

Abstract
BACKGROUND AND PURPOSE
Nitrate tolerance, the loss of vascular responsiveness with continued use of nitrates, remains incompletely understood and is a limitation of these therapeutic agents. Vascular superoxide, generated by uncoupled endothelial NOS (eNOS), may play a role. As arginase competes with eNOS for L-arginine and may exacerbate the production of reactive oxygen species (ROS), we hypothesized that arginase inhibition might reduce nitrate tolerance.
EXPERIMENTAL APPROACH
Vasodilator responses were measured in aorta from C57Bl/6 and arginase II knockout (argII –/–) mice using myography. Uncoupling of eNOS, determined as eNOS monomer : dimer ratio, was assessed using low-temperature SDS-PAGE and ROS levels were measured using L-012 and lucigenin-enhanced chemiluminescence.
KEY RESULTS
Repeated application of glyceryl trinitrate (GTN) on aorta isolated from C57Bl/6 mice produced a 32-fold rightward shift of the concentration–response curve. However this rightward shift (or resultant tolerance) was not observed in the presence of the arginase inhibitor (s)-(2-boronethyl)-L-cysteine HCl (BEC; 100 µM) nor in aorta isolated from argII –/– mice. Similar findings were obtained after inducing nitrate tolerance in vivo. Repeated administration of GTN in human umbilical vein endothelial cells induced uncoupling of eNOS from its dimeric state and increased ROS levels, which were reduced with arginase inhibition and exogenous L-arginine. Aortae from GTN tolerant C57Bl/6 mice exhibited increased arginase activity and ROS production, whereas vessels from argII –/– mice did not.
CONCLUSION AND IMPLICATIONS
Arginase II removal prevents nitrate tolerance. This may be due to decreased uncoupling of eNOS and consequent ROS production.
Keywords: nitric oxide, reactive oxygen species, glyceryl trinitrate, nitrate tolerance
Introduction
Nitrate tolerance describes the loss of vessel responsiveness to the continued use of drugs, which actions mimic the endothelial endogenous vasodilator NO. It is a serious limitation in an otherwise highly useful class of drugs, collectively referred to as nitrates, in the treatment of ischaemia and angina. Although these compounds have been used for well over a century, the mechanisms underlying the phenomenon of tolerance remain incompletely understood. One of the best-studied and most widely accepted postulates involves the production of reactive oxygen species (ROS). The essential hypothesis is that nitrate tolerance results from an increase in vascular superoxide due to uncoupled endothelial NOS (eNOS) and increased activity of NADPH oxidase(s) (Münzel et al., 1995). As well, there are studies that demonstrate abnormalities in the biotransformation and in particular, the de-nitrification of nitrates by the enzyme aldehyde dehydrogenase (ALDH-2) within the mitochondria (Sage et al., 2000; Chen et al., 2002). Importantly, the problem of mitochondrial de-nitrification has since been linked with increased mitochondrial free radical bioavailability during sustained nitrate therapy (Sydow et al., 2004).
Pertaining to the L-arginine-NO pathway, the enzyme arginase, originally identified for its role in the hepatic urea cycle, has been found in the vasculature (see Morris, 2009). Now known to exist in at least two isoforms, arginase I and II, up-regulation of this enzyme suppresses NO synthesis in endothelial cells (Li et al., 2001; Berkowitz et al., 2003; Chicoine et al., 2004; White et al., 2006), most likely by depleting L-arginine stores in competition with eNOS. Arginase II, in particular, appears to be the predominant form in endothelial cells (Gerzanich et al., 2003; Bachetti et al., 2004; Ming et al., 2004) and is confined to the mitochondria where it modulates eNOS activity by regulating intracellular L-arginine levels (Lim et al., 2007). Here, we have hypothesized that, as arginase contributes to the depletion of intracellular L-arginine stores and to downstream events including eNOS uncoupling and ROS production, inhibition of arginase, and specifically of arginase II, might prevent nitrate tolerance.
Methods
Animals and treatments
All animal care and experimental procedures were approved by the AMREP Institutional Animal Ethics Committee (Approval #E/0665/2008/B), which adheres to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Arginase II knockout (argII –/–) mice on a C57BL/6 (WT) background [a gift from Prof William O'Brien (Shi et al., 2001)] and WT mice were studied at 10 and 15 weeks old. Animals were housed under standard laboratory conditions and food and water was available ad libitum. Mice were killed by asphyxiation in 80% CO2 and 20% O2.
Vascular reactivity studies
Thoracic aorta from 10 and 15 week old WT and argII –/– mice were isolated and placed in ice-cold carbogenated (95% O2; 5% CO2) Krebs solution (composition in mM: NaCl 119, KCl 4.7, MgSO4 1.17, NaHCO3 25, KH2PO4 1.18, CaCl2 2.5 and glucose 11 and EDTA 0.03); and subsequently cleared of connective tissue and mounted in Mulvany myographs bubbled with carbogen (95% O2 and 5% CO2-) at 37°C (model 610 M, JP Trading), for isometric tension recording as described previously (Kimura et al., 2003). Vessel viability was assessed by maximally contracting the aorta with a K+-depolarizing solution (KPSS; composition in mM: KCl 123, MgSO4 1.17, KH2PO4 1.18, CaCl2 2.5 and glucose 11 and EDTA 0.03).
In vitro tolerance model
Full concentration response curves to glyceryl trinitrate (GTN; 0.1 nM–10 µM) were obtained in vessels preconstricted to approximately 50% of the KPSS maximum with phenylephrine (0.1–1 µM). A repeat curve was constructed 30 min later in the absence (control) or presence of the arginase inhibitor (s)-(2-boronethyl-L-cysteine) HCl (BEC; 100 µM). Repeat responses to GTN were also obtained in vessels from argII –/– mice. Relaxation responses are expressed as a percentage reversal of the PE precontraction. Individual relaxation curves were fitted to a sigmoidal logistic equation (GraphPad Prism 5.0, GraphPad Software, Inc., San Diego, CA, USA) and the corresponding negative log EC50 values (pEC50) calculated.
