

Abstract
Patients with psoriasis have systemic and vascular inflammation and are at increased risk for myocardial infarction, stroke, and cardiovascular death. However, the underlying mechanism(s) mediating the link between psoriasis and vascular disease is incompletely defined. This study sought to determine whether chronic skin-specific inflammation has the capacity to promote vascular inflammation and thrombosis. Using the KC-Tie2 doxycycline-repressible (Dox-off) murine model of psoriasiform skin disease, spontaneous aortic root inflammation was observed in 33% of KC-Tie2 compared to 0% of control mice by 12 months of age (P=0.04) and was characterized by the accumulation of macrophages, T-lymphocytes and B-lymphocytes and reduced collagen content and increased elastin breaks. Importantly, aortic inflammation was preceded by increases in serum TNF-α, IL-17A, VEGF, IL-12, MCP-1 and S100A8/A9 as well as splenic and circulating CD11b+Ly-6Chi pro-inflammatory monocytes. Doxycycline treatment of old mice with severe skin disease eliminated skin inflammation and aortic root lesion presence in 1 year old KC-Tie2 animals. Given the bi-directional link between inflammation and thrombosis, arterial thrombosis was assessed in KC-Tie2 and control mice; mean time to occlusive thrombus formation was shortened by 64% (P=0.002) in KC-Tie2 animals; doxycycline treatment returned thrombosis clotting times to control mouse levels (P=0.69). These findings demonstrate that sustained skin-specific inflammation promotes aortic root inflammation and thrombosis and suggest that aggressive treatment of skin inflammation may attenuate pro-inflammatory and prothrombotic pathways that produce cardiovascular disease in psoriasis patients.
Keywords: Psoriasis, Atherosclerosis, Inflammation, Monocytes, Thrombosis
INTRODUCTION
Psoriasis is a chronic inflammatory skin disease affecting between 2.5–6 million patients in the United States. Clinical data have convincingly demonstrated that psoriasis patients have an increased risk for developing cardiovascular disease (CVD), including myocardial infarction and stroke (Ahlehoff et al., 2011a; Gelfand et al., 2009; Gelfand et al., 2006; Mehta et al., 2010; Mehta et al., 2011a; Prodanovich et al., 2009) and an increased risk of thromboembolic events (Ahlehoff et al., 2011b). Psoriasis patients have more established CVD risk factors, including higher levels of serum cholesterol and triglycerides coupled with low HDL (Pietrzak et al., 2009; Toker et al., 2009), elevated pro-inflammatory mediators, (i.e. IL-17, IL-23 (Kryczek et al., 2008) and S100A8/A9 (Benoit et al., 2006)), and decreases in anti-inflammatory mediators such as IL-10 and adiponectin (Kaur et al., 2008; Takahashi et al., 2008) and these correlate with disease severity (Boehncke et al., 2011c). Psoriasis is accompanied by impaired endothelial function (Balci et al., 2009) and increased subclinical atherosclerosis as measured by carotid intimal-medial thickness measurements (Balci et al., 2009), arterial stiffness (Gisondi et al., 2009), and coronary calcium scores (Ludwig et al., 2007). More recent work suggests that psoriasis confers an additional 6.2% absolute risk of a 10-year rate of major adverse cardiac events compared with the general population (Mehta et al., 2011a). However, these epidemiological studies do not provide insight as to the etiology of this elevated risk and rely on adjusting for confounders such as hyperlipidemia, hypertension, and diabetes and thus cannot demonstrate causality.
Common inflammatory cascades play critical roles in the initiation and maintenance of psoriasis and CVD (Alexandroff et al., 2009; Libby, 2002; Lowes et al., 2007; Spah, 2008), including activation of antigen presenting cells and macrophages, involvement of Th1, Th17 and regulatory T cells, and a critical role for IL-12p40 and TNF-α. Psoriasis patients have significant inflammation not only in the skin, but also subclinical inflammation in the liver, joints, tendons and vascular tree even after adjusting for traditional cardiovascular risk factors (Mehta et al., 2011b), suggesting that psoriasis itself predisposes to pro-inflammation pathways independent of traditional risk factors. Others have suggested a more direct hypothesis that psoriasis-initiated skin and systemic inflammation cause insulin resistance, which promotes endothelial cell dysfunction, subsequent atherosclerosis and ultimately myocardial infarction or stroke (Boehncke et al., 2011c).
Despite the epidemiological association between psoriasis and CVD, the underlying mechanism(s) mediating the link between psoriasis and vascular disease is incompletely defined. Whether localized, chronic cutaneous inflammation directly promotes vascular inflammation and thrombosis is unknown. To address this question, we used the KC-Tie2 murine model of psoriasiform skin inflammation, in which Tie2 transgene expression is confined to keratinocytes and which recapitulates many characteristics of human plaque psoriasis (Johnston et al., 2011; Ostrowski et al., 2011; Swindell et al., 2011; Ward et al., 2011; Wolfram et al., 2009). We hypothesized that the presence of sustained skin-confined inflammation would predispose animals to vascular inflammation and thrombosis.
RESULTS
Lesion characterization
At 12 months of age on a standard chow diet, spontaneous development of robust aortic root inflammation was observed in 33% of KC-Tie2 mice with psoriasiform skin disease compared to 0% of control mice (P=0.04) (Figure 1a–d). Lesion formation, quantified by measuring vessel wall area of the aortic root at the level of the aortic valve was higher in the KC-Tie2 mice compared to control mice (325 ± 48 μm2 vs. 221 ± 12 μm2, n=18–19 per group; P=0.04) (Figure 1e). No lesions were detected in the descending thoracic or abdominal aorta. Aortic root lesions were also stained with oil-red O and Sudan IV for lipid deposition. There was no significant lipid staining in the inflamed aortic roots of KC-Tie2 mice on a standard chow diet.
