

Abstract
Recent discoveries concerning the architecture and cellular dynamics of the developing human brain are revealing new differences between mouse and human cortical development. In mice, neurons are produced by ventricular radial glial (RG) cells, or subventricular zone intermediate progenitor (IP) cells. In the human cortex, both ventricular RG and highly motile outer RG cells generate IP cells, which likely undergo multiple rounds of transit amplification in the outer subventricular zone before producing neurons. This creates a more complex environment for neurogenesis and neuronal migration, adding new arenas in which neurodevelopmental disease gene mutation could disrupt corticogenesis. A more complete understanding of disease mechanisms will involve use of emerging model systems with developmental programs more similar to that of the human neocortex.
Introduction
Our knowledge of the cellular basis of human neocortical development and the spatial organization of the developing neocortex has recently expanded considerably [1–8]. This challenges our basic assumptions about neurogenesis and, importantly, neurodevelopmental disease. What we have learned from model systems about the molecular mechanisms of disorders of the developing nervous system can now be reexamined from this perspective. Here we discuss some of the known mechanisms of neurodevelopmental disorders, what has changed in our understanding of primate neocortical development, and the implications of this new knowledge for understanding diseases of the developing human brain. We will concentrate on excitatory neurogenesis and related disorders in the dorsal cortex, a region where much recent work has been focused.
Neural progenitors in the rodent neocortex
During embryonic development, excitatory neurons of the mammalian neocortex originate from a proliferative epithelium of radial glial (RG) cells that line the cerebral ventricles [9] (Fig. 1a). Much of our understanding of the events intervening between RG cell proliferation and neuronal production is based on rodent studies, where RG cells are primarily restricted to the ventricular zone (VZ) and undergo multiple rounds of asymmetric divisions to produce intermediate progenitor (IP) cells. While RG cells display the apical-basal polarity typical of a neuroepithelium, IP cells appear to lack apical-basal polarity. IP cells subsequently occupy the subventricular zone (SVZ) and divide symmetrically to generate neurons that migrate along RG fibers towards the pia, populating the cortical plate (CP) [10]. Because RG cells primarily undergo asymmetric self-renewing divisions, and IP cells primarily undergo only one round of transit-amplifying division to produce two neurons, neurogenesis in the rodent involves a stable-sized population of progenitor cells bordering the lateral ventricles.
Figure 1. Increased spatial and cellular complexity of the developing human cortex.
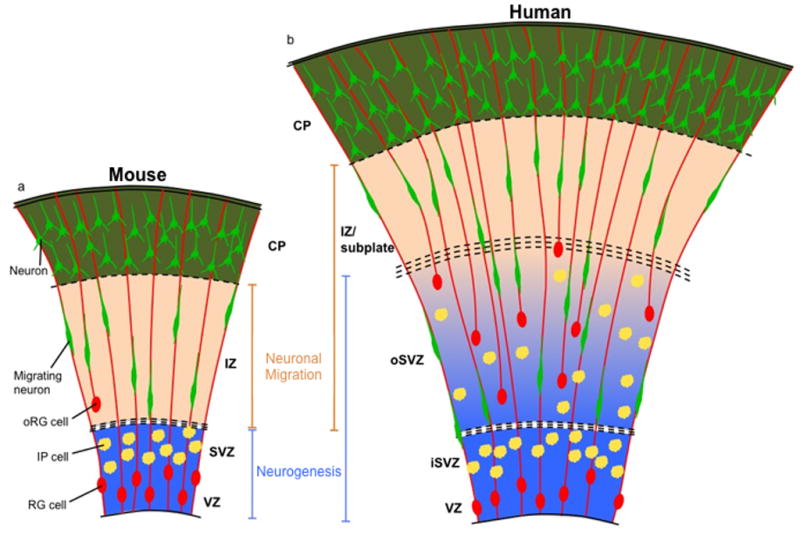
(A) Current model of mouse neocortical development. Primary zones of neurogenesis (blue) are the VZ and SVZ, where RG cells and IP cells reside, respectively. Neurons born in the VZ or SVZ migrate along the RG radial fiber scaffold found in the intermediate zone (IZ), the primary zone of neuronal migration (orange) in the mouse, to reach the CP. oRG cells are infrequent and not located in a distinct progenitor zone. (B) Expanded model of human neocortical development. RG cells, IP cells, and oRG cells are found in neurogenic zones (blue), which are the VZ, the inner SVZ (iSVZ), and the outer SVZ (oSVZ). Neurons migrate through the oSVZ and IZ/subplate (zones of neuronal migration, orange) to populate the CP. Neurons must navigate a larger distance than in the mouse, and a radial fiber scaffold of increased complexity, to reach the CP.
