.2) | −3.2 (0.3) |
| N7(G23) | −2.2 (0.3) | −2.3 (0.3) |
| O6(G23) | −0.9 (0.3) | −1.9 (0.5) |
| N7(A24) | −1.7 (0.3) | −1.7 (0.3) |
| p22 | −3.9 (0.3) | −8.3 (0.3) |
| O3′ (p22) | −0.6 (0.2) | −2.1 (0.3) |
| O2P(p22) | −1.1 (0.3) | −2.2 (0.5) |
ΔEp represents the electrostatic effect of Mg2+ on various DNA atoms or groups of atoms. Mg2+ binds to A22 and G23 (in bold) in the C21p22 A22p23 G23p24A24 fragment. ΔEp values were calculated for p22 either in BI (ε-ζ<−50°) or BII (ε-ζ>75°). T7 and T5 are the partners of A22 and A24, respectively. The phosphate group p22 is comprised of six atoms, O3′, O1P, P, O2P and O5′. A total of 90 snapshots extracted from S1 were examined. Standard deviations are given in parentheses.
These calculations create a credible scenario for Mg2+ interacting with the DNA major groove. Hydrated Mg2+ takes advantage of the negative pocket in the major groove of two successive bases, one guanine and one purine, to bridge them by water-mediated hydrogen bonds. When the 5′ phosphate group spontaneously shifts towards BII, its O2P and O3′ atoms move closer to the bound Mg2+, increasing their mutual electrostatic attractiveness. This additional interaction therefore helps stabilize the cation within the major groove and increases the BII population of pi–1.
Discussion
Instigated by a body of crystallographic evidence, our modeling investigations show that Mg2+ is able to form stable intra-strand cross-links in the major groove of the ApG step in solution. The interaction network involves the first hydration shell of Mg2+ and the pi–1Ai–1piGi fragment. Ai–1piGi adopts a conformation characterized by positive roll and BI phosphate linkages. Our molecular dynamics approach demonstrates that the counterion increases the percentage of BII conformers of pi–1, such that Cpi–1Ai–1 more frequently explores negative roll conformations. MD trajectories associated with Poisson-Boltzmann calculations helped reveal the mechanism underpinning the interaction of divalent cations with the DNA major groove. Cations are attracted to the major groove of Ai–1piGi, which is structurally well-adapted to cation binding and forms a strong and well-defined electronegative pocket. Both O2P and O3′ atoms of pi–1 benefit from a favorable electrostatic interaction with the cation, further enhanced when pi–1 is in BII. This latter interaction helps stabilize both the cation and BII conformers of pi–1.
Divalent cations bridging two successive guanines in the major groove have been suspected to induce local bends in X-ray structures [7]. Such cation-induced curvature is supported by indirect indications gleaned from observations in solution made on DNAs containing (G)n tracts [28]–[30]. The Jun-Fos oligomer in the present study bends towards the major groove with an average curvature of 16° [34]. Our observations here show that this bending is not significantly affected by one bound Mg2+. Nevertheless, from a general point of view, curvature intensity depends on the succession of roll signs along the sequence, which are correlated with BI/BII conformations. Accordingly, it cannot be ruled out that Mg2+ influences the global curvature of particular sequences, for instance in (G)n tracts, by accumulating modulations of their BI/BII ratio.
From a structural point of view, CpApG appears to form a very favorable cation binding site. In this trinucleotide fragment, an intrinsically BII-conducive phosphate group in CpA precedes an ApG step, ready to coordinate a hydrated cation. Our analysis of crystallographic structures reveals that a TpGpG tract is also an attractive site, likely because of the non negligible BII propensity of TpG and the strength of the electrostatic potential in the GpG major groove. Other trinucleotides, made of BII-rich dinucleotides followed by ApG, GpG or GpA, may also fix cations. In this line, another caveat concerns the balance between the Mg2+ residence times near the charged phosphate groups and near the major groove electronegative atoms. This point was previously addressed by a modeling study [46], which demonstrated that Mg2+ were more frequently found near phosphates than in the major groove. Yet, this study also suggested that the presence of Mg2+ in the major groove affects the bending occurring at A-tract flanking sequences. Given this context, systematic studies should to be done to inventory the most propitious sites for harboring cations.