Figure 1.
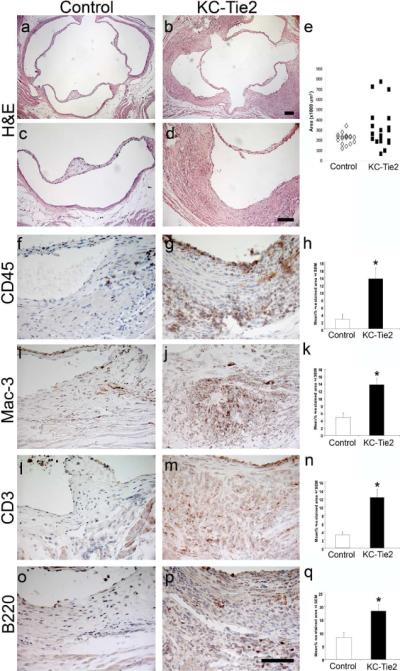
Aortic roots develop spontaneous vascular inflammation by 1 year of age in KC-Tie2 and not control animals. Aortic roots from control (a, c) and KC-Tie2 (b, d) animals stained with H&E. Quantitation of aortic root vessel wall area (e) demonstrates significant increases in area of KC-Tie2 mice (n=19) compared to control littermates (n=18). Immunohistological staining and quantitative analyses of positively stained CD45+ (f–h), Mac-3+ (i–k), CD3+ (l–n) and B220+ cells (o–q) (% positively stained area) within the aortic roots of representative control (f, l, i, o; n = 5) and KC-Tie2 mice with vascular inflammation (g, j, m, p; n=5) reveals significant increases in inflammatory cell infiltrates. * P <0.05 vs control animals. Scale bar = 100 μm.
Examination of the aortic root revealed significant accumulation of inflammatory cells (CD45-positive leukocytes; Figure 1f–h) in KC-Tie2 mice (13.5 ± 2.9% CD45-poitive area) compared to control animals (2.5 ± 1.4%, P<0.001) and affected all layers of the vessel wall. Leukocyte subset analysis was performed by staining for macrophages (Figure 1i–k), T-cells (Figure 1l–n), and B-cells (Figure 1o–q). Macrophage accumulation was increased 2.8-fold in KC-Tie2 mice (13.8 ± 1.7% Mac-3-positive area) compared to controls (4.9 ± 1.2%, P<0.0001). Both T-cell and B-cell accumulation in the aortic root were enhanced in KC-Tie2 mice with a 3.9-fold increase in T-cells (12.2 ± 2.0% vs. 3.2 ± 0.7% CD3-positive area, P=0.0004) and a 2.2-fold increase in B-cells (18.4 ± 2.4% vs 8.3 ± 1.9% B220-positive area, P=0.0003).
Examination of the extracellular matrix (ECM) using Trichrome staining revealed decreased collagen content in the lesions in KC-Tie2 animals compared to the dense collagen content observed in control mice (Figure 2a–d). Aortic elastin integrity was investigated by Verhoeff elastin staining and revealed abundant elastin breaks and fragmentation in the aortic root lesions of KC-Tie2 mice (Figure 2e–h).
Figure 2.
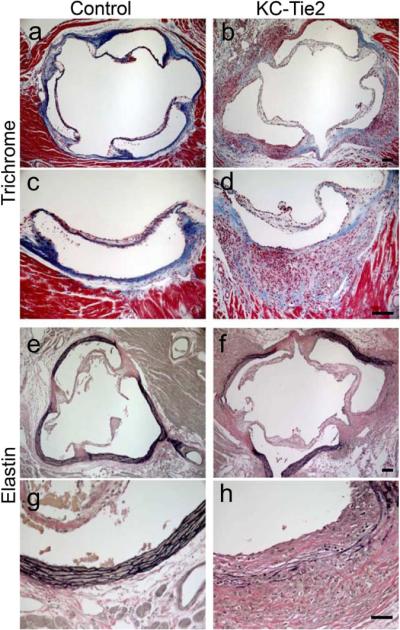
Anatomical characterization of aortic arch lesions. Aortic roots from control (a, c, e, g) and KC-Tie2 (b, d, f, h) animals stained with Trichrome (a–d) or Verhoeff–van Gieson elastin (e–h) reveals decreased expression of collagen in Trichrome stained aortic arch tissue from KC-Tie2 mice and increases in the numbers of elastin breaks in Verhoeff–van Gieson elastin stained aortic root tissues. Scale bar = 100 μm.
To verify that transgene expression was limited to the skin, we performed additional matings between the K5tTA driver mouse and nuclear LacZ reporter mouse lines and analyzed LacZ expression in the skin, spleen, brain, liver, kidney, lung, heart, and aorta of the of KC-LacZ mice (Supplemental Figure 1). Robust LacZ expression was observed in the skin with little to no expression in the other tissues including the aorta. In addition, we verified the absence of aberrant Tie2 expression in the aortic roots of KC-Tie2 mice that developed vascular inflammation (Supplemental Figure 1). Taken together, these observations indicate that chronic, skin-specific inflammation in KC-Tie2 mice promotes the de novo development of aortic root inflammation and alterations in the ECM.
Systemic inflammation
Biomarkers associated with CVD risk were examined in KC-Tie2 mice. Serum levels of MCP-1 (1.7-fold; P=0.038), IL-12p70 (4.3-fold; P=0.048), TNF-α (2.1-fold; P=0.05), IL17a (4.4-fold; P=0.038) and VEGF (1.6-fold; P=0.02) were all elevated in KC-Tie2 mice compared to age-matched littermate controls (Figure 3a). The expression of the pro-inflammatory mediator S100A8/A9 (myeloid related protein-8/14, MRP-8/14) was also increased in the skin and serum of KC-Tie2 compared to control mice (Figure 3b).
