

Abstract
Background
Most cardiovascular studies have implicated the central transcription factor nuclear factor kappa B (NF-κB) as contributing to the detrimental effects of cardiac injury. This ostensibly negative view of NF-κB competes with its important role in the normal host inflammatory and immune response. Pressure-overload, left ventricular hypertrophy (LVH), and heart failure represent a spectrum of disease that has both adaptive and maladaptive components. As opposed to its known effects related to myocardial ischemia-reperfusion, we hypothesized that NF-κB is necessary for the compensatory phase of cardiac remodeling.
Material and Methods
C57BL6 mice underwent minimally-invasive transverse aortic constriction (TAC) with or without inhibition of the proximal NF-κB kinase, inhibitory kappa B kinase-β (IKKβ). Isolated cardiomyocytes were cultured. Transthoracic echocardiography was performed on all animals.
Results
IKK-β inhibition successfully decreased cardiomyocyte expression of phosphorylated p65 NF-κB and decreased expression of hypertrophic markers with stimulation in vitro. Three weeks after TAC, mice treated with IKK-β inhibition more aggressively developed LVH as measured by heart weight/body weight, LV mass, and wall thickness. These animals also demonstrated functional decline as measured by decreased fractional shortening and ejection fraction. These findings were associated with decreased protein expression of p65 NF-κB.
Conclusions
Whereas short-term pressure-overload results in compensatory LVH with normal cardiac function, NF-κB inhibition resulted in increased LVH that was associated with functional deterioration. These observations suggest that NF-κB is an important part of the adaptive phase of LVH, and its inhibition detrimentally impacts cardiac remodeling.
Keywords: Heart failure, left ventricular hypertrophy, cardiac remodeling, nuclear factor kappa B, IKK
Introduction
Nuclear factor kappa-B (NF-κB) is a well-known pluripotent transcription factor involved in the inflammatory and immune response [1,2]. The majority of studies within the cardiovascular literature focus on the undesirable aspects of NF-κB-driven transcriptional events. Various methods of inhibiting the NF-κB signaling pathways have been utilized in order to attenuate an undesirable response to injury [3–6]. Imbedded within these reports is the concept that NF-κB is responsible for creating a bad phenotype (i.e. an injured heart). However, this pattern overlooks that fact that NF-κB is a vital part of our innate immunity and promotes survival signals. Indeed, some evidence suggests that NF-κB promotes cardioprotection in models of ischemic preconditioning and coronary ligation [2,7]. As such, understanding the adaptive and maladaptive balance of NF-κB activation remains a largely unanswered question [8].
Whether or not NF-κB promotes survival or death pathways is, in part, related to the environment and stimulus to which a cell or tissue is exposed. As opposed to the very noxious stimuli associated with acute ischemia-reperfusion injury, other relevant cardiovascular events are less dramatic such as those associated with hypertension, valvular disease, and heart failure. We have developed a murine model of pressure-overload that allows us to evaluate the development of compensatory, physiologic left ventricular hypertrophy (LVH) and its eventual, maladaptive progression to heart failure (HF) [9]. By removing a constricting band on the thoracic aorta we can also assess the physiologic and molecular aspects of myocardial recovery [10]. Within this model system, we have also shown that various components of NF-κB signaling are activated in both the development and regression of heart failure [11]. Within the paradigm of “good” versus “bad” NF-κB, we hypothesized that NF-κB is necessary for the development of compensated, adaptive hypertrophy. The logical follow-up will be that a molecular switch must occur that transitions to where excessive NF-κB stimulation actually becomes detrimental. This study will evaluate the former hypothesis by examining the influence of proximal NF-κB kinase blockade on mice subjected to pressure overload.