Are our findings applicable to cations other than Mg2+? The examination of the six Ca2+ present in the crystallographic dataset strongly suggests that this physiological cation behaves similarly to Mg2+. Nevertheless, the stability of Ca2+ in the DNA major groove remains to be confirmed, given that its first solvation shell, crucial for the interaction, may be more labile than the Mg2+ solvation shell [9]. Regarding monovalent cations, 31P NMR results [33] show the differential influence of Na+ and K+ on the BI↔BII equilibrium of several phosphate linkages, unambiguously demonstrating that DNA in solution is influenced by the nature of the monovalent cation. The examination of the two crystallographic structures that contain bound monovalent cations in the major groove (Rb+, PDB code 3GG; Tl+, PDB code 3GGI) reveals an interaction scheme identical to the one observed here for divalent cations. We carried out several MD simulations not presented here, replacing Mg2+ with K+. Nevertheless, the residence times of K+ at the ApG step were very limited (at best 1.5 ns) compared to those observed with Mg2+. Such instability, also noticed in other DNA sequences [47], [48], hinders the acquisition of significant statistics on ion coordination and structural effects. Thus, the mechanism by which monovalent cations influence DNA behavior remains an open question.
Although divalent cations do not radically change DNA structure and dynamics, they appear to modulate DNA behavior and they may thereby help stabilize sharp DNA deformations in DNA-protein complexes. Ions in DNA wrapped around a histone core offer a good system through which the potential role of cations in DNA-protein interactions could be addressed. The nucleosome core particle (NCP) consists of 146–147 bp of DNA wound twice around an octameric core of four histone proteins. This DNA superhelical path induces a periodic alternation of positive and negative roll tracts [49], mainly associated with BI and BII phosphates, respectively [42]. In the best resolved NCP structure (PDB code 1KX5), 12 Mn2+ (mimicking Mg2+) are observed in the DNA major groove [50], [51]. Two of them form inter-strand bridges. The 10 remaining cations coordinate GpG or ApG, characterized by positive rolls and BI phosphates. The 5′-TpG or CpA neighbors of these cation-binding dinucleotides exhibit marked negative rolls (-13±8° on average) associated with BII phosphates, and their O2P atoms are involved in a water-mediated hydrogen bond with Mn2+. These features are surprisingly similar to those observed on free DNA. Hence, divalent cations may fix particular DNA conformations required by interacting proteins, limiting the energetic and entropic costs of protein binding.
In conclusion, the present study suggests that DNA structure and dynamics are sensitive to divalent cations in the major groove. Cation binding is sequence-dependent and modulates the intrinsic sequence-dependent properties of DNA in terms of the population of conformational states, including the BI↔BII equilibrium of phosphate groups. Although the role of cation-mediated modulation in DNA packaging and readout is largely speculative at this stage, it is an exciting hypothesis to test in regard to DNA-protein processes.
Materials and Methods
Crystallographic dataset
DNA crystal structures were selected based on the following two criteria: high or very high resolution and the presence of divalent cations in the DNA major groove. The dataset comprises 11 B-DNA decamers with a resolution of ≤1.65 Å (PDB codes: 3GGI, 3GGK, 3GGB, 1ZF7, 1ZFB, 1D23, 1ENE, 1EN9, 1EN8, 1EN3, 1D8G).
DNA sequence
The double-stranded DNA oligomer studied using molecular dynamics simulations has the following 14 bp sequence: 5′-d(G1 C2 A3 T4 T5 C6 T7 G8 A9 G10 T11 C12 A13 G14 )-3′•5′-d(C15 T16 G17 A18 C19 T20 C21 A22 G23 A24 A25 T26 G27 C28 )-3′. This oligomer, called the Jun-Fos oligomer, has been exhaustively characterized by NMR and modeling [31]–[34].