Figure 3.
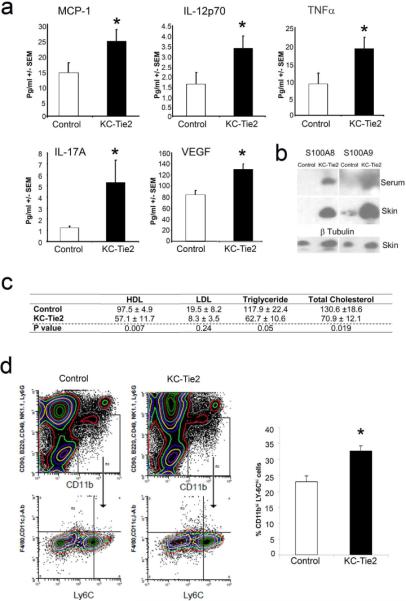
Aortic root vascular inflammation develops in the presence of elevated systemic inflammation and monocytosis and independent of lower lipid levels. Proinflammatory cytokines are significantly elevated in KC-Tie2 mouse sera, including levels of MCP-1, IL-12p70, TNFα, IL-17A and VEGF (a; mean ± SEM; n=5–11 per group) and S100A8/A9 expression is increased in skin and serum of KC-Tie2 animals (b). KC-Tie2 animals have significantly less total cholesterol, triglycerides and HDL compared to control mice (c; mean ± SEM; n=8 per group; p values indicated for each lipid in table). 4-colour flow cytometry reveals significant increases in splenic proatherogenic monocytes (CD90loB220loCD49bloNK1.1loLy6GloCD11cloIAbloF4/80loCD11bhiLy-6Chi). Representative flow cytometry and quantification of CD11b+F4/80loLy-6Chi cells (d; mean ± SEM; n =4 spleens per group).* P <0.05 vs. control animals.
Total cholesterol, triglyceride, and LDL cholesterol levels were lower in KC-Tie2 compared to control mice, although not all reached statistical significance. Reduced HDL levels were observed in KC-Tie2 mice (Figure 3c).
Specific monocyte sub-populations (i.e., Ly-6Chi) have been implicated in monocyte/macrophage vessel wall infiltration and atherosclerotic lesion formation. Therefore, we evaluated splenic and circulating CD11b+Ly-6Chi monocytes in KC-Tie2 mice at 6 months of age prior to aortic root inflammation. Splenic CD11b+Ly-6Chi monocytes levels were increased by 42% (P=0.009) in KC-Tie2 compared to control mice (Figure 3d). Similar results were observed in circulating blood (Supplemental Figure 2).
Inflammation and thrombosis
Given the bi-directional link between inflammation and thrombosis, we assessed arterial thrombosis in KC-Tie2 compared to control mice using a photochemical (Rose Bengal-green light laser) carotid artery injury model, which produces thrombosis due to local free radical release and oxidative endothelial cell injury (Falati et al., 2004; Furie and Furie, 2005; Watson et al., 1985). The time to occlusive thrombus formation was shortened significantly in KC-Tie2 compared to control mice (n=9–10; 19 ± 3 vs. 53 ± 8 min, P=0.002) (Figure 4).
Figure 4.
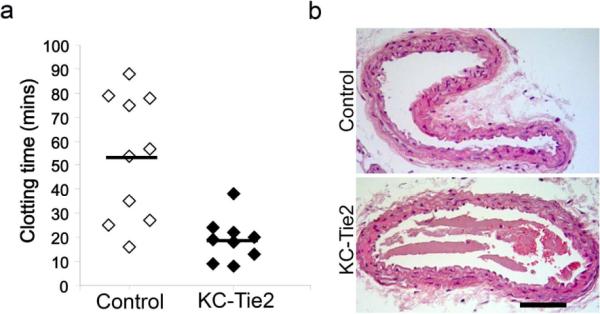
KC-Tie2 mice are pro-thrombotic. KC-Tie2 and control mice underwent experimental thrombosis; carotid artery blood flow was monitored continuously with a vascular flow probe, time to occlusion, defined as cessation of blood flow for 20 min, was recorded (a). Mean time (± SEM) to occlusive thrombus formation in control mice was 53 ± 8 minutes (n=10), and was shortened significantly in KC-Tie2 mice (19 ± 3 minutes; n=9; P=0.002). H&E stained carotid arteries from control and KC-Tie2 mice following Rose Bengal laser treatment. Note the fibrin-platelet rich thrombi with some red blood cells in the lumen of carotid arteries of KC-Tie2 and not control mice (b). *P<0.05 vs control animals. Scale bar = 100 μm.
Skin disease remission
To examine whether aggressive targeting of skin inflammation would reverse the presence of aortic root vascular lesions, 7 month old mice with severe skin disease were fed doxycycline-supplemented food to repress gene expression and reverse skin disease (Silver et al., 2011; Wolfram et al., 2009). At 12 months of age both skin inflammation (data not shown) and aortic root lesions were eliminated in KC-Tie2 mice (0%; n=9; Figure 5a–f).
Figure 5.

Repressing skin inflammation improves aortic root lesions and thrombosis clotting times. Seven month old KC-Tie2 mice (n=9) and control littermates (n=9) were treated with doxycycline to repress transgene expression and reverse skin inflammation. Aortic roots from representative 1 year old control (a, c, e) and KC-Tie2 (b, d, f) animals stained with H&E (a–b), Trichrome (c–d) and elastin (e–f). No aortic root lesions were observed in any control or KC-Tie2 doxycycline treated mice. (g) KC-Tie2 and control mice treated with doxycycline for 6 weeks underwent experimental thrombosis; mean time (± SEM) to occlusive thrombus formation was not significantly different between control mice treated with doxycycline (31 ± 8 minutes; n=10) and KC-Tie2 mice treated with doxycycline (36 ± 9 minutes; n=10; P=0.69). Scale bar = 100 μm.