Methods
Surgical model
In accordance with an institutionally approved IACUC protocol, minimally-invasive transverse arch banding and debanding were performed in 10-week-old C57BL6 male mice (Charles River Laboratories, Wilmington, MA) as previously described [9]. In brief, animals were anesthetized using inhaled isoflurane by facemask. A midline neck incision was used to approach the anterior mediastinum. The transverse arch was identified and a constrictive band was placed and tightened to the approximate diameter of a 27-gauge needle. Appropriate placement of the band was verified by evaluation of carotid artery dopplers both before and after placement of the aortic band. Adequate banding was accepted when the Doppler velocity ratio doubled from right to left carotid arteries. Prior to any experimentation the mice were housed in the same cubicle in a DLAM facility with 12 hour light cycling and ad libitum access to water and chow.
Mice were treated with the IKKβ inhibitor, Bay (15mg/kg intraperitoneally, generously supplied by Dr. AS Baldwin, University of North Carolina) or vehicle every other day during the experimental period. After 3 weeks, mice were euthanized and heart weight/body weights were recorded.
Transthoracic echocardiography
Transthoracic echocardiography was performed using the Vevo 660 High Resolution Biomicroscopy System equipped with a 30 Mhz transducer (Visual Sonics, Toronto, CA). During examination, mice were anesthetized with 1–1.5% inhaled isoflurane. Depth of anesthesia was standardized by recording images at heart rates of 480–520 bpm. Images were recorded in all animals before surgery and at 3 weeks, prior to necropsy. Two technicians, blinded to the animals’ experimental status, performed exams and measurements. Standard measurements were recorded including ventricular dimensions, LV mass, and ejection fraction.
Left ventricular protein expression
Left ventricles were dissected at necropsy and homogenized in whole cell lysis buffer (Cell Signaling Technology, Danvers, MA). Homogentaes were then centrifuged for 30 minutes at 18,000g. The Bradford assay (Bio-Rad Laboratories, Hercules, CA) was used to quantify protein concentration. Western blots were then run on polyvinylidene membranes. Membranes were incubated with phosphorylated (phospho) p65 (Ser 536), total endogenous p65 (Sigma-Aldrich, St Louis, MO) or β-tubulin (Chemicon International, Temecula, CA) primary antibodies. Visualization of protein bands was performed with an Invitrogen kit (Invitrogen, Carlsbad, CA).
Cardiomyocyte protein expression
Murine cardiomyocytes were cultured as previously described [12]. Protein fractions were isolated in ice cold lysis buffer during dounce homogenization and denatured in loading buffer. Concentrations were determined using the Bradford assay. 30 μg of each sample was then loaded into alternating lanes for gel β-tubulin was used as the loading control.
Statistical Analysis
Comparisons between experimental groups were made using one-way ANOVA with Newnab-Keuls multiple comparison post hoc testing using the statistical package, Prism 4 (GraphPad, San Diego, CA). Two-tailed Student’s t-test was used for echocardiographic comparisons. Data are expressed as mean ± SEM. Statistical significance was accepted at the 95% confidence interval.
Results
The best characterized signaling pathway, or canonical pathway, for NF-κB activation involves phosphorylation of the beta-subunit of the proximal IKK complex (IKK- β), thereby allowing for subsequent phosphorylation and degradation of the inhibitory kappa B alpha brake on translocation of the active p65 NF-κB subunit into the nucleus [3]. In order to validate our IKK-β inhibitor (Bay) within this model system, we tested the effect of Bay on isolated neonatal rat cardiomyocytes (Figure 1). Both leukemia inhibitory factor and phenylephrine are known hypertrophic agonists [13]. As expected, each agent increased protein expression of the hypertrophic marker, β-myosin heavy chain (β-MHC) as well as phosphorylated p65 NF-κB. IKK-β inhibition decreased β-MHC expression that was associated with a marked decrease in p65 NF-κB expression, thereby validating the quality of our inhibitor.
Figure 1.

IKK-β inhibition of cardiomyocyte hypertrophy and p65 expression in vitro. Murine cardiomyocytes were cultured and stimulated with hypertrophic agonists, leukemia inhibitory factor (LIF) and phenylephrine (PE). Western blot was performed for the hypertrophic marker, β-myosin heavy chain (β-MHC) and total and phosphorylated p65-NF-κB. Blot is representative of 3 separate mouse cardiomyocyte procurements. β-tubulin serves as the loading control.