In vivo tolerance model
In vivo tolerance to GTN was induced by subcutaneous injections of GTN (20 mg·kg−1) three times a day (0700, 1500, 2300 h) for 3 days (total 60 mg·kg−1·day−1) (Wang et al., 2002). Control animals received vehicle (5% glucose) diluted in saline over the same period. At the end of the 72 h period on day 4 mice were killed and the thoracic aorta removed for further study. These studies were performed in both WT and argII –/– mice.
Determination of ROS
In endothelial cells
Human umbilical vein endothelial cells (HUVECs) were seeded onto white 96 well plates and grown to confluence. 24 h prior to experimentation, cells were serum-starved and either treated with GTN (0.5 mM; diluted in Krebs-HEPES; composition in mM: NaCl 111.2, D-glucose 11.1, NaHCO3 25.0, HEPES 10.1, MgSO4·7H20 1.3, CaCl2 2.5, KH2PO4 1.2 and KCl 4.8) or vehicle (Krebs-HEPES) as previously described (Dikalov et al., 1998). Cells were washed and serum-free media was replaced with pre-warmed Krebs-HEPES and treated with GTN (0.5 mM) for a total of 60 min. 30 min from the start of GTN treatment, cells were treated with L-arginine (100 µM), BEC (100 µM) or tempol (1 mM) for a total of 30 min, after which L-012 (100 µM) was added to each well and fluorescence measurements obtained at 37°C (excitation 485 nm, emission 520 nm) and expressed as relative light units per second (RLU·s−1) as previously described (Sohn et al., 1999; Daiber et al., 2004).
In intact vessels
Superoxide formation in thoracic aortae from 15 week-old argII –/– and WT mice was measured using NADPH (100 µM)-driven lucigenin-enhanced chemiluminescence (5 µM) using a luminometer as previously described (Ritchie et al., 2007). Aortae were incubated at 37°C in the dark for 1 h either in the absence (control) or presence of GTN (100 µM). For vessels obtained from animals receiving GTN (or vehicle) in vivo only baseline superoxide formation was obtained. Background luminescence was subtracted from an average of 10 readings and each measurement was expressed as counts per mg per second (counts·mg−1·s−1).
eNOS monomer/dimer detection by Western blot analysis
Aortic lysates were obtained by homogenization in a glass homogenizer in cold lysis buffer while HUVECs were sonicated in ice-cold lysis buffer, and both were centrifuged at 13 000× g for 10 min to remove unlysed cells and cell fragments. Protein content was measured by the Lowry method (Lowry et al., 1951). 20 µg of protein per sample were dissolved in 0.225 M Tris-HCl (pH 6.8), 50% glycerol, 5% SDS, and 0.05% bromophenol blue; and loaded onto a 6% gel. Low-temperature SDS-PAGE (LT-PAGE) was performed using previously reported methods (Klatt et al., 1996; Yang et al., 2009). Gels and buffers were kept at 4°C prior to and during electrophoresis. Following LT-PAGE, gels were transferred on to a PVDF membrane at 4°C and the blots were probed as routine Western blots for eNOS monomer and dimer (both BD Transduction, Franklin Lakes, NJ, USA; 1:10 000) and β-tubulin (as the loading control; Bio-Rad, Hercules, CA, USA, 1:1000). Densitometry was used to quantitate the bands and the dimer/monomer calculated.
L-arginine uptake
Cellular 3H-L-arginine uptake was measured as previously described (Chin-Dusting et al., 2003; Parnell et al., 2004; Zhang et al., 2006). Briefly, EA.hy926 cells were plated in 24-well, flat bottom, tissue culture plates and grown to ≍ 90% confluence at 37 °C in 5% CO2. Two hours prior to treatment, cells were deprived of L-arginine by washing cells twice and replacing media with pre-warmed (37 °C) phosphate buffered saline (PBS, composition in mM; CaCl2 0.901, MgCl2 0.495, KCl 2.97, KH2PO4 1.47, NaCl 137.93, Na2HPO4 8.10). Cells were treated with GTN (100nM) for 30 min after an initial 30 min pre-treatment period with L-arginine (200µM), L-NOHA (10µM), BEC (100µM) or an untreated control. Treatments remained present throughout the remainder of the experiment. Cells were then incubated in 500µl of total uptake solution containing 3H-L-arginine (100nM) and unlabelled L-arginine (100 nM) prepared in Locke's buffer (composition in mM: NaCl 154, KCl 5.6, CaCl2 1, MgCl2 1, HEPES 10, NaHCO3 3.6 and D-glucose 5.6) or non-specific uptake solution which comprised of the total uptake solution with additional unlabelled L-arginine (10mM) for 20 min at 37 °C in triplicate. At the conclusion of the incubation period, each culture well was washed twice with ice cold PBS and then lysed in the endothelial cell lysis buffer [0.2% sodium dodecyl sulphate (SDS) and 0.2 N NaOH] at room temperature for 1 hour. Counts were then obtained by liquid scintillation spectroscopy, whereby 400 µl of the lysate was added to 4 ml of scintillation fluid and 3H was counted for 2 min on the Liquid Scintillation analyzer. The remaining 100µl, containing 3H-L-arginine, was used for determination of protein content. Specific arginine uptake over 20 min was calculated by subtracting averaged counts of the non-specific from total uptake mixture. All counts (specific 3H-L-arginine uptake) were normalised as dpm per mg total cellular proteins and normalised to the control (% control dpm.mg-1 of protein).