Molecular dynamics (MD) simulations
All simulations were performed using the AMBER 10 program [52], with the Parm98 force field [53] and under NMR restraints. Actually, in absence of experimental restraints, notorious shortcomings occur with AMBER force fields, especially regarding the backbone behavior [34]. Parm98 does not implement realistic BI↔BII equilibrium (related to ε-ζ) [34] and drives α/γ angles in unusual conformations [54]. Parmbsc0 [55] overly suppresses BII conformers [34] but improves the α/γ behavior, without totally avoiding undesirable flips [56]. Unfortunately, NMR restraints associated to Parmbsc0 do not rescue BI/BII populations [34]. In contrast, the same restraints used in conjunction with Parm98 restitute more acceptable behaviors for both ε-ζ and α/γ [31], [34]. It is the reason why we chose Parm98, supplemented by NMR restraints.
The starting point was a canonical B-DNA, neutralized with either 26 Na+ or 24 Na+ and one Mg2+ and hydrated with TIP3P [57] water molecules in a truncated octahedron simulation box. The distance between the center of DNA periodic images is ∼90 Å, which allows for a solvent shell extending at least 15 Å around DNA. Simulations were performed at constant temperature (300 K) and pressure (1 bar) using a Berendsen coupling algorithm [58]. The integration time step was 2 fs and covalent bonds involving hydrogen were constrained using SHAKE [59]. Long-range electrostatic interactions were treated using the particle mesh Ewald approach [60] with a 9 Å direct space cut-off and a 0.00001 Ewald convergence tolerance for the long range electrostatic interactions. The non-bonded pair-list was updated heuristically and the center-of-mass motion was removed every 10 ps.
Water molecules and cations were energy-minimized and equilibrated in the NVT ensemble at 100 K for 100 ps, with the DNA constrained. The entire system (DNA, water molecules and ions) was then heated from 100 to 300 K in 10 ps in 5 K increments with harmonic restraints of 5.0 kcal mol−1 Å−2 on the solute atoms. The simulations were continued in the NPT ensemble, without a noticeable change in volume. The positional restraints were gradually removed over 250 ps and followed by the production phases.
All MD simulations were run with a set of restraints consisting in three NMR internucleotide distances per dinucleotide, H2′i-H6/8i+1, H2″i-H6/8i+1, and H6/8i-H6/8i+1, inferred from 31P chemical shifts [31], [32]. These restraints were applied instantaneously via a mixed parabolic (for distance −10%) and hyperbolic (for distance +10%) potential with a force constant of 10 Kcal.mol−1.Å−2 around a central, flat-bottomed shape covering the experimental range of distances (estimated at 10% of the distances). We previously showed that this protocol best reproduces the exhaustive NMR data measured on the studied oligomer, while preserving the DNA dynamics [31]. Removing these NMR restraints in an additional 25 ns trajectory ensured that the main conclusions presented here were robust.
Four trajectories were considered (Table 1). S0, done in presence of Na+ counterions, will be the reference simulation. Three simulations, S1–S3, were carried out with Na+ and Mg2+, keeping the same starting point but reinitializing velocities. Mg2+ was initially restrained close to two bases of the Jun-Fos oligomer, A22 and G23, by three distance restraints (d(Mg2+ – N7(A22)) <4.8 Å, d(Mg2+ -N7(G23)) <6.0 Å and d(Mg2+ -O6(G23)) <5.0 Å), according to the preferred major groove location observed in X-ray DNA structures. In S1 and S2, the restraints were then gradually removed before running trajectories in which Mg2+ was thus free to escape from its initial position. To ensure robust statistics on DNA-ion interactions, an additional trajectory, S3, was carried out maintaining the restraints. The analysis covered a total of 200 ns of production runs.
The default Mg2+ parameters provided in AMBER were used in S1 and S3. S2 was carried out using Mg2+ parameters given by a recent study [15]. These parameters (r* = 0.6245 Å and ε = 28.4444 kcal mol−1) reinforce the van der Waals potential, and accurately reproduce the free energy of Mg2+ hydration and the radial distribution function of water.