In order for these events to unfold, a number of important cell movements must be properly regulated. First, RG cells exhibit a highly stereotyped behavior known as interkinetic nuclear migration (INM), where the cell body shuttles up and down in the VZ in coordination with its cell cycle phase. The nucleus ascends to the upper VZ during G1 phase, and after passing through S phase descends during G2 to undergo M phase at the ventricular surface [11]. RG cells also control their cleavage plane such that the self-renewed cell retains both apical and basal compartments, whereas the daughter cell delaminates from the epithelial structure to become an IP cell. Finally, once neurons are born in the SVZ, they must be able to migrate long distances in the radial direction [12].
Current molecular understanding of neurodevelopmental diseases
Much of our current understanding of human disease comes from combining knowledge of human genetics with mouse models. Cell cycle regulation, neurogenesis, and the origin of human developmental diseases were linked by the identification of genes associated with cortical malformations such as microcephaly and lissencephaly [13]. Many of these genes encode centrosomal and microtubule-related proteins important for cell division (neurogenesis) and/or cell dynamics including cell cycle related motion such as INM, and neuronal migration. Therefore, we will begin with a brief discussion of centrosome and microtubule functions in INM, mitosis, and neuronal migration in the developing neocortex.
INM precedes mitosis in RG cells and involves the centrosome and microtubule-based motors, as well as the actomyosin system [11] (Fig. 2a,b). During the G1 phase of INM, apical-to-basal nuclear movement is directed away from the centrosome, which is located at the base of the primary cilium and protrudes into the cerebral ventricle. Microtubules are oriented parallel to the apical-basal axis with the minus ends closer to the ventricle, and plus-end microtubule associated motor proteins (kinesins) such as Kif1a are involved in pulling the nucleus in a basal direction [11, 14]. The actomyosin motor system is also involved in basal-to-apical INM, as myosin II inhibition selectively retards this phase of INM in mouse cortex [15]. During the G2 phase of INM, basal-to-apical nuclear movement is directed toward the centrosome. The minus-end microtubule associated motor protein dynein, and the dynein-interacting protein Lis1 control apical-to-basal nuclear movements during INM [14, 16], and the microtubule-associated protein Tpx2 moves into the apical process from the nucleus during this phase, further modulating microtubule dynamics [17]. Recent studies examining the role of the actomyosin motor system in basal-to-apical INM have shown that this behavior is specifically affected due to inhibition of the small GTPase Rac in developing mouse brain [18], or due to myosin II inhibition in zebrafish retina [19]. These findings suggest that the degree to which actomyosin motors contribute to apical-to-basal INM is not completely clear and may be species-specific. As M phase proceeds, the centrosome continues to play important roles as both the location of checkpoint proteins and a physical organizer of cell division (Fig. 2c). The G2 to M transition involves centrosome-mediated microtubule nucleation [20]. The centrosome during metaphase organizes the spindle and microtubule attachment to kinetochores, and the centrosome further functions in cytokinesis and mitotic exit [21]. Differential inheritance of mother and daughter centrosomes suggests that these organelles also play a role in asymmetric inheritance of cellular components and subsequent determination of daughter cell fates [22, 23]. After RG cells divide at the VZ, newborn cortical neurons migrate to the CP along RG fibers (Fig. 2d). The two main dynamic events during neuronal locomotion are leading process growth and nucleokinesis. Centrosome movement into the leading process precedes nucleokinesis, and a microtubule lattice originating from the centrosome surrounds the nucleus and forms a cage the functions to pull the nucleus up into the leading process [24]. The dynein complex, including the regulatory protein Lis1, controls centrosome movement into the leading process, and coupling of centrosomal and nuclear movements [16]. Doublecortin (dcx), another microtubule-binding protein, has also been implicated in radial neuronal migration [25, 26].
Figure 2. Molecular mechanisms of centrosome and microtubule-related cell behaviors in the developing neocortex.