Determination of arginase activity in intact vessels
Arginase activity was determined in aortic lysates following GTN (or vehicle) treatment in vivo using a previously reported method (Corraliza et al., 1994). Briefly, aortas were homogenized in ice-cold lysis buffer and the homogenate (100 µL) added to Tris-HCl (50 mM, pH 7.5, 150 µL), which contained 10 mM MnCl2. To activate arginase, samples were heated to 56°C for 10 min, L-arginine (100 µL of 0.5 mM) was added and samples incubated at 37°C for a further 60 min. To halt the reaction, a mixture of acids (H2SO4 (17.8M): H3PO4 (14.6M): H2O = 1:3:7; 800 µL) was added. α-Isonitrosopropriophenone (50 µL, 9% in absolute ethanol, w/v) was added and the samples heated at 100°C for 45 min followed by 10 min in darkness at room temperature. The colorimetric determination of urea was achieved spectrophotometrically at an absorbance of 550 nm. Urea production was calculated from a standard curve and expressed as pmol of urea per minute per milligram of tissue.
Statistical analysis
Results are expressed as mean ± SEM. A two way anova with Bonferroni post hoc analysis was used to compare all vascular reactivity responses. Where more than two groups were studied, a one-way anova with Bonferroni's multiple comparisons post hoc analysis in GraphPad Prism (v 5.0) was used, where P < 0.05 was considered statistically significant. For experiments using mice, n refers to the number of animals and for cell experiments n refers to independent experiments.
Materials
The following suppliers were used: [3H]L-arginine from Perkin Elmer Life Sciences, Boston, USA; BEC and L-NOHA from Calbiochem, USA; Tempol and α-isonitrosopropriophenone from Sigma, USA; L-012 from Wako Pure Chemicals, USA.
Results
Arginase inhibition or arginase II deletion abolishes in vitro nitrate tolerance
A significant rightward shift was evident in the repeated concentration response curve to GTN (Figure 1A pEC50 1st vs. 2nd curve: 7.5 ± 0.1 vs. 6.0 ± 0.4 n= 8, P < 0.01; these values represent about 30-fold difference in molar concentrations), establishing that tolerance had occurred. Non-specific arginase inhibition by BEC prevented tolerance (Figure 1B). GTN (pEC50= 7.7 ± 0.1 M, Rmax = 91 ± 3% relaxation) caused concentration-dependent relaxation responses in aorta from argII –/– mice (Figure 1C) comparable with that observed on aorta from WT mice (Figure 1A). However, unlike aorta from WT mice, subsequent administration of GTN after a 30 min washout period did not result in the development of tolerance (n= 5–8; P > 0.05).
Figure 1.
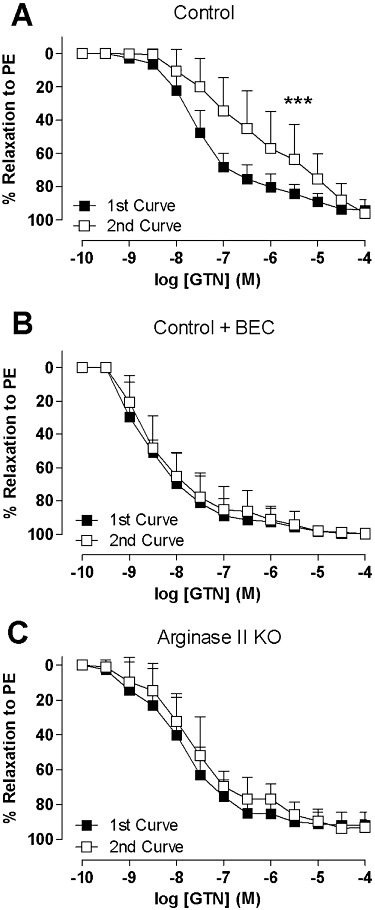
Two concentration response curves were obtained 30 min apart in aorta from WT mice (A, B) and argII –/– mice (C) to GTN either (A, C) alone or in the presence of (B) BEC (100 µM). The aorta was pre-contracted by phenylephrine (PE). Data are presented as mean ± SEM; n= 5–8 mice. ***P < 0.001, significant difference between curves; two-way anova with Bonferroni post hoc analysis.
In vivo tolerance to GTN was not evident in aorta from argII –/– mice
Aorta isolated from WT mice treated with GTN in vivo showed a significant rightward shift compared to vehicle-treated animals confirming that nitrate tolerance had occurred (Figure 2A: vehicle vs. GTN treated: pEC50= 8.2 ± 0.3 vs. 7.4 ± 0.3, n= 9–11; P < 0.01). Tolerance however was not apparent in aorta from argII –/– mice (Figure 2B: vehicle vs. GTN treated: pEC50= 7.9 ± 0.2 vs. 7.4 ± 0.2 M, n= 7–9, P > 0.05), nor was there any significant differences in GTN-induced relaxation between vehicle-treated WT and argII –/– mice (P > 0.05). Cross-tolerance between nitrates is well established (see Münzel et al., 2005a). Diethylamine NONOate (DEA/NO), like GTN, is an exogenous NO donor. In aorta from WT mice (Figure 2C), a significant shift to the right to DEA/NO is seen in GTN tolerant vessels (pEC50= 7.8 ± 0.1, n= 5) compared with vehicle-treated non-tolerant vessels (pEC50= 8.2 ± 0.1, n= 8; P < 0.001). Cross-tolerance was not observed in aorta from argII –/– mice (Figure 2D; vehicle vs. GTN treated: pEC50= 8.1 ± 0.1 vs. 8.4 ± 0.2, n= 6–8; P > 0.05).
Figure 2.