Electrostatic Calculations
Electrostatic potential maps were calculated with the Adaptive Poisson-Boltzmann Solver (APBS) [61] using a nonlinear solution. These calculations were done on snapshots extracted from trajectories, using APBS default parameters (physiological NaCl concentration of 150 mM, temperature of 298 K, solvent dielectric of 78.4, and solute dielectric of 2). Van der Waals radii and partial charges for DNA were those of the Parm98 force field. Solute charges were distributed onto grid points using a cubic B-spline discretization. The molecular surface was defined by the interface between a 1.4 Å solvent probe, corresponding to the radius of a water molecule, and the solute van der Waals radii.
Structure analysis
X-ray and MD DNA structures were analyzed using the Curves+ algorithm [62]. The phosphate conformations were analyzed in terms of BI and BII states, defined by the value of the pseudo-angle (ε-ζ): BI corresponds to (ε-ζ) <0° and BII to (ε-ζ) >0°. The alternative atomic positions proposed in the PDB files of some X-ray structures (PDB codes: 3GGI, 3GGK, 3GGB, 1EN3, 1D8G, 1EN9) were analyzed separately. The first nanosecond of each MD was discarded from the analysis.
For clarity, we consider that pi phosphate linkage is composed of the atoms O3′, O1P, P, O2P and O5′, noted X(pi).
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: Funding was provided by the Institut National de la Transfusion Sanguine, 6 rue Alexandre Cabanel, 75015 Paris, France. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Egli M. DNA-cation interactions: quo vadis? Chem Biol. 2002;9:277–286. doi: 10.1016/s1074-5521(02)00116-3. [DOI] [PubMed] [Google Scholar]
- 2.Hud NV, Polak M. DNA-cation interactions: The major and minor grooves are flexible ionophores. Curr Opin Struct Biol. 2001;11:293–301. doi: 10.1016/s0959-440x(00)00205-0. [DOI] [PubMed] [Google Scholar]
- 3.Subirana JA, Soler-Lopez M. Cations as hydrogen bond donors: a view of electrostatic interactions in DNA. Annu Rev Biophys Biomol Struct. 2003;32:27–45. doi: 10.1146/annurev.biophys.32.110601.141726. [DOI] [PubMed] [Google Scholar]
- 4.Ponomarev SY, Thayer KM, Beveridge DL. Ion motions in molecular dynamics simulations on DNA. Proc Natl Acad Sci U S A. 2004;101:14771–14775. doi: 10.1073/pnas.0406435101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai Y, Greenfeld M, Travers KJ, Chu VB, Lipfert J, et al. Quantitative and comprehensive decomposition of the ion atmosphere around nucleic acids. J Am Chem Soc. 2007;129:14981–14988. doi: 10.1021/ja075020g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abrescia NG, Malinina L, Fernandez LG, Huynh-Dinh T, Neidle S, et al. Structure of the oligonucleotide d(CGTATATACG) as a site-specific complex with nickel ions. Nucleic Acids Res. 1999;27:1593–1599. doi: 10.1093/nar/27.7.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu TK, Dickerson RE. 1 A crystal structures of B-DNA reveal sequence-specific binding and groove-specific bending of DNA by magnesium and calcium. J Mol Biol. 2000;301:915–945. doi: 10.1006/jmbi.2000.4012. [DOI] [PubMed] [Google Scholar]
- 8.Grzeskowiak K, Goodsell DS, Kaczor-Grzeskowiak M, Cascio D, Dickerson RE. Crystallographic analysis of C-C-A-A-G-C-T-T-G-G and its implications for bending in B-DNA. Biochemistry. 1993;32:8923–8931. doi: 10.1021/bi00085a025. [DOI] [PubMed] [Google Scholar]
- 9.Kielkopf CL, Ding S, Kuhn P, Rees DC. Conformational flexibility of B-DNA at 0.74 A resolution: d(CCAGTACTGG)(2). J Mol Biol. 2000;296:787–801. doi: 10.1006/jmbi.1999.3478. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Subirana JA. Structure of d(CGCGAATTCGCG) in the presence of Ca(2+) ions. J Biol Chem. 1999;274:24749–24752. doi: 10.1074/jbc.274.35.24749. [DOI] [PubMed] [Google Scholar]