To assess the effect of skin disease remission on occlusive thrombosis formation, KC-Tie2 and control animals were fed doxycycline-supplemented food for 6 weeks and then underwent photochemical carotid artery injury to produce thrombosis. Doxycycline treatment returned KC-Tie2 thrombosis clotting times to control mouse levels (n=10; 31 ± 8 controls + doxycycline vs 36 ± 9 KC-Tie2 + doxycycline; P=0.69; Figure 5g). Control animals treated with doxycycline had slightly shorter clotting times than untreated control mice, however this difference was not significant (P=0.07).
DISCUSSION
In this study, we have identified that sustained skin inflammation itself is sufficient to promote vascular inflammation and thrombosis. This conclusion is supported by the following data: 1) KC-Tie2 mice develop aortic root inflammatory lesions 2) Collagen content is decreased and elastin fragmentation is increased in the aortic root of KC-Tie2 mice; 3) Pro-inflammatory cytokines and chemokines are increased in the skin and peripheral blood of KC-Tie2 mice that precede the development of aortic root inflammatory lesions; 4) Inflammatory CD11b+ Ly-6Chi monocytes are elevated in KC-Tie2 animals prior to lesion development; 5) Thrombotic occlusion time after photochemical carotid injury is shortened in KC-Tie2 mice; and 6) Aortic root inflammatory lesions and thrombosis clotting times significantly improve in KC-Tie2 mice following skin disease repression.
Psoriasis patients have an increased risk for developing CVD, including myocardial infarction and stroke. However the high prevalence of standard CVD risk factors in patients with psoriasis required statistical adjustments for the confounding effects of these variables in psoriasis epidemiological studies (Gelfand et al., 2009; Gelfand et al., 2006; Mehta et al., 2010; Mehta et al., 2011a). Thus, the question of whether inflammatory hyperplastic disease confined to the skin has the capacity to directly cause vascular inflammation and thrombosis was, heretofore, unknown. It was highly beneficial to employ a murine model of psoriasis that recapitulates many aspects of the human disease (Johnston et al., 2011; Ostrowski et al., 2011; Swindell et al., 2011; Ward et al., 2011; Wolfram et al., 2009), including the characteristic histologic, immunologic, and pro-inflammatory cytokine profiles as well as disease attenuation/clearance in response to clinically efficacious therapeutics, but without standard CVD risk factors (i.e., hyperlipidemia, hypertension, diabetes, obesity) to elucidate the effect of the disease itself. Using this model, our findings provide direct evidence linking skin inflammation with the development of vascular inflammation as well as an increased propensity for arterial thrombosis after endothelial redox-mediated injury.
Chronic inflammation and pro-inflammatory cytokines play significant roles in the pathogenesis of both psoriasis and vascular disease. Evidence suggests that psoriasis and CVD share common pathogenic features (Alexandroff et al., 2009; Spah, 2008), including immunological processes, inflammatory cytokine profiles, and the presence of local and systemic inflammatory markers (Alexandroff et al., 2009; Boehncke et al., 2011c; Federman et al., 2009; Gisondi and Girolomoni, 2009; Hansson, 2005; Libby, 2002; Lowes et al., 2007; Ross, 1999; Spah, 2008). Activation of these inflammatory cells (dendritic cells, macrophages and T cells) together with the release of pro-inflammatory cytokines (e.g., TNF-α, IFN-γ, IL-12) contribute to the development of psoriatic lesions (Lowes et al., 2007) and play a major role in the development and vulnerability of atherosclerotic plaque (Hansson, 2005; Libby, 2002; Libby and Simon, 2001; Ross, 1999). The idea that similar mechanisms underlie the development of both psoriasis and CVD is supported by findings of others (Shepherd et al., 2004) such that sustained IL-1 signaling, accomplished via genomic deletion of the IL-1RA, resulted in three types of strain-dependent inflammatory changes, including a psoriasiform dermatitis in ear skin, arthritis-like inflammation in the joints, and large vessel arterial inflammation. However, this experimental approach resulted in increased IL-1 signaling throughout the body. The KC-Tie2 mouse has confined gene expression of the membrane-bound Tie2 receptor to keratinocytes, thus our findings provide experimental evidence that keratinocyte signaling and cell-cell interactions within the cutaneous environment can initiate inflammation capable of causing vascular inflammatory foci formation in distant vessels, perhaps as a result of changes in both circulating proinflammatory cytokines and leukocytes (CD11b+ Ly-6Chi monocytes). KC-Tie2 mice have increases in pro-inflammatory cytokines and chemokines and increases in splenic and circulating pro-inflammatory CD11b+Ly-6Chi monocytes that precede and are associated with the development of vascular inflammation and disease and occur independent of changes in lipids. The clinical relevance of our observations in KC-Tie2 mice is supported by highly sensitive, metabolic imaging (i.e., [18F]-fluorodeoxyglucose positron emission tomography-computed tomography, FDG-PET/CT) in psoriasis patients showing concomitant aortic inflammation. Coupled with our thrombosis observations, this suggests that elevated levels of inflammation could potentially be used to predict poor outcomes and adverse cardiovascular events in patients with psoriasis. Taken together, these findings provide insight into skin-specific contributions underlying prior epidemiological reports that psoriasis patients are at increased risk for MI, stroke, venous thromboembolism and cardiovascular mortality (Ahlehoff et al., 2011a; Ahlehoff et al., 2011b; Gelfand et al., 2009; Gelfand et al., 2006; Mehta et al., 2010; Mehta et al., 2011a; Prodanovich et al., 2009).