(A) An RG cell is depicted undergoing apical-to-basal INM during the G1 phase of the cell cycle. The basal direction is up, apical is down, and the centrosome (red) is located at the ventricular surface at the base of the primary cilium (not shown). Kinesin and actomyosin motors control nuclear movement, which is ab-centrosomal and toward the microtubule plus ends. (B) RG cell undergoing basal-to-apical INM during the G2 phase of the cell cycle. Nuclear movement is ad-centrosomal and towards microtubule minus ends. The process is controlled by dynein and associated proteins such as Lis1, and possibly by actomyosin motors. The centrosome has already replicated at this time. (C) RG cell division at the ventricular surface. The centrosomes move to opposite poles of the cell and function throughout mitosis in microtubule nucleation, spindle and microtubule attachment to kinetochores, cytokinesis, and mitotic exit. The centrosome also plays a role in asymmetric inheritance of cell fate components in daughter cells. (D) Radial neuronal migration along an RG fiber. The centrosome moves into the leading process prior to nucleokinesis, and a microtubule lattice originating from the centrosome forms a cage around the nucleus. Dynein and associated proteins function to pull the nucleus into the leading process. (E) oRG cell mitotic somal translocation (MST) is depicted with possible molecular mechanisms for controlling this cell behavior. Analogous to neuronal migration, movement of the centrosome precedes the nucleus into the basal process; therefore MST likely involves nuclear movement toward microtubule minus ends. MST may utilize minus-end directed motors such as dynein, and/or actomyosin motors.
A clear indication that mutation of centrosomal proteins can cause defects in neurogenesis comes from the genes responsible for autosomal recessive primary microcephaly (MCPH) in humans. Five of the eight known MCPH genes encode proteins that localize to the centrosome during all or part of the cell cycle: microcephalin (MCPH1) [27, 28], centromere-associated protein J (Cenpj) [29, 30], abnormal spindle-like microcephaly-associated protein (Aspm) [31–33], cyclin-dependent kinase 5 regulatory-associated protein 2 (Cdk5rap2) [29, 34], and the pericentriolar gene STIL [35]. This disease is characterized by a decrease in brain volume with normal brain architecture, and patients can display various neurological and psychiatric features including seizures (most common), mental retardation, delayed motor and speech function, hyperactivity and attention deficit, and balance and coordination difficulties [36]. Mouse models have indicated that ASPM is involved in maintaining symmetric divisions of RG cells, and may be involved in completion of cytokinesis [37–39]. In the mouse brain, Cdk5rap2 is highly expressed in the neural progenitor pool, and its loss results in a depletion of RG cells and increased cell-cycle exit leading to premature neuronal differentiation [40]. This protein has also recently been shown to stimulate microtubule nucleation [41] and regulate centriole replication [42]. Cdk5rap2 function was linked to the pericentriolar protein, pericentrin, as depletion of pericentrin in neural progenitor cells phenocopies the effects of Cdk5rap2 knockdown and results in decreased recruitment of Cdk5rap2 to the centrosome [43]. Similar to MCPH genes, pericentrin mutation in human patients causes reduced brain and body size [44]. Microdeletions encompassing human NDE1, also a centrosomal gene, are risk factors for microcephaly, mental retardation, and epilepsy [45]. Mutations in NDE1 can also result in a condition called microlissencephaly, characterized by extreme microcephaly and grossly simplified cortical gyral structure [46]. Following knockout of Nde1 in mice, dramatic defects in mitotic progression, mitotic orientation, and mitotic chromosome localization in RG were observed, as well as altered neuronal cell fates. Mutated NDE1 proteins were found to be unstable, incapable of binding cytoplasmic dynein, and had aberrant centrosomal localization [46–48]. Other centrosome-associated proteins implicated in neurodevelopmental disease include WDR62, which causes microcephaly with simplified gyri and abnormal cortical architecture [49], and CEP152, the putative mammalian ortholog of Drosophila asterless which affects mitosis in the fly and results in primary microcephaly in humans [50].
Defects in microtubules and associated proteins have primarily been hypothesized to affect dynamic cellular processes such as INM and neuronal migration. Mutation of Lis1 causes lissencephaly in the human brain, while knockdown of Lis1 in rodents causes arrest of centrosomal and nuclear movements during neuronal migration, and disruption of basal-to-apical INM [16]. Furthermore, knockout of Lis1 in neural progenitors disrupts regulation of RG cleavage plane angle, leading to widespread apoptosis of neuroepithelial cells and culling of the progenitor pool [51]. This phenotype is due to an inability of astral microtubules to be captured by the cell cortex, resulting in a failure of cell division. Tubulin-related disorders likely affect similar cellular processes; for example, mutations in alpha and beta tubulins, commonly in domains necessary for microtubule polymerization of association with motor proteins, can cause severe lissencephaly and microcephaly in human patients, among other malformations [52]. In a mouse model of tubulin diseases, a mutation in Tuba1 affecting tubulin heterodimer formation was shown to cause defective cortical lamination and neuronal migration in mice [53]. It is thought that neuronal migration is similarly disrupted in humans with tubulin mutations.