- 11.Mack DR, Chiu TK, Dickerson RE. Intrinsic bending and deformability at the T-A step of CCTTTAAAGG: a comparative analysis of T-A and A-T steps within A-tracts. J Mol Biol. 2001;312:1037–1049. doi: 10.1006/jmbi.2001.4994. [DOI] [PubMed] [Google Scholar]
- 12.Millonig H, Pous J, Gouyette C, Subirana JA, Campos JL. The interaction of manganese ions with DNA. J Inorg Biochem. 2009;103:876–880. doi: 10.1016/j.jinorgbio.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Minasov G, Tereshko V, Egli M. Atomic-resolution crystal structures of B-DNA reveal specific influences of divalent metal ions on conformation and packing. J Mol Biol. 1999;291:83–99. doi: 10.1006/jmbi.1999.2934. [DOI] [PubMed] [Google Scholar]
- 14.Soler-Lopez M, Malinina L, Subirana JA. Solvent organization in an oligonucleotide crystal. The structure of d(GCGAATTCG)2 at atomic resolution. J Biol Chem. 2000;275:23034–23044. doi: 10.1074/jbc.M002119200. [DOI] [PubMed] [Google Scholar]
- 15.Varnai P, Timsit Y. Differential stability of DNA crossovers in solution mediated by divalent cations. Nucleic Acids Res. 2010;38:4163–4172. doi: 10.1093/nar/gkq150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hud NV, Schultze P, Sklenar V, Feigon J. Binding sites and dynamics of ammonium ions in a telomere repeat DNA quadruplex. J Mol Biol. 1999;285:233–243. doi: 10.1006/jmbi.1998.2327. [DOI] [PubMed] [Google Scholar]
- 17.Hud NV, Sklenar V, Feigon J. Localization of ammonium ions in the minor groove of DNA duplexes in solution and the origin of DNA A-tract bending. J Mol Biol. 1999;286:651–660. doi: 10.1006/jmbi.1998.2513. [DOI] [PubMed] [Google Scholar]
- 18.Quintana JR, Grzeskowiak K, Yanagi K, Dickerson RE. Structure of a B-DNA decamer with a central T-A step: C-G-A-T-T-A-A-T-C-G. J Mol Biol. 1992;225:379–395. doi: 10.1016/0022-2836(92)90928-d. [DOI] [PubMed] [Google Scholar]
- 19.Pullman A, Pullman B. Molecular electrostatic potential of the nucleic acids. Quarterly reviews of biophysics. 1981;14:289–380. doi: 10.1017/s0033583500002341. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasan AR, Sauers RR, Fenley MO, Boschitsch AH, Matsumoto A, et al. Properties of the Nucleic-acid Bases in Free and Watson-Crick Hydrogen-bonded States: Computational Insights into the Sequence-dependent Features of Double-helical DNA. Biophys Rev. 2009;1:13–20. doi: 10.1007/s12551-008-0003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moldrheim E, Andersen B, Froystein NA, Sletten E. Interaction of manganese(II), cobalt(II) and nickel(II) with DNA oligomers studied by 1H NMR spectroscopy. Inorganica Chimica Acta. 1998;273:41–46. [Google Scholar]
- 22.Froystein N, Davis J, Reid B, Sletten E. Sequence-selective metal ion binding to DNA oligonucleotides. Acta Chem Scand. 1993;47:49–57. doi: 10.3891/acta.chem.scand.47-0649. [DOI] [PubMed] [Google Scholar]
- 23.Froystein N, Sletten E. The Binding of Manganese and Zinc to the Synthetic Oligonucleotide d(CGCGAATTCGCG). A 1H NMR Study. Acta chemica scandinavica. 1991;45:219–225. doi: 10.3891/acta.chem.scand.45-0219. [DOI] [PubMed] [Google Scholar]
- 24.Steinkopf S, Sletten E. Sequence-selective metal ion binding to DNA hexamers. Acta Chem Scand. 1994;48:388–392. doi: 10.3891/acta.chem.scand.48-0388. [DOI] [PubMed] [Google Scholar]
- 25.Tereshko V, Minasov G, Egli M. The Dickerson-Drew B-DNA Dodecamer Revisited at Atomic Resolution. Journal of the American Chemical Society. 1998;121:470–471. [Google Scholar]