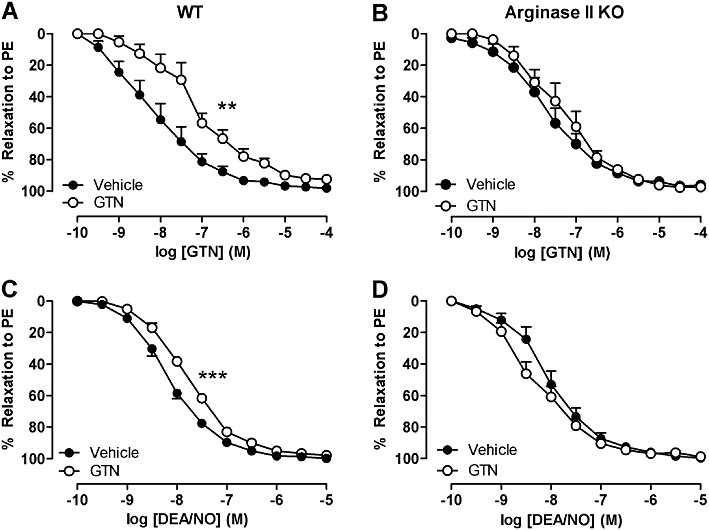
Concentration response curves to (A and B) GTN and (C and D) DEA/NO obtained from aorta from (A and C) WT and (B and D) argII –/– mice after in vivo treatment with GTN or vehicle for 3 days. The aorta was pre-contracted by phenylephrine (PE). Data are presented as mean ± SEM, n= 5–8. **P < 0.01 ***P < 0.001, significant difference between curves; two-way anova with Bonferroni post hoc analysis.
GTN induces ROS production in HUVECs and in intact vessels
Recent findings show that GTN can induce both increased ROS production and eNOS uncoupling (Sage et al., 2000; Chen et al., 2002; Münzel et al., 2005b), particularly when the endothelium is in an L-arginine-deprived state (Abou-Mohamed et al., 2000). The effect of GTN tolerance on acute ROS generation was investigated in the presence of exogenously added L-arginine, superoxide scavenging with tempol and arginase inhibiton with BEC. Addition of L-arginine and BEC by themselves did not significantly affect basal ROS production (Figure 3A), compared with control. Addition of the antioxidant tempol did reduce basal ROS levels (Figure 3A). GTN significantly increased ROS generation (Figure 3B), which was significantly reduced by pre-treatment of the endothelial cells with BEC, L-arginine and tempol (Figure 3C).
Figure 3.
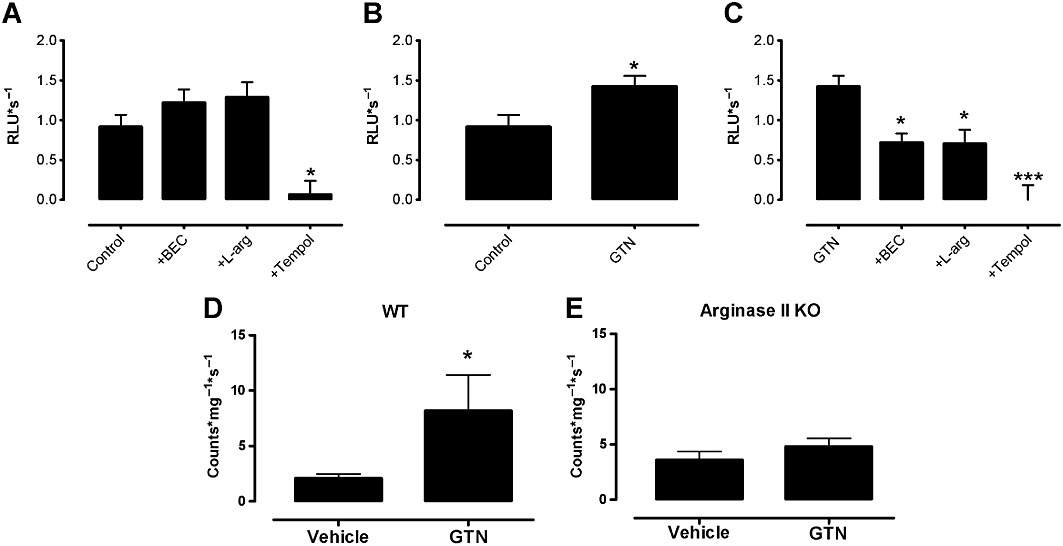
Basal levels of superoxide production were measured in HUVECs using a L-012-enhanced chemiluminescence assay receiving either (A) no treatment (control) or pretreatment with the arginase inhibitor BEC (100 µM), L-arginine (L-arg, 100 µM) or tempol (1 mM) or (B) no treatment or the NO donor, GTN (0.5 mM) or (C) GTN in the presence of either L-arginine, BEC or tempol. Superoxide levels were measured using a lucigenin-enhanced chemiluminescence assay in aorta from (D) WT and (E) argII –/– mice after in vivo treatment with GTN or vehicle for 3 days. Data are presented as mean ± SEM, n= 5–10. Data analysis used either (A, C) one-way anova with Bonferroni's post hoc analysis or (B, D, E) Student's t-test. *P < 0.05, ***P < 0.001, significantly different from (A, B, D) control or (C) from GTN alone.
ROS production in aorta from WT mice treated with GTN in vivo was significantly higher compared with vessels from vehicle-treated WT mice (Figure 3D). ROS production in aorta from argII –/– mice treated with GTN in vivo was not different from vehicle-treated argII –/– mice nor WT mice (Figure 3E).
GTN induces eNOS uncoupling
HUVECs, tolerant to GTN, showed a significant increase in eNOS present in the monomeric compared with dimeric state when compared with control non-tolerant cells (Figure 4A). Similarly, WT aorta tolerant to GTN show a trend towards an increase in monomer : dimer ratio (Figure 4C), whereas GTN and vehicle-treated argII –/– mice were not different (Figure 4C), but significantly lower than that observed in WT aorta tolerant to GTN.