There is increasing evidence for an important link between inflammation and thrombosis. Leukocyte-platelet interactions induce bi-directional signals that amplify pro-inflammatory and pro-thrombotic cellular responses (Libby and Simon, 2001). Activated platelets and platelet-released mediators (e.g., PDGF and PAF) activate leukocytes, thereby enhancing their responses such as chemotaxis, ROS generation, phagocytosis, and pro-coagulant activity (Bazzoni et al., 1991; Kuijper et al., 1997; Lindemann et al., 2001; Nagata et al., 1993). Conversely, activated leukocytes induce platelet activation as evidenced by increased platelet P-selectin and activated glycoprotein IIb/IIIa expression (Li et al., 2000). Psoriasis patients have elevated P-selectin and levels correlate with disease severity (Garbaraviciene et al., 2010). The most compelling observation of this study relates to the finding that occlusive thrombus formation was shortened significantly in KC-Tie2 compared to control mice and supports recent epidemiological reports demonstrating psoriasis patients had higher incident rates of venous thromboembolism (Ahlehoff et al., 2011b). Acute myocardial infarction typically results from atherosclerotic plaque disruption or superficial endothelial cell erosion and thrombosis that cause coronary arterial occlusion (Davies and Thomas, 1985; Farb et al., 1996). Yet, molecular events that precede acute myocardial infarction in patients with psoriasis remain uncertain. This mouse model provides valuable clues addressing this gap area in at-risk psoriasis patients. For example, both S100A8/A9 (Figure 3B) and MPO (data not shown), which are elevated in the skin and peripheral blood of KC-Tie2 mice and psoriasis patients (Benoit et al., 2006), have been implicated in acute coronary syndromes, including unstable angina and myocardial infarction (Baldus et al., 2003; Healy et al., 2006; Morrow et al., 2008).
Interestingly, most of the literature examining psoriasis and CVD report increases in either cardiovascular risk factors (diabetes, hypertension, hyperlipidemia)(Gelfand et al., 2006; Mehta et al., 2010; Prodanovich et al., 2009), changes in CVD surrogate markers (MPO, adiponectin, S100A8/A9) (Benoit et al., 2006; Kaur et al., 2008; Takahashi et al., 2008); or indirect measures of cardiovascular disease via endothelial function, carotid artery intimal-medial thickness (IMT)(Balci et al., 2009) and stiffness measurements(Gisondi et al., 2009). KC-Tie2 animals have elevated systemic inflammation independent of obesity, hyperglycemia and hyperlipidemia, demonstrating that skin inflammation alone can elicit systemic levels of inflammation critical for inflammatory lesion formation in the aortic root and promotion of thrombosis. Our observation of localized inflammation in the aortic root is consistent with the anatomical localization of aortic inflammation previously observed in psoriasis patients (Mehta et al., 2011b); and this foci of inflammation appears to be specific rather than representative of an overall generalized vascular inflammatory effect affecting all vessels of the body. Moreover, the location of vascular inflammatory lesions also correlates highly with sites that develop atherosclerosis in humans. Whether atherosclerosis is in fact elicited by psoriasis, remains unclear. We can not rule out that with time, these mice would develop atherosclerosis; however on a non-atherosusceptible genetic background strain, and with the short lifespan of a mouse, these experiments are not likely to yield insightful data addressing this issue. Rather, the usefulness of the murine model lends itself to modeling experiments whereby variables known to be increased in psoriasis patient's (i.e. dietary choices, V-LDL levels, ApoB, diabetic state, hypertension) could be manipulated in KC-Tie2 animals and the synergistic or additive effects of vascular inflammation coupled with the experimental manipulation on atherosclerotic plaque development and thrombosis could be examined.
Our observations of aortic root vascular lesion resolution and improved thrombosis outcomes following skin-specific transgene repression provide further evidence demonstrating cutaneous inflammation promotes vascular inflammation and thrombosis and suggest that aggressive treatment of skin inflammation may attenuate pro-inflammatory and pro-thrombotic pathways that produce CVD in psoriasis patients. Although not skin-specific, recent prospective reports document significant improvement in endothelial vasodilator function (Boehncke et al., 2011a) and CVD biomarker levels (Boehncke et al., 2011b) in psoriasis patients following 24 weeks of systemic therapeutic treatment.
Evidence linking chronic inflammation to the development of vascular inflammation is not limited to psoriasis. For example, periodontal disease is an important independent causal risk factor for atherosclerotic disease, including coronary heart disease and ischemic stroke (DeStefano et al., 1993; Humphrey et al., 2008). Importantly, periodontal therapy has been shown to alter gene expression of peripheral blood monocytes, suggesting that local therapies could have a systemic anti-inflammatory effect (Papapanou et al., 2007). Similar to periodontal disease, rheumatoid arthritis is associated with increased carotid artery intimal:medial thickening as well as increased cardiovascular events independent of traditional cardiac risk factors. Prospective clinical trials are examining the effect of disease modifying therapies, such as anti-TNFα, on cardiovascular complications of rheumatoid arthritis.
The results of our study provide strong evidence of remote, extravascular tissue inflammation promoting induction of CVD and suggest that suppression of skin inflammation is a viable approach worthy of investigation to favorably impact CVD complications of psoriasis.
METHODS
Animals
The KC-specific (K5-tTA) driver line and the TetosTek/Tie2 responder lines have been described previously (Diamond et al., 2000; Jones et al., 2001). Matings were performed between the K5tTA line and the TetosTek/Tie2 line and offspring were genotyped by polymerase chain reaction (PCR) using DNA extracted from ear biopsies as previously described (Wolfram et al., 2009). We have previously demonstrated that animals inheriting a single copy of K5tTA and Tetos Tie2 (KC-Tie2 bi-transgenic mice) have ~50-fold increase in Tie2 mRNA and spontaneously develop a psoriasisform skin phenotype (Wolfram et al., 2009). Male and female KC-Tie2 animals were used in the current studies and littermates inheriting one or no transgenes served as experimental controls. Transgene repression was completed as previously described (Silver et al., 2011; Wolfram et al., 2009).