- 26.Ahmad R, Arakawa H, Tajmir-Riahi HA. A comparative study of DNA complexation with Mg(II) and Ca(II) in aqueous solution: major and minor grooves bindings. Biophys J. 2003;84:2460–2466. doi: 10.1016/S0006-3495(03)75050-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiu TK, Kaczor-Grzeskowiak M, Dickerson RE. Absence of minor groove monovalent cations in the crosslinked dodecamer C-G-C-G-A-A-T-T-C-G-C-G. J Mol Biol. 1999;292:589–608. doi: 10.1006/jmbi.1999.3075. [DOI] [PubMed] [Google Scholar]
- 28.Brukner I, Susic S, Dlakic M, Savic A, Pongor S. Physiological concentration of magnesium ions induces a strong macroscopic curvature in GGGCCC-containing DNA. J Mol Biol. 1994;236:26–32. doi: 10.1006/jmbi.1994.1115. [DOI] [PubMed] [Google Scholar]
- 29.Han W, Dlakic M, Zhu YJ, Lindsay SM, Harrington RE. Strained DNA is kinked by low concentrations of Zn2+. Proc Natl Acad Sci U S A. 1997;94:10565–10570. doi: 10.1073/pnas.94.20.10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jerkovic B, Bolton PH. Magnesium increases the curvature of duplex DNA that contains dA tracts. Biochemistry. 2001;40:9406–9411. doi: 10.1021/bi010853j. [DOI] [PubMed] [Google Scholar]
- 31.Abi-Ghanem J, Heddi B, Foloppe N, Hartmann B. DNA structures from phosphate chemical shifts. Nucleic Acids Res. 2010;38:e18. doi: 10.1093/nar/gkp1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heddi B, Foloppe N, Bouchemal N, Hantz E, Hartmann B. Quantification of DNA BI/BII backbone states in solution. Implications for DNA overall structure and recognition. J Am Chem Soc. 2006;128:9170–9177. doi: 10.1021/ja061686j. [DOI] [PubMed] [Google Scholar]
- 33.Heddi B, Foloppe N, Hantz E, Hartmann B. The DNA structure responds differently to physiological concentrations of K(+) or Na(+). J Mol Biol. 2007;368:1403–1411. doi: 10.1016/j.jmb.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Heddi B, Foloppe N, Oguey C, Hartmann B. Importance of accurate DNA structures in solution: the Jun-Fos model. J Mol Biol. 2008;382:956–970. doi: 10.1016/j.jmb.2008.07.047. [DOI] [PubMed] [Google Scholar]
- 35.Djuranovic D, Hartmann B. Conformational characteristics and correlations in crystal structures of nucleic acid oligonucleotides: evidence for sub-states. J Biomol Struct Dyn. 2003;20:771–788. doi: 10.1080/07391102.2003.10506894. [DOI] [PubMed] [Google Scholar]
- 36.Djuranovic D, Hartmann B. DNA fine structure and dynamics in crystals and in solution: the impact of BI/BII backbone conformations. Biopolymers. 2004;73:356–368. doi: 10.1002/bip.10528. [DOI] [PubMed] [Google Scholar]
- 37.Grzeskowiak K, Yanagi K, Prive GG, Dickerson RE. The structure of B-helical C-G-A-T-C-G-A-T-C-G and comparison with C-C-A-A-C-G-T-T-G-G. The effect of base pair reversals. Journal of Biological Chemistry. 1991;266:8861–8883. doi: 10.2210/pdb1d23/pdb. [DOI] [PubMed] [Google Scholar]
- 38.Hartmann B, Piazzola D, Lavery R. BI-BII transitions in B-DNA. Nucleic Acids Res. 1993;21:561–568. doi: 10.1093/nar/21.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oguey C, Foloppe N, Hartmann B. Understanding the sequence-dependence of DNA groove dimensions: implications for DNA interactions. PLoS One. 2010;5:e15931. doi: 10.1371/journal.pone.0015931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Dam L, Levitt MH. BII nucleotides in the B and C forms of natural-sequence polymeric DNA: A new model for the C form of DNA. J Mol Biol. 2000;304:541–561. doi: 10.1006/jmbi.2000.4194. [DOI] [PubMed] [Google Scholar]
