

Abstract
The first gold-catalyzed addition of N-arylhydroxylamines to aliphatic terminal alkynes is developed to access O-alkenyl-N-arylhydroxylamines, which undergo facile in situ sequential 3,3-rearrangements and cyclodehydrations to afford 2-alkylindoles with regiospecificity and under exceptionally mild reaction conditions.
The Fischer indole synthesis1 is perhaps the most versatile method for the construction of indole rings2 and has been applied extensively in the synthesis of various indole alkaloids3 since the original report by Fischer4 more than one hundred years ago. This annulation between an arylhydrazine and a ketone relies on a key 3,3-sigmatropic rearrangement of an N-alkenyl-N’-arylhydrazine intermediate. While it has been subjected to various modifications/improvements,5 there are still notable deficiencies including the often difficulty in achieving excellent regioselectivities with non-symmetric ketones and demanding reaction conditions such as strong acidities and/or elevated reaction temperatures.
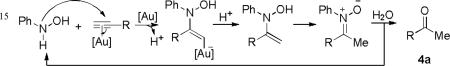
Similarly, 3, 3-rearrangements of O-vinyl-N-arylhydroxylamines or their derivatives6 can lead to indole synthesis upon subsequent cyclodehydration. A notable example is the Bartoli indole synthesis.7 While this rearrangement typically proceeds at much lower temperatures than in the case of the Fischer indole synthesis, there is a lack of general and straigthforward methods6 for the generation of O-alkenyl-N-arylhydroxyamines. We envisioned that these intermediates could be formed via metal-promoted additions of the HO groups of N-arylhydroxylamines onto C-C triple bonds (Scheme 1). By using alkynes as substrates, this indole synthesis may provide solutions to some of the deficiencies in the Fischer indole synthesis. Surprisingly, this strategy has not been realized although related ones using propiolates and hydroxamic acids in the presence of bases have been reported.8
Scheme 1.

Formation of O-alkenyl-N-phenylhydroxylamines via HO addition to alkynes en route to indoles
Efficient additions of various nucleophiles (NuH) to C-C triple bonds have been realized in gold catalysis;9 however, hydroxyamine have not been used as nucleophiles.10 In continuation of our research on gold catalysis, we anticipated that gold complexes could be employed in Scheme 1 to promote the formation of O-alkenyl-N-arylhydroxylamine 1 via activation of alkynes toward nucleophilic attack. Herein, we disclose a regiospecific synthesis of 2-alkylindoles via the annulation of N-arylhydroxylamines and terminal alkynes under mild reaction conditions.
We started by using N-phenylhydroxylamine and 1-dodecyne (2 equiv) as substrates and Ph3PAuNTf2 as the catalyst. To our delight, 2-n-decylindole 3a11 was indeed formed after reacting at room temperature for 18 h (Table 1, entry 1), and methyl ketone 4a was the major side product. Due to the disproportionation of the hydroxylamine substrate, some aniline and trace amount of diazene N-oxide 5a were observed. We attributed the formation of ketone 4a partly to the competitive N-H addition of the hydroxylamine to 2a followed by subsequent hydrolysis (Eq. 1) and partly to direct gold-catalyzed hydration. H2O consumed in these processes should mostly come from the cyclodehydrative indole formation. Screening different gold catalysts (entries 2-7) revealed that phosphite-based cationic gold(I) complex, (ArO)3PAuNTf2 (Ar = 2,4-di-tert-butylphenyl), gave the best result (entry 4). Somewhat to our surprise, DCE was a better solvent than DCM, and the yield was up to 94% (entry 8). Additional solvents (entries 10-14) were examined, and both diethyl ether (entry 10) and toluene (entry 13) were excellent choices as well. The reaction yield decreases as the amount of the alkyne decreases (entries 15 and 16). With 1.8 equivalents of 1-dodecyne (entry 15), the reaction yield was still very good, and the isolated yield was 84%.
 |
(1) |
Table 1.
Screening gold catalysts and reaction conditions.a
| ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | solvent | yieldb | |||
| 3a | 4a | 5a | 6a | |||
| 1 | Ph3PAuNTf2 | DCM | 57% | 62% | trace | 9% |
| 2 | (4-CF3Ph)3PAuNTf2 | DCM | 74% | 72% | trace | 6% |
| 3 | IPrAuNTf2 | DCM | 63% | 55% | trace | 8% |
| 4 | (ArO)3PAuNTf2c | DCM | 83% | 74% | 1% | 3% |
| 5 | Et3PAuNTf2 | DCM | 46% | 61% | trace | 11% |
| 6 | BrettphosAuNTf2 | DCM | 21% | 67% | - | 10% |
| 7 | (F5C6)3PAuNTf2 | DCM | 4% | 8% | 9% | 7% |
| 8 | (ArO)3PAuNTf2c | DCE | 94% | 71% | 2% | 4% |
| 9 | (ArO)3PAuOTfc | DCE | 55% | 64% | 3% | 4% |
| 10 | (ArO)3PAuNTf2c | Et2O | 94% | 80% | 3% | 1% |
| 11 | (ArO)3PAuNTf2c | THF | 41% | 80% | 6% | 2% |
| 12 | (ArO)3PAuNTf2c | CH3CN | 38% | 24% | 4% | 5% |
| 13 | (ArO)3PAuNTf2c | toluene | 93% | 65% | 1% | 2% |
| 14 | (ArO)3PAuNTf2c | hexanes | 28% | 66% | 1% | 4% |
| 15d | (ArO)3PAuNTf2c | DCE | 86%e | 75% | 1% | 2% |
| 16f | (ArO)3PAuNTf2c | DCE | 76% | 63% | 2% | 4% |
Anhydrous solvents were used; [N-phenylhydroxylamine] = 0.1 M; under nitrogen.