The increased cellular complexity of human neocortical development
Recent studies have indicated that neurogenesis in humans occurs in a greater number of germinal zones and involves a larger number of cell types and behaviors than in mice [1, 2, 54] (Fig. 1b). We must consider these differences when applying knowledge of the molecular mechanisms of disease in mice to our understanding of disease mutations in humans. The human neocortex, like the mouse and rat, also begins its development from an epithelium of RG cells. However, the lineage relationship between human RG cells and neurons is much more protracted than in the mouse, containing a diversity of progenitor cell types, and resulting in an accumulation of progenitor cells before neuronal differentiation. Most notably, there is an expanded proliferative region termed the outer SVZ (OSVZ) in the developing human neocortex that contains the majority of progenitor cells during peak periods of neurogenesis [1, 2].
Unlike the rodent SVZ, which contains primarily IP cells, the human OSVZ contains a highly heterogeneous population of progenitor cells [1]. In particular, recent studies have focused on a novel class of neural stem cell found in the OSVZ, termed outer radial glia (oRG). Timelapse imaging and fate analysis in human show that oRG cells divide asymmetrically to self-renew, and give rise to an extended lineage of transit amplifying cells [1]. Unlike RG cells, oRG cells are located far from the ventricle, with no apical contact to the luminal surface, but they possess a long basal fiber that often extends to the pial surface. Similar but less numerous oRG and oRG-like cells have recently been characterized in other species, including mouse [7, 8], ferret [2, 55, 56] and marmoset [4, 5]. These cells may represent an avenue for inquiry into the function and fate of oRG cells that is technically difficult using human tissue. For instance, the oRG cells in mice were shown to be the progeny of ventricular RG cells [7, 8], and to undergo similar mitotic movements [8]. A similar lineage relationship may exist in the human.
While RG cells in the VZ undergo INM during mitosis, oRG cells undergo mitotic somal translocation (MST), a distinctive behavior where the cell soma rapidly ascends along its own radial fiber toward the CP a distance of several cell diameters during the hour before cytokinesis [1, 8]. The MST of oRG cells contrasts with the INM of RG cells, in which the nucleus moves apically and mitosis occurs at the ventricular surface [11]. However, the direction of nuclear movement just prior to cytokinesis is towards the centrosome in INM [11], and recent evidence in the mouse has shown that the centrosome moves into the leading process prior to nuclear translocation during MST [8]. It is therefore likely that the two behaviors involve conserved gene function (Fig. 2e). oRG cells use multiple rounds of cell division involving MST to simultaneously amplify neuron number and extend the boundary of the OSVZ outwards [54]. Both INM and MST differ from the far less dynamic IP cell mitosis, where the cell divides in place without nuclear translocation [10].
The OSVZ and oRG cells in human disease
We are now in a position to begin applying our expanded knowledge of human brain development to insights about disease mechanisms gleaned from animal models and from patients with genetic mutations. This will involve formulating new hypotheses concerning the effect of disease mutations on the developing human brain, and ultimately testing these hypotheses in emerging model systems or in the human neocortex itself. For example, it is possible that diseases of cell motility and neuronal migration may have partially or entirely different manifestations in rodent and human. Neurons not only have to navigate a vastly greater distance to reach the CP in the developing human cortex, but they are also born in two distinct locations, with OSVZ-born neurons arising at a distance from the ventricle and already closer to their final destination. Furthermore, the basal processes of oRG cells both increase the number of glial guides for neuronal migration and modify the structure of the radial migration scaffold by providing non-continuous radial fibers [54, 56]. We must also consider the possibility that disease mutations that disrupt cell motility could affect not just INM and neuronal migration, but also MST and possibly other unidentified novel behaviors required for OSVZ growth. Evidence for this comes from recent findings showing that centrosomal behaviors during MST mirror those during neuronal migration [8]. Possibly, the modest microcephalic phenotype found in Tuba1 mutant mice (discussed above) could be partially due to defects in oRG cell MST in addition to neuronal migration. We currently do not have a clear understanding of the molecular machinery involved in MST, but conservation of gene function and the similarity of MST to neuronal migration suggest that MST also involves tubulin. Double cortex syndrome in humans, which involves a subcortical band of misplaced neurons, is thought to be due to disrupted neuronal migration caused by a mutation in the microtubule-binding protein Dcx [25, 57, 58]. However, the displaced neurons could be due in part to a defect in MST, or could involve neurons that arise in the OSVZ and are arrested in migration. Clearly, abnormal MST could play a role in diseases involving disrupted neuronal layering, and new experimental systems could be utilized to explore this possibility.