Figure 4.
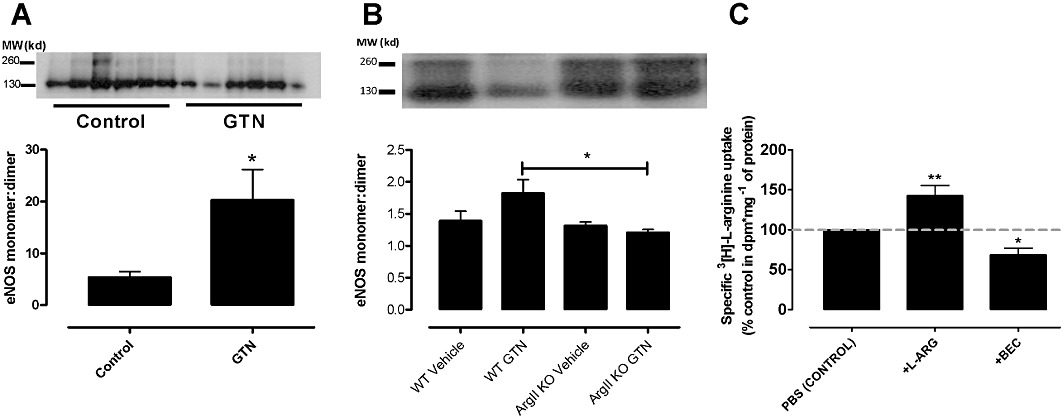
Ratios of eNOS dimer: monomer in tolerant and non-tolerant HUVECS (A) and aorta (B) was assessed by Western blotting and the densitometric ratio of eNOS monomer and dimer expression were normalized to β-tubulin expression. Data are presented as mean ± SEM, n= 3–6. *P < 0.05, significantly different from control; Student's t-test. (C) Basal levels of L-arginine uptake were measured in EA.hy926 cells receiving either no treatment (control) or pre-treatment with L-arginine (L-arg, 200 µM) or the arginase inhibitor, BEC (100 µM). Data are presented as % control uptake (mean ± SEM; n= 5–6 experiments, each performed in triplicate). *P < 0.05; **P < 0.01, significantly different from control; one-way anova with Bonferroni's post hoc analysis.
L-arginine uptake is increased with L-arginine and reduced by arginase inhibition
Using radioactively labelled L-arginine, exogenous L-arginine significantly increased L-arginine uptake into endothelial cells (Figure 4B) whereas BEC significantly reduced basal L-arginine uptake.
Arginase activity is increased in WT aorta after in vivo induction of nitrate tolerance
The arginase activity of aorta from tolerant WT mice was increased by 330% above that observed in non-tolerant WT aorta (Figure 5A). In contrast there was no significant increase in arginase activity in GTN-treated aorta from argII –/– mice compared with vehicle-treated aorta from the same strain (Figure 5B), nor was there any significant differences in aortic arginase activity between vehicle-treated WT and argII –/– mice (Figure 5). BEC (100 µM) significantly reduced arginase activity in WT vehicle-treated aorta (control = 7095 ± 1208 vs. +BEC = 0 ± 0 pmol·mg−1·min−1; n= 3–6; P < 0.05) suggesting specificity for arginase.
Figure 5.
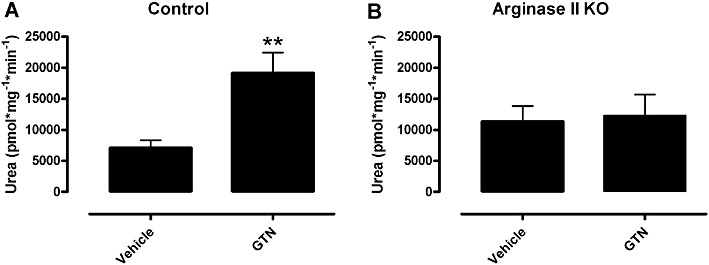
Arginase activity levels were measured in aorta of (A) WT and (B) argII –/– mice after in vivo treatment with GTN or vehicle for 3 days. Data are presented as mean ± SEM, n= 5–7. **P < 0.01, significantly different from vehicle; Student's t-test.
Discussion and conclusions
Our major new finding is that inhibition of arginase, and specifically of arginase II, prevents nitrate tolerance. We showed that increased arginase activity together with excessive ROS production and uncoupled eNOS was induced by repeated exposure to nitrates and propose that arginase inhibition prevents nitrate tolerance by reducing ROS production possibly through increasing intracellular L-arginine (Chicoine et al., 2004; White et al., 2006). Our finding is consistent with previous work and is the first to show that arginase inhibition has the beneficial, functional result of preventing nitrate tolerance.
In the current study we demonstrated a reduction in responsiveness upon repeated application of the organic nitrate GTN in an in vitro model of tolerance. Importantly, pharmacological blockade of arginase using the non-specific arginase inhibitor BEC prevented tolerance to GTN. In endothelial cells, inhibition of arginase stimulates NO production (Chicoine et al., 2004), whereas overexpression of arginase I or II decreases intracellular L-arginine concentrations and suppresses NO synthesis (Li et al., 2001). It has recently been proposed that arginase II is confined to the mitochondria where it regulates eNOS and therefore NO bioavailability (Lim et al., 2007). Although eNOS is predominantly localized to the caveolae, microdomains located in the plasma membrane, internalization of eNOS from the caveolae to the cytoplasm is now thought to regulate its function. Indeed, in HUVECs eNOS has been reported to be co-localized with the mitochondria (Gao et al., 2004), rendering a direct modulation of eNOS via arginase likely. Of interest in this context, and particularly as the arginase II isoform is located in the mitochondria, is that mitochondrial de-nitrification abnormalities and increased mitochondrial ROS have previously been identified in previous studies of nitrate tolerance (Chen et al., 2002; Sydow et al., 2004) suggesting potential advantages of arginase inhibition, related to cellular compartmentalization.