All animal protocols were approved by the Case Western Reserve University institutional animal care and use committee (IACUC) and conformed to the American Association for Accreditation of Laboratory Animal Care guidelines.
Protein analysis
See Online Supplement.
Flow cytometry
4-color flow cytometry was used to stain neutrophils, monocytes, and macrophages. Cells were incubated with a cocktail of mAbs against T cells (CD90-PE, 53–2.1), B cells (B220-PE, RA3-6B2), NK cells (CD49b-PE, DX5 and NK1.1-PE, PK136), neutrophils (Ly-6G–PE, 1A8), myeloid cells (CD11b-APC, M1/70) and monocyte subsets (Ly-6C–FITC, AL-21), as described previously (Shi et al., 2008; Swirski et al., 2007). F4/80 (BM8)-biotin-strep-PerCP, I-Ab (AF6-120.1)-biotin-strep-PerCP and CD11c (HL3)-biotin-strep-PerCP mAbs (BD Biosciences) also served to determine macrophage and dendritic cell differentiation (see also Online Supplement).
Histology and lesion analysis
Mice were perfused transcardially as previously described (Croce et al., 2009) prior to dissection of the heart and aorta. For analysis of the aortic root, the bottom half of the heart was cut off in a plane parallel to the left and right atria. The top half of heart was embedded in paraffin after dehydration in ethanol and xylene. Serial 5μm heart sections were obtained and stained with Hematoxylin and Eosin (Sigma). Trichrome staining (Sigma) was performed to visualize collagen. Elastic laminae were visualized by staining the sections with Verhoeff–van Gieson (Sigma) as recommended by the manufacturer. Immune cell staining was completed as described in the Online Supplement.
To quantitate the size of aortic root lesions, vessel wall area (including lesional area, if present) was measured using a computer-assisted image analysis program (Zeiss Axiovision software, Rel 4.5).
Photochemical carotid artery thrombosis
Ten week-old male and female KC-Tie2 transgenic and littermate controls mice were anesthetized by intraperitoneal injection with sodium pentobarbital (62.5 mg/kg) and placed in the supine position on a dissecting microscope. Animals at this time point have established skin disease with abundant inflammatory cell infiltrate (Ostrowski et al., 2011; Ward et al., 2011; Wolfram et al., 2009). Animals had a midline surgical incision made to expose the right common carotid artery and a Doppler flow probe (MC 0.5PSL Nanoprobe, Model 0.5 VB, Transonic Systems, Ithaca, NY) was placed under the exposed artery. The probe was connected to a flow meter (Transonic Systems Model TS420). Flow data was interpreted with a computerized data acquisition program (Windaq, DATAQ Instruments, Akron, OH). Rose Bengal at a concentration of 10 mg/mL in phosphate-buffered saline was then injected into the tail vein to administer a dose of 50 mg/kg. Following Rose Bengal injection, the mid portion of the common carotid artery was illuminated with a 1.5-mW green light laser source (540 nm; Melles Griot, Carlsbad, CA) 5 cm from the artery. Blood flow was monitored continuously from the onset of injury. The time to occlusion, determined only after the vessel remained closed with a cessation of blood flow for 20 min, was recorded.
Statistical analysis
All data are represented as mean ± standard error of the mean (SEM). Between group comparisons were analyzed using either a Student's T test or a Mann Whitney U test and statistical significance was defined as P<0.05.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by grants to N.L.W., T.S.M. and K.D.C. from the National Institutes of Health (P30AR39750 and P50AR05508) and the Murdough Family Center for Psoriasis; to N.L.W. and Y.W. from the National Psoriasis Foundation; to NLW from the American Heart Association and the Juvenile Diabetes Research Foundation and to D.I.S. from the National Institutes of Health (HL85816, HL57506 MERIT Award, and HL73852). The authors would like to thank Dr. Nehal N. Mehta for his helpful discussions and critical reading of the manuscript.
ABBREVIATIONS
- TNF-α
Tumour necrosis factor alpha
- IL
interleukin
- KC
keratinocyte
- CVD
cardiovascular disease
- HDL
high density lipoprotein
- MCP
monocyte chemoattractant protein
- VEGF
vascular endothelial growth factor
Footnotes
CONFLICT OF INTEREST The authors have no conflicts of interest to disclose.