- 41.Winger RH, Liedl KR, Pichler A, Hallbrucker A, Mayer E. Helix morphology changes in B-DNA induced by spontaneous B(I)< = = >B(II) substrate interconversion. J Biomol Struct Dyn. 1999;17:223–235. doi: 10.1080/07391102.1999.10508355. [DOI] [PubMed] [Google Scholar]
- 42.Heddi B, Oguey C, Lavelle C, Foloppe N, Hartmann B. Intrinsic flexibility of B-DNA: the experimental TRX scale. Nucleic Acids Res. 2010;38:1034–1047. doi: 10.1093/nar/gkp962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Auffinger P, Grover N, Westhof E. Metal ion binding to RNA. Met Ions Life Sci. 2011;9:1–35. [PubMed] [Google Scholar]
- 44.Draper DE, Grilley D, Soto AM. Ions and RNA folding. Annual review of biophysics and biomolecular structure. 2005;34:221–243. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 45.Joli F, Bouchemal N, Laigle A, Hartmann B, Hantz E. Solution structure of a purine rich hexaloop hairpin belonging to PGY/MDR1 mRNA and targeted by antisense oligonucleotides. Nucleic Acids Res. 2006;34:5740–5751. doi: 10.1093/nar/gkl617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young MA, Beveridge DL. Molecular dynamics simulations of an oligonucleotide duplex with adenine tracts phased by a full helix turn. J Mol Biol. 1998;281:675–687. doi: 10.1006/jmbi.1998.1962. [DOI] [PubMed] [Google Scholar]
- 47.Auffinger P, Westhof E. Water and ion binding around RNA and DNA (C, G) oligomers. J Mol Biol. 2000;300:1113–1131. doi: 10.1006/jmbi.2000.3894. [DOI] [PubMed] [Google Scholar]
- 48.Varnai P, Zakrzewska K. DNA and its counterions: a molecular dynamics study. Nucleic Acids Res. 2004;32:4269–4280. doi: 10.1093/nar/gkh765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 50.Davey CA, Richmond TJ. DNA-dependent divalent cation binding in the nucleosome core particle. Proc Natl Acad Sci U S A. 2002;99:11169–11174. doi: 10.1073/pnas.172271399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu B, Davey CA. Using soft X-rays for a detailed picture of divalent metal binding in the nucleosome. J Mol Biol. 2010;398:633–640. doi: 10.1016/j.jmb.2010.03.038. [DOI] [PubMed] [Google Scholar]
- 52.Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheatham TE 3rd, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J Biomol Struct Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 54.Dixit SB, Beveridge DL, Case DA, Cheatham TE 3rd, Giudice E, et al. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys J. 2005;89:3721–3740. doi: 10.1529/biophysj.105.067397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE 3rd, et al. Refinement of the AMBER force field for nucleic acids: improving the description of [alpha]/[gamma] conformers. Biophysical journal. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lankas F, Spackova N, Moakher M, Enkhbayar P, Sponer J. A measure of bending in nucleic acids structures applied to A-tract DNA. Nucleic Acids Res. 2010;38:3414–3422. doi: 10.1093/nar/gkq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jorgensen WL, Chandrasekhar J, Madura JD. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 58.Berendsen HJC, Postma WF, Van Gunsteren A, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 59.Ryckaert J, Ciccotti G, Berendsen H. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J comput Phys. 1977;23:327–341. [Google Scholar]
- 60.Cheatham TE, III, Miller JL, Fox T, Darden TA, Kollman PA. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. Journal of the American Chemical Society. 1995;117:4193–4194. [Google Scholar]
- 61.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lavery R, Moakher M, Maddocks JH, Petkeviciute D, Zakrzewska K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]