As such we examined whether arginase II inhibition may be particularly effective in preventing nitrate tolerance. As there are no selective inhibitors of the different enzyme isoforms, we also studied nitrate tolerance in argII –/– mice. Nitrate tolerance was not observed in aortae from argII –/– mice. We thus speculate that inhibition of arginase II may be particularly effective in the prevention of nitrate tolerance, due at least in part to its subcellular localization concurrently enabling mitochondrial eNOS stabilization as well as preventing mitochondrial ROS production, which has a key role in the de-nitrification process.
To confirm the key finding that elimination of arginase II prevents nitrate tolerance and as in vitro induction of tolerance has been reported to lack certain physiological features such as the activation of the renin-angiotensin system, these studies were repeated in vivo. Using the commonly reported model of inducing tolerance by repeated in vivo application of GTN (Wang et al., 2002) we observed a shift to the right of the concentration response curve to GTN of similar magnitude to that reported in mice previously (Wang et al., 2002). Further, we confirmed that elimination of arginase II not only prevented tolerance to GTN but also to DEA/NO indicating that cross-tolerance, the phenomenon of tolerance to other nitrovasodilators, did not develop in the absence of arginase.
Under conditions of reduced L-arginine or co-factors such as tetrahydrobiopterin, eNOS is ‘uncoupled’ from its dimeric state and, rather than producing NO, the superoxide anion •O2- is formed (Vasquez-Vivar et al., 1998; Ozaki et al., 2002; Münzel et al., 2005b). Increased eNOS expression and uncoupling of eNOS have been reported to contribute to nitrate tolerance in several studies (Abou-Mohamed et al., 2000; Kaesemeyer et al., 2000; Münzel et al., 2000; Gori et al., 2001a,b) and although inhibition of eNOS has been shown to decrease ROS (Münzel et al., 2000), a definitive role is still somewhat controversial. As our findings were consistent with the hypothesis (Caramori et al., 1998; Gori et al., 2001b) that GTN uncouples NOS causing an increase in ROS production and that arginase activity appears to further contribute to the phenomenon, the monomer : dimer ratio of eNOS and ROS production were determined in the setting of tolerance. Similar to previous reports (Kaesemeyer et al., 2000; Schmidt et al., 2010), we also showed that GTN uncoupled eNOS and increased ROS production in cultured endothelial cells and in aortae made tolerant in vivo. In addition, this increase in ROS was prevented by treatment with the arginase inhibitor BEC and L-arginine supplementation. ROS was increased in vessels from WT mice treated with repeated doses of GTN over 3 days but was not increased in vessels from arg II –/– mice, consistent with studies by others showing a reduction in GTN tolerance in the presence of an antioxidant (Münzel et al., 2000; Abou-Mohamed et al. 2004). It has been shown that in vitro and in vivo tolerances to GTN have different underlying mechanisms, where only in vivo tolerant aorta display increased production of superoxide (Münzel et al., 1999). In the current paper, the concentration of GTN used in the in vitro cell experiments, although similar to those previously reported by Dikalov et al. 1998, were higher than those used in the vascular reactivity studies. Similarly incubation periods differed under the two conditions. Although providing some mechanistic insight, direct comparison between these studies thus need to be viewed with some caution.
In our study, exogenous L-arginine supplementation increased cellular L-arginine uptake. This is as expected as one of the main factors in the regulation of L-arginine transport is intracellular substrate availability. In addition BEC reduced L-arginine uptake in endothelial cells. Although BEC can be postulated to increase intracellular L-arginine levels, whether this increase was sufficient to saturate NOS activity is not known. An alternative postulate might be that BEC has direct interaction with cationic amino acid transporters as has been reported for NOS inhibitors (Bogle et al., 1992).
Finally, the arginase activity in WT and argII –/– mice treated with GTN for 3 days was investigated. The aorta from GTN tolerant mice had significantly increased arginase activity. However, arginase activity in argII –/– mice (presumably due to arginase I) was not increased in aortic homogenates from mice treated with GTN, providing further evidence that arginase II was the isoform involved in tolerance to GTN. Increased arginase activity has been reported in many vascular pathologies involving endothelial dysfunction including atherosclerosis, hypertension, diabetes and ageing (Berkowitz et al., 2003; Johnson et al., 2005; Romero et al., 2008; Ryoo et al., 2008). Further, inhibition of arginase has been shown to be highly beneficial in treating in vitro endothelial dysfunction (Huynh et al., 2009) and can also restore NOS coupling and reduce the production of ROS from uncoupled eNOS (Ryoo et al., 2008; Kim et al., 2009; Zhang et al., 2009). Increased mitochondrial and NADPH oxidase produced ROS have been consistently reported in tolerant vessels (Münzel et al., 1995; Sydow et al., 2004; Szöcs et al., 2007) and this is also true in disease states where there is increased arginase activity (Romero et al., 2008; Ryoo et al., 2008). Indeed, ROS can mediate the induction of arginase in the vasculature after only 1 h of treatment (Thengchaisri et al., 2006) and reduce L-arginine levels (Gao et al., 2007) suggesting that arginase may be an early target of increased ROS. However, NO has also been shown to activate arginase by the nitrosylation of Cys303, stabilizing the arginase trimer and increasing its activity (Santhanam et al., 2007) and subsequently leading to a decrease in L-arginine, the uncoupling of eNOS and increased ROS production. From the current studies, it is not possible to explain exactly how arginase activity is increased in tolerant vessels, although such an increase is clearly demonstrable.