Minimally-invasive transverse aortic constriction (TAC) was then performed and animals were followed for 3 weeks. We have previously demonstrated that after 3 weeks of banding, mouse hearts develop significant left ventricular hypertrophy that remains functionally compensated [9]. Figure 2 demonstrates the velocity ratios between the right and left carotid arteries that are used as a surrogate for the quality and persistence of our aortic obstruction. Both control and treated animals had significant gradients across the aortic arch that persisted after 3 weeks of banding. Therefore, bands remained intact (did not slip).
Figure 2.

Velocities in the carotid arteries as measured by Doppler. Right/left ratios prior to banding, immediately after banding, and after 3 weeks of banding are recorded. *p<0.05 versus sham and pre-banded mice (n=5/group).
After 3 weeks of banding, echocardiography was performed and hearts were recovered to determine their size. Compared to sham animals, banded animals had a slight increase in heart weight to body weight ratio (4.17 ± 0.06 vs 4.58 ± 0.17). After administration of our IKK-β inhibitor, hearts were sizably enlarged compared to both sham and vehicle animals (5.39 ±0.30, p=0.012). In contrast to the weight-based studies, banded animals given vehicle had increased echocardiographic evidence of LVH as measured by posterior wall thickness and left ventricular mass. As depicted in Figure 3, these echocardiographic features were even more pronounced in animals treated with IKK-β inhibition
Figure 3.

Assessment of left ventricular hypertrophy as measured by heart weight/body weight ratios, left ventricular posterior wall thickness, and left ventricular mass. Respective lines represent differences between groups (n= 5/group) at p<0.05.
Echocardiography was furthermore used to functionally analyze treated animals. Commiserate with the increased LVH and compared to baseline and vehicle animals, mice banded in the presence of IKK-β inhibition had decreases in both fractional shortening and ejection fraction (Figure 4). In order to demonstrate in vivo activity of our inhibitor, Western blots on mouse hearts from the respective groups demonstrate similar amounts of p65 NF-κB protein after three weeks of banding which was expectantly suppressed with chronic administration of IKK-β blockade (Figure 5).
Figure 4.

Echocardiographic assessment of ventricular function as measured by fractional shortening and ejection fraction. Respective lines represent differences between groups (n=5/group) at p<0.05.
Figure 5.


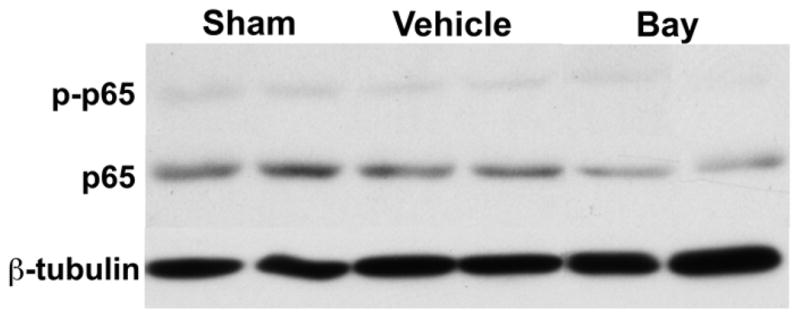
Western blot for p65 expression in sham, banded, and banded with inhibitor mice. Blot is representative of 4 separate groups of animals. β-tubulin serves as the loading control.
Discussion
Pressure-overload induced by transverse aortic constriction is a well-known model for promoting LVH. The Law of LaPlace demands that as wall stress increases with enhanced afterload, the heart will increase its thickness. With ongoing overload (>4 weeks), the ventricle begins to dilate and enters a maladaptive phase including the development of heart failure [11,14]. In the present study, we demonstrate that proximal kinase blockade of NF-κB during acute pressure-overload results in the early development of decompensated LVH. As opposed to the preponderance of studies that suggest that NF-κB mediates myocardial injury (i.e. related to ischemia-reperfusion), this report adds to the evidence that NF-κB actually plays an important homeostatic role in cardiac physiology.