An increase in the diversity of neural progenitor types between mice and humans similarly invites a re-exploration of diseases of neurogenesis. Proteins important for mitosis, such as the centrosome-associated primary microcephaly genes described above, could be involved in cell division of many different progenitor types. A defect in mitosis of RG cells suggests the possibility of a similar defect in IP and/or oRG cells; conversely, lack of a phenotype in one progenitor cell type does not preclude a mitosis-related role in a different progenitor cell type.
OSVZ evolution and emerging model systems
The mouse has provided us with a good foundation to formulate specific hypotheses concerning the molecular and cellular biology of development and developmental disease in the human brain. However, we may need new experimental systems that more fully recapitulate human development and disease. Following the recent characterization of the human OSVZ, a number of studies have extended these analyses to other species that share features of human cortical development and may represent potential model systems for exploring disease mechanisms [2–5, 8, 55, 56].
The human OSVZ contains the largest proportion of progenitor cells of the species examined so far, but oRG cells are present during development in many species throughout the evolutionary tree [54]. The amount of OSVZ proliferation appears to correlate with brain size and degree of folding. The lissencephalic brain of mice represents one extreme with no identifiable OSVZ, though recent evidence suggests that the developing rat cortex contains some OSVZ cytoarchitectural features [3]. The ferret brain constitutes an intermediate level of gyrencephaly and OSVZ size, while the highly folded human cortex sits at the other extreme with a notably developed OSVZ [54]. This is only a trend and not a rule, however, as the marmoset, a lissencephalic primate, exhibits a large OSVZ containing oRG cells, but may have lost its ancestral gyrencephaly [4, 5]. These findings suggest that oRG cells may be necessary, but not sufficient for developing a large folded brain.
Conclusion
The appreciation that OSVZ progenitor cells could be responsible for generating the majority of cortical neurons invites a re-interpretation of the mechanisms behind a wide range of neurodevelopmental diseases, especially those that broadly affect neuronal number and brain size such as lissencephaly and microcephaly. In the context of the human neocortex, the function of disease genes could extend to the OSVZ and specifically to oRG cells, since their MST, division, and derivation from ventricular RG cells are all processes likely to utilize related centrosome, actomyosin, and microtubule-based mechanisms [11, 21]. Pursuing a cell biological understanding of oRG cell behavior will be an exciting future direction that may reveal unexpected links to other neurodevelopmental disorders where cortex is affected, such as autism and schizophrenia.
Highlights.
Mechanisms of neurodevelopmental diseases have primarily been modeled in the mouse.
Neurodevelopmental disease genes often impact centrosome and microtubule dynamics.
Human neocortex development involves a novel lineage of neural stem cells in the OSVZ.
OSVZ cell behaviors likely also involve the centrosome and microtubule dynamics.
The role of disease genes in OSVZ development must now be considered.
Acknowledgments
Support was provided, in part, by grants from the NINDS of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Bridget E. LaMonica, Email: bridget.lamonica@ucsf.edu.
Jan H. Lui, Email: jan.lui@ucsf.edu.
Xiaoqun Wang, Email: WangX2@stemcell.ucsf.edu.