In summary, we confirm the hypothesis that nitrate tolerance is at least in part due to increased ROS production. It is likely that this occurs due to increased arginase activity limiting cellular L-arginine and causing an uncoupling of eNOS. We show for the first time that arginase, and more specifically arginase II, can influence nitrate tolerance and that inhibition of this enzyme increases L-arginine levels, diminishes ROS production and ameliorates tolerance. As such we suggest that arginase inhibition should be investigated for possible prevention of nitrate tolerance in man.
Acknowledgments
The authors would like to gratefully acknowledge and thank Emma Harris and Margaret Vincent for their technical assistance during this study. This work was supported by a grant from the National Health and Medical Research Council of Australia (52664) and supported in part by the Victorian Government's OIS Program.
Glossary
- ALDH-2
aldehyde dehydrogenase 2
- argII
–/–, arginase II knockout
- BEC
(s)-(2-boronethyl)-L-cysteine HCl
- DEA/NO
diethylamine NONOate
- eNOS
endothelial NOS
- GTN
glyceryl trinitrate
- HUVEC
human umbilical vein endothelial cell
- KPSS
potassium depolarizing solution
- ROS
reactive oxygen species
Conflict of interest
None declared.
References
- Abou-Mohamed G, Kaesemeyer WH, Caldwell RB, Caldwell RW. Role of L-arginine in the vascular actions and development of tolerance to nitroglycerin. Br J Pharmacol. 2000;130:211–218. doi: 10.1038/sj.bjp.0703293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abou-Mohamed G, Johnson JA, Jin L, El-Remessy AD, Do K, Kaesemeyer WH, et al. Roles of superoxide, peroxynitrite, and protein kinase C in the development of tolerance to nitroglycerin. J Pharmacol Exp Ther. 2004;308:289–299. doi: 10.1124/jpet.103.056119. [DOI] [PubMed] [Google Scholar]
- Bachetti T, Comini L, Francolini G, Bastianon D, Valetti B, Cadei M, et al. Arginase pathway in human endothelial cells in pathophysiological conditions. J Mol Cell Cardiol. 2004;37:515–523. doi: 10.1016/j.yjmcc.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- Bogle RG, Moncada S, Pearson JD, Mann GE. Identification of inhibitors of nitric oxide synthase that do not interact with the endothelial cell L-arginine transporter. Br J Pharmacol. 1992;105:768–770. doi: 10.1111/j.1476-5381.1992.tb09053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramori PR, Adelman AG, Azevedo ER, Newton GE, Parker AB, Parker JD. Therapy with nitroglycerin increases coronary vasoconstriction in response to acetylcholine. J Am Coll Cardiol. 1998;32:1969–1974. doi: 10.1016/s0735-1097(98)00456-2. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicoine LG, Paffett ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L60–L68. doi: 10.1152/ajplung.00194.2003. [DOI] [PubMed] [Google Scholar]
- Chin-Dusting JP, Ahlers BA, Kaye DM, Kelly JJ, Whitworth JA. L-arginine transport in humans with cortisol-induced hypertension. Hypertension. 2003;41:1336–1340. doi: 10.1161/01.HYP.0000070024.59313.AE. [DOI] [PubMed] [Google Scholar]
- Corraliza IM, Campo ML, Soler G, Modolell M. Determination of arginase activity in macrophages: a micromethod. J Immunol Methods. 1994;174:231–235. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Daiber A, Oelze M, August M, Wendt M, Sydow K, Wieboldt H, et al. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue L-012. Free Radic Res. 2004;38:259–269. doi: 10.1080/10715760410001659773. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Fink B, Skatchkov M, Stalleicken D, Bassenge E. Formation of reactive oxygen species by pentaerithrityltetranitrate and glyceryl trinitrate in vitro and development of nitrate tolerance. J Pharmacol Exp Ther. 1998;286:938–944. [PubMed] [Google Scholar]
- Gao S, Chen J, Brodsky SV, Huang H, Adler S, Lee JH, et al. Docking of endothelial nitric oxide synthase (eNOS) to the mitochondrial outer membrane: a pentabasic amino acid sequence in the autoinhibitory domain of eNOS targets a proteinase K-cleavable peptide on the cytoplasmic face of mitochondria. J Biol Chem. 2004;279:15968–15974. doi: 10.1074/jbc.M308504200. [DOI] [PubMed] [Google Scholar]
- Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, et al. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2007;27:1269–1275. doi: 10.1161/ATVBAHA.107.142521. [DOI] [PubMed] [Google Scholar]
- Gerzanich V, Ivanov A, Ivanova S, Yang JB, Zhou H, Dong Y, et al. Alternative splicing of cGMP-dependent protein kinase I in angiotensin-hypertension: novel mechanism for nitrate tolerance in vascular smooth muscle. Circ Res. 2003;93:805–812. doi: 10.1161/01.RES.0000097872.69043.A0. [DOI] [PubMed] [Google Scholar]
- Gori T, Burstein JM, Ahmed S, Miner SE, Al-Hesayen A, Kelly S, et al. Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: a human in vivo study. Circulation. 2001a;104:1119–1123. doi: 10.1161/hc3501.095358. [DOI] [PubMed] [Google Scholar]
- Gori T, Mak SS, Kelly S, Parker JD. Evidence supporting abnormalities in nitric oxide synthase function induced by nitroglycerin in humans. J Am Coll Cardiol. 2001b;38:1096–1101. doi: 10.1016/s0735-1097(01)01510-8. [DOI] [PubMed] [Google Scholar]
- Huynh NN, Harris EE, Chin-Dusting JFP, Andrews KL. The vascular effects of different arginase inhibitors in rat isolated aorta and mesenteric arteries. Br J Pharmacol. 2009;156:84–93. doi: 10.1111/j.1476-5381.2008.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FK, Johnson RA, Peyton KJ, Durante W. Arginase inhibition restores arteriolar endothelial function in Dahl rats with salt-induced hypertension. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1057–R1062. doi: 10.1152/ajpregu.00758.2004. [DOI] [PubMed] [Google Scholar]