REFERENCES
- Ahlehoff O, Gislason GH, Jorgensen CH, Lindhardsen J, Charlot M, Olesen JB, et al. Psoriasis and risk of atrial fibrillation and ischaemic stroke: a Danish Nationwide Cohort Study. Eur Heart J. 2011a doi: 10.1093/eurheartj/ehr285. [DOI] [PubMed] [Google Scholar]
- Ahlehoff O, Gislason GH, Lindhardsen J, Charlot MG, Jorgensen CH, Olesen JB, et al. Psoriasis carries an increased risk of venous thromboembolism: a Danish nationwide cohort study. PLoS One. 2011b;6:e18125. doi: 10.1371/journal.pone.0018125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandroff AB, Pauriah M, Camp RD, Lang CC, Struthers AD, Armstrong DJ. More than skin deep: atherosclerosis as a systemic manifestation of psoriasis. Br J Dermatol. 2009;161:1–7. doi: 10.1111/j.1365-2133.2009.09281.x. [DOI] [PubMed] [Google Scholar]
- Balci DD, Balci A, Karazincir S, Ucar E, Iyigun U, Yalcin F, et al. Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J Eur Acad Dermatol Venereol. 2009;23:1–6. doi: 10.1111/j.1468-3083.2008.02936.x. [DOI] [PubMed] [Google Scholar]
- Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Dejana E, Del Maschio A. Platelet-neutrophil interactions. Possible relevance in the pathogenesis of thrombosis and inflammation. Haematologica. 1991;76:491–499. [PubMed] [Google Scholar]
- Benoit S, Toksoy A, Ahlmann M, Schmidt M, Sunderkotter C, Foell D, et al. Elevated serum levels of calcium-binding S100 proteins A8 and A9 reflect disease activity and abnormal differentiation of keratinocytes in psoriasis. Br J Dermatol. 2006;155:62–66. doi: 10.1111/j.1365-2133.2006.07198.x. [DOI] [PubMed] [Google Scholar]
- Boehncke S, Fichtlscherer S, Salgo R, Garbaraviciene J, Beschmann H, Diehl S, et al. Systemic therapy of plaque-type psoriasis ameliorates endothelial cell function: results of a prospective longitudinal pilot trial. Arch Dermatol Res. 2011a;303:381–388. doi: 10.1007/s00403-010-1108-6. [DOI] [PubMed] [Google Scholar]
- Boehncke S, Salgo R, Garbaraviciene J, Beschmann H, Hardt K, Diehl S, et al. Effective continuous systemic therapy of severe plaque-type psoriasis is accompanied by amelioration of biomarkers of cardiovascular risk: results of a prospective longitudinal observational study. J Eur Acad Dermatol Venereol. 2011b;25:1187–1193. doi: 10.1111/j.1468-3083.2010.03947.x. [DOI] [PubMed] [Google Scholar]
- Boehncke WH, Boehncke S, Tobin AM, Kirby B. The 'psoriatic march': a concept of how severe psoriasis may drive cardiovascular comorbidity. Exp Dermatol. 2011c;20:303–307. doi: 10.1111/j.1600-0625.2011.01261.x. [DOI] [PubMed] [Google Scholar]
- Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–436. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MJ, Thomas AC. Plaque fissuring--the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J. 1985;53:363–373. doi: 10.1136/hrt.53.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeStefano F, Anda RF, Kahn HS, Williamson DF, Russell CM. Dental disease and risk of coronary heart disease and mortality. British Medical Journal. 1993;306:688–691. doi: 10.1136/bmj.306.6879.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115:788–794. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- Falati S, Gross PL, Merrill-Skoloff G, Sim D, Flaumenhaft R, Celi A. In vivo models of platelet function and thrombosis: study of real-time thrombus formation. Methods Mol Biol. 2004;272:187–197. doi: 10.1385/1-59259-782-3:187. [DOI] [PubMed] [Google Scholar]
- Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, et al. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–1363. doi: 10.1161/01.cir.93.7.1354. [DOI] [PubMed] [Google Scholar]
- Federman DG, Shelling M, Prodanovich S, Gunderson CG, Kirsner RS. Psoriasis: an opportunity to identify cardiovascular risk. Br J Dermatol. 2009;160:1–7. doi: 10.1111/j.1365-2133.2008.08874.x. [DOI] [PubMed] [Google Scholar]
- Furie B, Furie BC. Thrombus formation in vivo. J Clin Invest. 2005;115:3355–3362. doi: 10.1172/JCI26987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbaraviciene J, Diehl S, Varwig D, Bylaite M, Ackermann H, Ludwig RJ, et al. Platelet P-selectin reflects a state of cutaneous inflammation: possible application to monitor treatment efficacy in psoriasis. Exp Dermatol. 2010;19:736–741. doi: 10.1111/j.1600-0625.2010.01095.x. [DOI] [PubMed] [Google Scholar]
- Gelfand JM, Dommasch ED, Shin DB, Azfar RS, Kurd SK, Wang X, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol. 2009;129:2411–2418. doi: 10.1038/jid.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand JM, Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB. Risk of myocardial infarction in patients with psoriasis. Jama. 2006;296:1735–1741. doi: 10.1001/jama.296.14.1735. [DOI] [PubMed] [Google Scholar]
- Gisondi P, Fantin F, Del Giglio M, Valbusa F, Marino F, Zamboni M, et al. Chronic plaque psoriasis is associated with increased arterial stiffness. Dermatology. 2009;218:110–113. doi: 10.1159/000182256. [DOI] [PubMed] [Google Scholar]
- Gisondi P, Girolomoni G. Psoriasis and atherothrombotic diseases: disease-specific and non-disease-specific risk factors. Semin Thromb Hemost. 2009;35:313–324. doi: 10.1055/s-0029-1222610. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, et al. Platelet Expression Profiling and Clinical Validation of Myeloid-Related Protein-14 as a Novel Determinant of Cardiovascular Events. Circulation. 2006;113:2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J Gen Intern Med. 2008;23:2079–2086. doi: 10.1007/s11606-008-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol. 2011;186:2613–2622. doi: 10.4049/jimmunol.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N, Voskas D, Master Z, Sarao R, Jones J, Dumont DJ. Rescue of the early vascular defects in Tek/Tie2 null mice reveals an essential survival function. EMBO Rep. 2001;2:438–445. doi: 10.1093/embo-reports/kve093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Zilmer K, Kairane C, Kals M, Zilmer M. Clear differences in adiponectin level and glutathione redox status revealed in obese and normal-weight patients with psoriasis. Br J Dermatol. 2008;159:1364–1367. doi: 10.1111/j.1365-2133.2008.08759.x. [DOI] [PubMed] [Google Scholar]
- Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181:4733–4741. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijper PH, Gallardo Torres HI, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet and fibrin deposition at the damaged vessel wall: cooperative substrates for neutrophil adhesion under flow conditions. Blood. 1997;89:166–175. [PubMed] [Google Scholar]
- Li N, Hu H, Lindqvist M, Wikstrom-Jonsson E, Goodall AH, Hjemdahl P. Platelet-leukocyte cross talk in whole blood. Arterioscler Thromb Vasc Biol. 2000;20:2702–2708. doi: 10.1161/01.atv.20.12.2702. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation. 2001;103:1718–1720. doi: 10.1161/01.cir.103.13.1718. [DOI] [PubMed] [Google Scholar]
- Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- Ludwig RJ, Herzog C, Rostock A, Ochsendorf FR, Zollner TM, Thaci D, et al. Psoriasis: a possible risk factor for development of coronary artery calcification. Br J Dermatol. 2007;156:271–276. doi: 10.1111/j.1365-2133.2006.07562.x. [DOI] [PubMed] [Google Scholar]
- Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. 2010;31:1000–1006. doi: 10.1093/eurheartj/ehp567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta NN, Yu Y, Pinnelas R, Krishnamoorthy P, Shin DB, Troxel AB, et al. Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med. 2011a;124:775, e771–776. doi: 10.1016/j.amjmed.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta NN, Yu Y, Saboury B, Foroughi N, Krishnamoorthy P, Raper A, et al. Systemic and Vascular Inflammation in Patients With Moderate to Severe Psoriasis as Measured by [18F]-Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography (FDG-PET/CT): A Pilot Study. Arch Dermatol:archdermatol.2011. 2011b:2119. doi: 10.1001/archdermatol.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DA, Wang Y, Croce K, Sakuma M, Sabatine MS, Gao H. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155:49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Tsuji T, Todoroki N, Katagiri Y, Tanoue K, Yamazaki H, et al. Activated platelets induce superoxide anion release by monocytes and neutrophils through P-selectin (CD62) J Immunol. 1993;151:3267–3273. [PubMed] [Google Scholar]
- Ostrowski SM, Belkadi A, Loyd CM, Diaconu D, Ward NL. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530–1538. doi: 10.1038/jid.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapanou PN, Sedaghatfar MH, Demmer RT, Wolf DL, Yang J, Roth GA, et al. Periodontal therapy alters gene expression of peripheral blood monocytes. Journal of Clinical Periodontology. 2007;34:736–747. doi: 10.1111/j.1600-051X.2007.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzak A, Kadzielewski J, Janowski K, Rolinski J, Krasowska D, Chodorowska G, et al. Lipoprotein (a) in patients with psoriasis: associations with lipid profiles and disease severity. Int J Dermatol. 2009;48:379–387. doi: 10.1111/j.1365-4632.2009.03994.x. [DOI] [PubMed] [Google Scholar]
- Prodanovich S, Kirsner RS, Kravetz JD, Ma F, Martinez L, Federman DG. Association of Psoriasis With Coronary Artery, Cerebrovascular, and Peripheral Vascular Diseases and Mortality. Arch Dermatol. 2009;145:700–703. doi: 10.1001/archdermatol.2009.94. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Shepherd J, Little MC, Nicklin MJ. Psoriasis-like cutaneous inflammation in mice lacking interleukin-1 receptor antagonist. J Invest Dermatol. 2004;122:665–669. doi: 10.1111/j.0022-202X.2004.22305.x. [DOI] [PubMed] [Google Scholar]
- Shi C, Sakuma M, Mooroka T, Liscoe A, Gao H, Croce KJ, et al. Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008;112:4699–4711. doi: 10.1182/blood-2008-01-137018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R, Helms A, Fu W, Wang H, Diaconu D, Loyd CM, et al. Using optical coherence tomography for the longitudinal noninvasive evaluation of epidermal thickness in a murine model of chronic skin inflammation. Skin Res Technology. 2011 doi: 10.1111/j.1600-0846.2011.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spah F. Inflammation in atherosclerosis and psoriasis: common pathogenic mechanisms and the potential for an integrated treatment approach. British Journal of Dermatology. 2008;159:10–17. doi: 10.1111/j.1365-2133.2008.08780.x. [DOI] [PubMed] [Google Scholar]
- Swindell WR, Johnston A, Carbajal S, Han G, Wohn C, Lu J, et al. Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS One. 2011;6:e18266. doi: 10.1371/journal.pone.0018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Tsuji H, Takahashi I, Hashimoto Y, Ishida-Yamamoto A, Iizuka H. Plasma adiponectin and leptin levels in Japanese patients with psoriasis. Br J Dermatol. 2008;159:1207–1208. doi: 10.1111/j.1365-2133.2008.08823.x. [DOI] [PubMed] [Google Scholar]
- Toker A, Kadi M, Yildirim AK, Aksoy H, Akcay F. Serum lipid profile paraoxonase and arylesterase activities in psoriasis. Cell Biochem Funct. 2009;27:176–180. doi: 10.1002/cbf.1553. [DOI] [PubMed] [Google Scholar]
- Ward NL, Loyd CM, Wolfram JA, Diaconu D, Michaels CM, McCormick TS. Depletion of antigen-presenting cells by clodronate liposomes reverses the psoriatic skin phenotype in KC-Tie2 mice. Br J Dermatol. 2011;164:750–758. doi: 10.1111/j.1365-2133.2010.10129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17:497–504. doi: 10.1002/ana.410170513. [DOI] [PubMed] [Google Scholar]
- Wolfram JA, Diaconu D, Hatala DA, Rastegar J, Knutsen DA, Lowther A, et al. Keratinocyte but not endothelial cell specific overexpression of Tie2 leads to the development of psoriasis. Am J Pathol. 2009;174:1443–1458. doi: 10.2353/ajpath.2009.080858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.