References
- **1.Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464(7288):554–61. doi: 10.1038/nature08845. Using histological labeling techniques and timelapse imaging, this paper describes the progenitor cell composition of the developing human neocortex. The authors show that a novel class of radial glia found in the OSVZ, termed oRG cells, are neural stem cells, and may be responsible for increasing neuronal number and cortical gyration in evolution. [DOI] [PubMed] [Google Scholar]
- **2.Fietz SA, Kelava I, Vogt J, Wilsch-Brauninger M, Stenzel D, Fish JL, Corbeil D, Riehn A, Distler W, Nitsch R, Huttner WB. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010;13(6):690–9. doi: 10.1038/nn.2553. This paper describes the presence of oRG cells in the human, and extends analysis of oRG cells analysis to another gyrencephalic mammal, the ferret. This is the first evidence that oRG cells are not specific to humans or primates, and may represent a more general mechanism for cortical expansion in many species. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Cerdeno V, Cunningham CL, Camacho J, Antczak JL, Prakash AN, Cziep ME, Walker AI, Noctor SC. Comparative analysis of the subventricular zone in rat, ferret and macaque: evidence for an outer subventricular zone in rodents. PLoS One. 2012;7(1):e30178. doi: 10.1371/journal.pone.0030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelava I, Reillo I, Murayama AY, Kalinka AT, Stenzel D, Tomancak P, Matsuzaki F, Lebrand C, Sasaki E, Schwamborn JC, Okano H, Huttner WB, Borrell V. Abundant Occurrence of Basal Radial Glia in the Subventricular Zone of Embryonic Neocortex of a Lissencephalic Primate, the Common Marmoset Callithrix jacchus. Cereb Cortex. 2012;22(2):469–81. doi: 10.1093/cercor/bhr301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Moreno F, Vasistha NA, Trevia N, Bourne JA, Molnar Z. Compartmentalization of cerebral cortical germinal zones in a lissencephalic primate and gyrencephalic rodent. Cereb Cortex. 2012;22(2):482–92. doi: 10.1093/cercor/bhr312. [DOI] [PubMed] [Google Scholar]
- 6.Smart IH, Dehay C, Giroud P, Berland M, Kennedy H. Unique morphological features of the proliferative zones and postmitotic compartments of the neural epithelium giving rise to striate and extrastriate cortex in the monkey. Cereb Cortex. 2002;12(1):37–53. doi: 10.1093/cercor/12.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shitamukai A, Konno D, Matsuzaki F. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J Neurosci. 2011;31(10):3683–95. doi: 10.1523/JNEUROSCI.4773-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **8.Wang X, Tsai JW, LaMonica B, Kriegstein AR. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat Neurosci. 2011;14(5):555–61. doi: 10.1038/nn.2807. This paper shows that oRG cells are present at small numbers in the mouse, opening the door to using mouse genetics for interrogating the mechanisms of oRG cell biology. Timelapse imaging shows that the centrosome moves into the leading process prior to mitotic somal translocation and cytokinesis, linking oRG cell behavior to centrosomal regulation and potentially to centrosomal disease genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–84. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7(2):136–44. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- 11.Taverna E, Huttner WB. Neural progenitor nuclei IN motion. Neuron. 2010;67(6):906–14. doi: 10.1016/j.neuron.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 12.Noctor G, Dutilleul C, De Paepe R, Foyer CH. Use of mitochondrial electron transport mutants to evaluate the effects of redox state on photosynthesis, stress tolerance and the integration of carbon/nitrogen metabolism. J Exp Bot. 2004;55(394):49–57. doi: 10.1093/jxb/erh021. [DOI] [PubMed] [Google Scholar]
- 13.Manzini MC, Walsh CA. What disorders of cortical development tell us about the cortex: one plus one does not always make two. Curr Opin Genet Dev. 2011;21(3):333–9. doi: 10.1016/j.gde.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai JW, Lian WN, Kemal S, Kriegstein AR, Vallee RB. Kinesin 3 and cytoplasmic dynein mediate interkinetic nuclear migration in neural stem cells. Nat Neurosci. 2010;13(12):1463–71. doi: 10.1038/nn.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schenk J, Wilsch-Brauninger M, Calegari F, Huttner WB. Myosin II is required for interkinetic nuclear migration of neural progenitors. Proc Natl Acad Sci U S A. 2009;106(38):16487–92. doi: 10.1073/pnas.0908928106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10(8):970–9. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 17.Kosodo Y, Suetsugu T, Suda M, Mimori-Kiyosue Y, Toida K, Baba SA, Kimura A, Matsuzaki F. Regulation of interkinetic nuclear migration by cell cycle-coupled active and passive mechanisms in the developing brain. EMBO J. 2011;30(9):1690–704. doi: 10.1038/emboj.