We have previously demonstrated that during the progression phase of TAC-induced LVH, a pattern of NF-κB-dependent survival genes are expressed [11]. This led us to examine the effect of NF-κB blockade on the early physiologic changes associated with pressure overload. Others have recently demonstrated the significant role of NF-κB signaling with this model system by constructing cardiomyocyte-specific deletions of IKK-β and NEMO/IKKγ [15,16]. Both of these proteins are part of the IKK complex and their conditional absence promoted an aggressive development of dilated cardiomyopathy mediated, in part, by dysregulation of the oxidant stress response. These transgenic studies are confounded by the propensity of the mouse hearts to naturally dilate as well as compensatory upregulation of other NF-κB related genes, potentially distorting the effects of induced stress (pressure-overload) that occurs to non-genetically manipulated animals. As such, we felt pharmacologic inhibition was an important element to decipher the role of NF-κB and compensatory LVH. Indeed, our IKK-β inhibitor, albeit not as potent as its transgenic ablation, successfully decreased adult mouse heart NF-κB activity both in vitro and in vivo.
Notwithstanding the ischemia-reperfusion studies – with the noxious stimulus of acutely interrupted blood supply to the heart - the current study using chronic pressure overload demonstrates disparate results to other models of LVH. In particular, we and others have shown that isoproterenol or angiotensin-II induced LVH is abrogated with NF-κB blockade [17,18]. A number of factors likely are in play including the microenvironment as well as the intensity and chronicity of stimulus. For example, phosphorylation of the natural NF-κB upstream brake, IκBα, can occur via serine and tyrosine phosphorylation pathways that are differentially activated depending on the pathophysiologic situation [19].
We recognize that more work will be necessary to determine the mechanism by which NF-κB favorably promotes adaptive LVH. Putative targets include its transcriptional influence over fibrosis genes, myocardial structural proteins, and cell death programs. As previously mentioned, NF-κB is an important and beneficial component of the innate immunity, including the host’s response to infection and inflammation [20]. These salubrious functions of NF-κB extend to its salubrious influence with regards to ischemic preconditioning and coronary ligation, perhaps through its interaction with heat shock proteins [2,7]. Finally, although we have focused on the classic, canonical pathway of NF-κB stimulation, we must acknowledge that other non-canonical signals are active. Indeed, the p50-p105 axis has been shown to alternatively protect from and promote cardiac remodeling in ischemic models [21,22]. These latter studies further support the previously mentioned issues related to the complexity of stimulus and environment.
Summarily, we demonstrate that NF-κB is an important part of the normal response to pressure overload. Within the realm of innate immunity, it makes sense, at some level, that NF-κB is necessary for adaptive LVH [20]. That said, reconciling when NF-κB is acting “good” versus “bad” will remain a challenge.
Acknowledgments
This work was funded in part by the National Institute of Health R01HL089592 (CHS) and American College of Surgeons (CHS).