Estimated by 1H NMR using diethyl phthalate as an internal reference; the yield for methyl ketone 4a is calculated based on N-phenylhydroxylamine for comparison purpose although it was formally formed via hydration of 2a.
Ar = 2,4-di-tert-butylphenyl.
1.8 Equiv of 1-dodecyne.
84% isolated yield.
1.6 Equiv of 1-dodecyne.
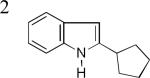
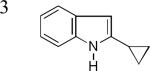
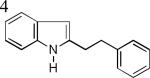
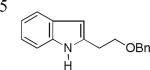
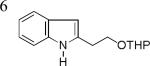
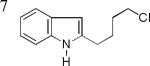
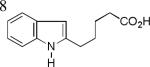
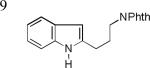
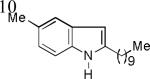
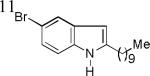
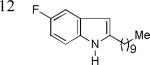
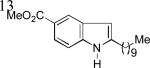
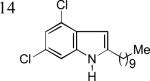
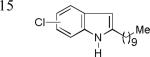
As shown in Table 2, this indole synthesis worked well with various cycloalkylacetylenes (entries 1-3) including cyclopropylacetylene (entry 3). Linear aliphatic terminal alkynes containing various functional groups also reacted well. These functional groups include phenyl (entry 4), protected HO groups (entries 5 and 6), chloro (entry 7), carboxylic acid (entry 8) and a protected amino group (entry 9). Of particular note is the tolerance of the acid labile THP group, confirming the exceptionally mild nature of this indole synthesis; in contrast, the Fischer indole synthesis is typically performed under highly acidic environment. These mild reaction conditions also permit the preparation of chloroindole 3h without affecting with the chloro moiety. A methyl (entry 10) or electron-withdrawing substituents (entries 11-15) at the benzene ring para or meta to the hydroxylamine moiety permitted good to efficient reactions; however, an ortho-Me led to little indole product (<10%), and N-(4-methoxyphenyl)hydroxylamine12 was not stable to try this chemistry. Attempts to extend this chemistry to aliphatic internal alkynes and arylacetylenes, however, yielded little products.
Table 2.
Reaction scope studiesa
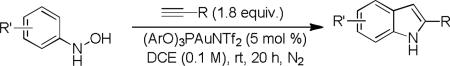
| ||
|
|
|
| 3b, 77% | 3c, 62%b | 3d, 70% |
|
|
|
| 3e, 89% | 3f, 84% | 3g, 67% |
|
|
|
| 3h, 74% | 3i, 73% | 3j, 72% |
|
|
|
| 3k, 64% | 31, 86% | 3m, 82% |
|
|
|
| 3n, 76% | 3o, 63% | 3p, 85%c |
[hydroxylamine] = 0.1 M; Ar = 2,4-di-tert-butyl
Toluene as solvent.
4-Cl/6-Cl = 1/1.3.
An important feature of all the above cases is the exclusive selectivity toward 2-alkylindoles, and no 3-alkylindoles were observed. This regioselectivity stems from the Markovnikov additions of hydroxamic acids to the gold-activated terminal alkynes. In contrast, with corresponding methyl ketones as substrates in the Fischer indole synthesis, strongly acidic mediums (e.g., 5 % P2O5/neat MsOH13 or neat PPA14) are necessary to favor 2-alkylindoles with low to moderate regioselectivities.
Conclusions
We have developed a novel access to O-alkenyl-N-arylhydroxylamines via the first gold-catalyzed addition of N-arylhydroxylamines to aliphatic terminal alkynes. This mild gold catalysis is coupled in situ with sequential facile 3,3-rearrangement and cyclodehydration, affording 2-alkylindoles regiospecifically in typically good yields and under exceptionally mild reaction conditions.
Supplementary Material
Acknowledgments
We gratefully thank NIH (R01 GM084254) and UCSB for generous financial support. LZ is a Alfred P. Sloan fellow.