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minobe S, Sakakibara A, Ohdachi T, Kanda R, Kimura M, Nakatani S, Tadokoro R, Ochiai W, Nishizawa Y, Mizoguchi A, Kawauchi T, Miyata T. Rac is involved in the interkinetic nuclear migration of cortical progenitor cells. Neurosci Res. 2009;63(4):294–301. doi: 10.1016/j.neures.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Norden C, Young S, Link BA, Harris WA. Actomyosin is the main driver of interkinetic nuclear migration in the retina. Cell. 2009;138(6):1195–208. doi: 10.1016/j.cell.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimmerman WC, Sillibourne J, Rosa J, Doxsey SJ. Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol Biol Cell. 2004;15(8):3642–57. doi: 10.1091/mbc.E03-11-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doxsey S, McCollum D, Theurkauf W. Centrosomes in cellular regulation. Annu Rev Cell Dev Biol. 2005;21:411–34. doi: 10.1146/annurev.cellbio.21.122303.120418. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Tsai JW, Imai JH, Lian WN, Vallee RB, Shi SH. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature. 2009;461(7266):947–55. doi: 10.1038/nature08435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita YM, Mahowald AP, Perlin JR, Fuller MT. Asymmetric inheritance of mother versus daughter centrosome in stem cell division. Science. 2007;315(5811):518–21. doi: 10.1126/science.1134910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Z. Molecular regulation of neuronal migration during neocortical development. Mol Cell Neurosci. 2009;42(1):11–22. doi: 10.1016/j.mcn.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6(12):1277–83. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- 26.Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Gleeson JG. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci. 2006;9(6):779–86. doi: 10.1038/nn1704. [DOI] [PubMed] [Google Scholar]
- 27.Jackson AP, McHale DP, Campbell DA, Jafri H, Rashid Y, Mannan J, Karbani G, Corry P, Levene MI, Mueller RF, Markham AF, Lench NJ, Woods CG. Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am J Hum Genet. 1998;63(2):541–6. doi: 10.1086/301966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, Karbani G, Jafri H, Rashid Y, Mueller RF, Markham AF, Woods CG. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71(1):136–42. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *29.Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO, Costa SM, Baralle D, Raponi M, Karbani G, Rashid Y, Jafri H, Bennett C, Corry P, Walsh CA, Woods CG. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37(4):353–5. doi: 10.1038/ng1539. Using genetic linkage analysis, this paper identifies two genes that cause microcephaly, Cdk5rap2 and Cenpj. The authors demonstrate the centrosomal expression pattern of the proteins encoded by these genes, implicating centrosomal functions as key aspects of congenital microcephaly. [DOI] [PubMed] [Google Scholar]
- 30.Leal GF, Roberts E, Silva EO, Costa SM, Hampshire DJ, Woods CG. A novel locus for autosomal recessive primary microcephaly (MCPH6) maps to 13q12.2. J Med Genet. 2003;40(7):540–2. doi: 10.1136/jmg.40.7.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattison L, Crow YJ, Deeble VJ, Jackson AP, Jafri H, Rashid Y, Roberts E, Woods CG. A fifth locus for primary autosomal recessive microcephaly maps to chromosome 1q31. Am J Hum Genet. 2000;67(6):1578–80. doi: 10.1086/316910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen J, Eyaid W, Mochida GH, Al-Moayyad F, Bodell A, Woods CG, Walsh CA. ASPM mutations identified in patients with primary microcephaly and seizures. J Med Genet. 2005;42(9):725–9. doi: 10.1136/jmg.2004.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *33.Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, Walsh CA, Woods CG. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32(2):316–20. doi: 10.1038/ng995. This paper identifies the disease gene Aspm to be responsible for the most prevalent form of MCPH, and demonstrates its expression in neural progenitors. Aspm regulates mitotic spindle function, implicating disrupted mitosis and cleavage plane formation as causes of microcephaly. [DOI] [PubMed] [Google Scholar]
- 34.Moynihan L, Jackson AP, Roberts E, Karbani G, Lewis I, Corry P, Turner G, Mueller RF, Lench NJ, Woods CG. A third novel locus for primary autosomal recessive microcephaly maps to chromosome 9q34. Am J Hum Genet. 2000;66(2):724–7. doi: 10.1086/302777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar A, Girimaji SC, Duvvari MR, Blanton SH. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. 2009;84(2):286–90. doi: 10.1016/j.ajhg.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P. Many roads lead to primary autosomal recessive microcephaly. Prog Neurobiol. 2009;90(3):363–83. doi: 10.1016/j.pneurobio.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Kouprina N, Pavlicek A, Collins NK, Nakano M, Noskov VN, Ohzeki J, Mochida GH, Risinger JI, Goldsmith P, Gunsior M, Solomon G, Gersch W, Kim JH, Barrett JC, Walsh CA, Jurka J, Masumoto H, Larionov V. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum Mol Genet. 2005;14(15):2155–65. doi: 10.1093/hmg/ddi220. [DOI] [PubMed] [Google Scholar]