Footnotes
The authors have no disclosures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baldwin AS., Jr Series introduction: The transcription factor NF-κB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tranter M, Ren X, Forde T, et al. NF-kappaB driven cardioprotective gene programs; Hsp70.3 and cardioprotection after late ischemic preconditioning. J Mol Cell Cardiol. 2010;49:664–672. doi: 10.1016/j.yjmcc.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moss NC, Stansfield WE, Willis MS, Tang RH, Selzman CH. IKKbeta inhibition attenuates myocardial injury and dysfunction following acute ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293:H2248–2253. doi: 10.1152/ajpheart.00776.2007. [DOI] [PubMed] [Google Scholar]
- 4.Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH. Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2007;84:120–125. doi: 10.1016/j.athoracsur.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 5.Valen G. Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res Cardiol. 2004;99:1–7. doi: 10.1007/s00395-003-0442-7. [DOI] [PubMed] [Google Scholar]
- 6.Pye J, Ardeshirpour F, McCain A, et al. Proteasome inhibition ablates activation of NF-kappa B in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol. 2003;284:H919–926. doi: 10.1152/ajpheart.00851.2002. [DOI] [PubMed] [Google Scholar]
- 7.Wilhide ME, Tranter M, Ren X, et al. Identification of a NF-kappaB cardioprotective gene program: NF-kappaB regulation of Hsp70.1 contributes to cardioprotection after permanent coronary occlusion. J Mol Cell Cardiol. 2011;51:82–89. doi: 10.1016/j.yjmcc.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–1132. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 9.Stansfield WE, Rojas M, Corn D, et al. Characterization of a model to independently study regression of ventricular hypertrophy. J Surg Res. 2007;142:387–393. doi: 10.1016/j.jss.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 10.Stansfield WE, Charles PC, Tang RH, et al. Regression of pressure-induced left ventricular hypertrophy is characterized by a distinct gene expression profile. J Thorac Cardiovasc Surg. 2009;137:232–238. 238e231–238. doi: 10.1016/j.jtcvs.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen NM, Stansfield WE, Tang RH, Rojas M, Patterson C, Selzman CH. Recovery from decompensated heart failure is associated with a distinct, phase-dependent gene expression profile. J Surg Res. 2012 doi: 10.1016/j.jss.2011.12.017. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang RH, Zheng XL, Callis TE, et al. Myocardin inhibits cellular proliferation by inhibiting NF-kappaB(p65)-dependent cell cycle progression. Proc Natl Acad Sci U S A. 2008;105:3362–3367. doi: 10.1073/pnas.0705842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng XF, Rokosh DG, Simpson PC. Autonomous and growth factor-induced hypertrophy in cultured neonatal mouse cardiac myocytes. Comparison with rat. Circ Res. 2000;87:781–788. doi: 10.1161/01.res.87.9.781. [DOI] [PubMed] [Google Scholar]
- 14.Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- 15.Hikoso S, Yamaguchi O, Nakano Y, et al. The I{kappa}B kinase {beta}/nuclear factor {kappa}B signaling pathway protects the heart from hemodynamic stress mediated by the regulation of manganese superoxide dismutase expression. Circ Res. 2009;105:70–79. doi: 10.1161/CIRCRESAHA.108.193318. [DOI] [PubMed] [Google Scholar]
- 16.Kratsios P, Huth M, Temmerman L, et al. Antioxidant amelioration of dilated cardiomyopathy caused by conditional deletion of NEMO/IKKgamma in cardiomyocytes. Circ Res. 2010;106:133–144. doi: 10.1161/CIRCRESAHA.109.202200. [DOI] [PubMed] [Google Scholar]
- 17.Kawano S, Kubota T, Monden Y, et al. Blockade of NF-kappaB ameliorates myocardial hypertrophy in response to chronic infusion of angiotensin II. Cardiovasc Res. 2005;67:689–698. doi: 10.1016/j.cardiores.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 18.Stansfield WE, Tang R, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294:H645–650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- 19.Brown M, McGuinness M, Wright T, et al. Cardiac-specific blockade of NF-kappaB in cardiac pathophysiology: differences between acute and chronic stimuli in vivo. Am J Physiol Heart Circ Physiol. 2005;289:H466–476. doi: 10.1152/ajpheart.00170.2004. [DOI] [PubMed] [Google Scholar]
- 20.Valen G. Innate immunity and remodelling. Heart Fail Rev. 2011;16:71–78. doi: 10.1007/s10741-010-9187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frantz S, Hu K, Bayer B, et al. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. Faseb J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 22.Timmers L, van Keulen JK, Hoefer IE, et al. Targeted deletion of nuclear factor kappaB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ Res. 2009;104:699–706. doi: 10.1161/CIRCRESAHA.108.189746. [DOI] [PubMed] [Google Scholar]