Footnotes
† Electronic Supplementary Information (ESI) available: experimental procedure, 1H and 13C NMR spectra, and the X-ray structure of compound 6a. See DOI: 10.1039/b000000x/
References
- 1.a Robinson B. The Fischer indole synthesis. Wiley; Chichester; New York: 1982. [Google Scholar]; b Downing RS, Kunkeler PJ. In: Fine chemicals through heterogenous catalysis. Sheldon RA, Bekkum H, editors. Wiley-VCH; Weinheim; New York: 2001. pp. 178–183. [Google Scholar]; c Hughes DL. Org. Prep. Proced. Int. 1993;25:607–632. [Google Scholar]
- 2.Humphrey GR, Kuethe JT. Chem. Rev. 2006;106:2875–2911. doi: 10.1021/cr0505270. [DOI] [PubMed] [Google Scholar]
- 3.a Bonjoch J, Catena J, Valls N. J. Org. Chem. 1996;61:7106–7115. doi: 10.1021/jo960848z. [DOI] [PubMed] [Google Scholar]; b Iyengar R, Schildknegt K, Aube J. Org. Lett. 2000;2:1625–1627. doi: 10.1021/ol005913c. [DOI] [PubMed] [Google Scholar]; c Roberson CW, Woerpel KA. J. Am. Chem. Soc. 2002;124:11342–11348. doi: 10.1021/ja012152f. [DOI] [PubMed] [Google Scholar]; d Gan T, Liu R, Yu P, Zhao S, Cook JM. J. Org. Chem. 1997;62:9298–9304. doi: 10.1021/jo971067g. [DOI] [PubMed] [Google Scholar]; e Ueda H, Satoh H, Matsumoto K, Sugimoto K, Fukuyama T, Tokuyama H. Angew. Chem., Int. Ed. 2009;48:7600–7603. doi: 10.1002/anie.200902192. [DOI] [PubMed] [Google Scholar]
- 4.a Fischer E, Jourdan F. Ber. 1883;16:2241–2245. [Google Scholar]; b Fischer E, Hess O. Ber. 1884;17:559–568. [Google Scholar]
- 5.a Lipin'ska T. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2001;37:231–236. [Google Scholar]; b Chen C-Y, Senanayake CH, Bill TJ, Larsen RD, Verhoeven TR, Reider PJ. J. Org. Chem. 1994;59:3738–3741. [Google Scholar]; c Katritzky AR, Rachwal S, Bayyuk S. Org. Prep. Proced. Int. 1991;23:357–363. [Google Scholar]; d Inman M, Moody CJ. Chem. Commun. 2011;47:788–790. doi: 10.1039/c0cc04306k. [DOI] [PubMed] [Google Scholar]; e Narayana B, Ashalatha BV, Vijaya RKK, Fernandes J, Sarojini BK. Bioorg. Med. Chem. 2005;13:4638–4644. doi: 10.1016/j.bmc.2005.04.068. [DOI] [PubMed] [Google Scholar]
- 6.Joule JA. Sci. Synth. 2000;10:380–383. [Google Scholar]
- 7.For a review, see: Dalpozzo R, Bartoli G. Curr. Org. Chem. 2005;9:163–178.
- 8.a Pereira MMA, Prabhakar S, Lobo AM. J. Nat. Prod. 1996;59:744–747. doi: 10.1021/np9601869. [DOI] [PubMed] [Google Scholar]; b Hwu JR, Patel HV, Lin RJ, Gray MO. J. Org. Chem. 1994;59:1577–1582. [Google Scholar]; c Toyota M, Fukumoto K. J. Chem. Soc., Perkin Trans. 1. 1992:547–552. [Google Scholar]; d Toyota M, Fukumoto K. Heterocycles. 1990;31:1431–1433. [Google Scholar]
- 9.For reviews, see: Corma A, Leyva-Pérez A, Sabater MJ. Chem. Rev. 2011;111:1657–1712. doi: 10.1021/cr100414u.Rudolph M, Hashmi ASK. Chem. Commun. 2011;47:6536–6544. doi: 10.1039/c1cc10780a.Wang S, Zhang G, Zhang L. Synlett. 2010;2010:692–706.Abu Sohel SM, Liu R-S. Chem. Soc. Rev. 2009;38:2269–2281. doi: 10.1039/b807499m.Arcadi A. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d.Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x.Fürstner A, Davis PW. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335.Zhang L, Sun J, Kozmin SA. Adv. Synth. Catal. 2006;348:2271–2296.
- 10.For an intramolecular case using hydroxamic acids, see: Yeom H-S, So E, Shin S. Chem. Eur. J. 2011:1764–1767. doi: 10.1002/chem.201002863.
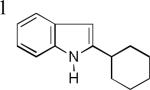
- 11.The regiochemistry is supported by the chemical shift at 6.24 ppm at its 1H NMR spectrum, and the observed nOe between the 3- and 4-protons in the NOESY1D spectrum of product 3n.
- 12.Ayyangar NR, Brahme KC, Kalkote UR, Srinivasan KV. Synthesis. 1984;1984:938, 941. [Google Scholar]
- 13.Hughes DL, Zhao D. J. Org. Chem. 1993;58:228–233. [Google Scholar]
- 14.Bosch J, Bonjoch J, Diez A, Linares A, Moral M, Rubiralta M. Tetrahedron. 1985;41:1753–1762. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.