- 38.Higgins J, Midgley C, Bergh AM, Bell SM, Askham JM, Roberts E, Binns RK, Sharif SM, Bennett C, Glover DM, Woods CG, Morrison EE, Bond J. Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol. 2010;11:85. doi: 10.1186/1471-2121-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci U S A. 2006;103(27):10438–43. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Megraw TL, Sharkey JT, Nowakowski RS. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 2011;21(8):470–80. doi: 10.1016/j.tcb.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi YK, Liu P, Sze SK, Dai C, Qi RZ. CDK5RAP2 stimulates microtubule nucleation by the gamma-tubulin ring complex. J Cell Biol. 2010;191(6):1089–95. doi: 10.1083/jcb.201007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barrera JA, Kao LR, Hammer RE, Seemann J, Fuchs JL, Megraw TL. CDK5RAP2 regulates centriole engagement and cohesion in mice. Dev Cell. 2010;18(6):913–26. doi: 10.1016/j.devcel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchman JJ, Tseng HC, Zhou Y, Frank CL, Xie Z, Tsai LH. Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron. 2010;66(3):386–402. doi: 10.1016/j.neuron.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 44.Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, van Essen AJ, Goecke TO, Al-Gazali L, Chrzanowska KH, Zweier C, Brunner HG, Becker K, Curry CJ, Dallapiccola B, Devriendt K, Dorfler A, Kinning E, Megarbane A, Meinecke P, Semple RK, Spranger S, Toutain A, Trembath RC, Voss E, Wilson L, Hennekam R, de Zegher F, Dorr HG, Reis A. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816–9. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 45.Ghannad M. The essential role of NDE1 in extreme microcephaly. Clin Genet. 2011;80(3):241–2. doi: 10.1111/j.1399-0004.2011.01753.x. [DOI] [PubMed] [Google Scholar]
- 46.Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, Kentab A, Jan M, Shaheen R, Feng Y, Walsh CA. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected] Am J Hum Genet. 2011;88(5):536–47. doi: 10.1016/j.ajhg.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44(2):279–93. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 48.Bakircioglu M, Carvalho OP, Khurshid M, Cox JJ, Tuysuz B, Barak T, Yilmaz S, Caglayan O, Dincer A, Nicholas AK, Quarrell O, Springell K, Karbani G, Malik S, Gannon C, Sheridan E, Crosier M, Lisgo SN, Lindsay S, Bilguvar K, Gergely F, Gunel M, Woods CG. The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet. 2011;88(5):523–35. doi: 10.1016/j.ajhg.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhat V, Girimaji SC, Mohan G, Arvinda HR, Singhmar P, Duvvari MR, Kumar A. Mutations in WDR62, encoding a centrosomal and nuclear protein, in Indian primary microcephaly families with cortical malformations. Clin Genet. 2011;80(6):532–40. doi: 10.1111/j.1399-0004.2011.01686.x. [DOI] [PubMed] [Google Scholar]
- 50.Guernsey DL, Jiang H, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau-Sturk T, Beis J, Dumas N, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Patry L, Rideout AL, Thomas A, Orr A, Hoffmann I, Michaud JL, Awadalla P, Meek DC, Ludman M, Samuels ME. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet. 2010;87(1):40–51. doi: 10.1016/j.ajhg.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yingling J, Youn YH, Darling D, Toyo-Oka K, Pramparo T, Hirotsune S, Wynshaw-Boris A. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;132(3):474–86. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tischfield MA, Cederquist GY, Gupta ML, Jr, Engle EC. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev. 2011;21(3):286–94. doi: 10.1016/j.gde.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, Oliver PL, Fray M, Harvey RJ, Molnar Z, Pinon MC, Dear N, Valdar W, Brown SD, Davies KE, Rawlins JN, Cowan NJ, Nolan P, Chelly J, Flint J. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128(1):45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;146(1):18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reillo I, Borrell V. Germinal Zones in the Developing Cerebral Cortex of Ferret: Ontogeny, Cell Cycle Kinetics, and Diversity of Progenitors. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr284. [DOI] [PubMed] [Google Scholar]
- *56.Reillo I, de Juan Romero C, Garcia-Cabezas MA, Borrell V. A role for intermediate radial glia in the tangential expansion of the mammalian cerebral cortex. Cereb Cortex. 2011;21(7):1674–94. doi: 10.1093/cercor/bhq238. This paper describes the presence of oRG cells in the ferret, and proposes the idea that expansion in cortical size is due to the tangential spreading of migrating neurons that follow the more complex migratory scaffold laid down by oRG cells. [DOI] [PubMed] [Google Scholar]
- 57.des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92(1):51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 58.Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb Cortex. 2006;16(9):1323–31. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]