- Kaesemeyer WH, Ogonowski AA, Jin L, Caldwell RB, Caldwell RW. Endothelial nitric oxide synthase is a site of superoxide synthesis in endothelial cells treated with glyceryl trinitrate. Br J Pharmacol. 2000;131:1019–1023. doi: 10.1038/sj.bjp.0703665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol. 2009;107:1249–1257. doi: 10.1152/japplphysiol.91393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Sudhir K, Jones M, Simpson E, Jefferis AM, Chin-Dusting JP. Impaired acetylcholine-induced release of nitric oxide in the aorta of male aromatase-knockout mice: regulation of nitric oxide production by endogenous sex hormones in males. Circ Res. 2003;93:1267–1271. doi: 10.1161/01.RES.0000103172.98986.25. [DOI] [PubMed] [Google Scholar]
- Klatt P, Schmidt K, Werner ER, Mayer B. Determination of nitric oxide synthase cofactors: heme, FAD, FMN, and tetrahydrobiopterin. Methods Enzymol. 1996;268:358–365. doi: 10.1016/s0076-6879(96)68038-0. [DOI] [PubMed] [Google Scholar]
- Li H, Meininger CJ, Hawker JR, Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab. 2001;280:E75–E82. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- Lim HK, Lim HK, Ryoo S, Benjo A, Shuleri K, Miriel V, et al. Mitochondrial arginase II constrains endothelial NOS-3 activity. Am J Physiol Heart Circ Physiol. 2007;293:H3317–H3324. doi: 10.1152/ajpheart.00700.2007. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Ming X-F, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T, Sayegh H, Freeman BA, Tarpey MM, Harrison DG. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T, Hink U, Yigit H, Macharzina R, Harrison DG, Mulsch A. Role of superoxide dismutase in in vivo and in vitro nitrate tolerance. Br J Pharmacol. 1999;127:1224–1230. doi: 10.1038/sj.bjp.0702622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münzel T, Li H, Mollnau H, Hink U, Matheis E, Hartmann M, et al. Effects of long-term nitroglycerin treatment on endothelial nitric oxide synthase (NOS III) gene expression, NOS III-mediated superoxide production, and vascular NO bioavailability. Circ Res. 2000;86:E7–E12. doi: 10.1161/01.res.86.1.e7. [DOI] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Mülsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005a;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol. 2005b;25:1551–1557. doi: 10.1161/01.ATV.0000168896.64927.bb. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Kawashima S, Yamashita T, Hirase T, Namiki M, Inoue N, et al. Overexpression of endothelial nitric oxide synthase accelerates atherosclerotic lesion formation in apoE-deficient mice. J Clin Invest. 2002;110:331–340. doi: 10.1172/JCI15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell MM, Chin-Dusting JP, Starr J, Kaye DM. In vivo and in vitro evidence for ACh-stimulated L-arginine uptake. Am J Physiol Heart Circ Physiol. 2004;287:H395–H400. doi: 10.1152/ajpheart.01094.2003. [DOI] [PubMed] [Google Scholar]
- Ritchie RH, Quinn JM, Cao AH, Drummond GR, Kaye DM, Favaloro JM, et al. The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo. J Mol Cell Cardiol. 2007;42:1119–1128. doi: 10.1016/j.yjmcc.2007.03.900. [DOI] [PubMed] [Google Scholar]
- Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res. 2008;102:95–102. doi: 10.1161/CIRCRESAHA.107.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, et al. Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res. 2008;102:923–932. doi: 10.1161/CIRCRESAHA.107.169573. [DOI] [PubMed] [Google Scholar]
- Sage PR, de la Lande IS, Stafford I, Bennett CL, Phillipov G, Stubberfield J, et al. Nitroglycerin tolerance in human vessels: evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Rehn M, Stessel H, Wolkart G, Mayer B. Evidence against tetrahydrobiopterin depletion of vascular tissue exposed to nitric oxide/superoxide or nitroglycerin. Free Radic Biol Med. 2010;48:145–152. doi: 10.1016/j.freeradbiomed.2009.10.038. [DOI] [PubMed] [Google Scholar]
- Shi O, Morris SM, Jr, Zoghbi H, Porter CW, O'Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol. 2001;21:811–813. doi: 10.1128/MCB.21.3.811-813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn HY, Gloe T, Keller M, Schoenafinger K, Pohl U. Sensitive superoxide detection in vascular cells by the new chemiluminescence dye L-012. J Vasc Res. 1999;36:456–464. doi: 10.1159/000025688. [DOI] [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöcs K, Lassègue B, Wenzel P, Wendt M, Daiber A, Oelze M, et al. Increased superoxide production in nitrate tolerance is associated with NAD(P)H oxidase and aldehyde dehydrogenase 2 downregulation. J Mol Cell Cardiol. 2007;42:1111–1118. doi: 10.1016/j.yjmcc.2007.03.904. [DOI] [PubMed] [Google Scholar]
- Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, et al. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang EQ, Lee W-I, Fung H-L. Lack of critical involvement of endothelial nitric oxide synthase in vascular nitrate tolerance in mice. Br J Pharmacol. 2002;135:299–302. doi: 10.1038/sj.bjp.0704532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AR, Ryoo S, Li D, Champion HC, Steppan J, Wang D, et al. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension. 2006;47:245–251. doi: 10.1161/01.HYP.0000198543.34502.d7. [DOI] [PubMed] [Google Scholar]
- Yang Y-M, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297:H1829–H1836. doi: 10.1152/ajpheart.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, et al. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53:532–538. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WZ, Venardos K, Chin-Dusting J, Kaye DM. Adverse effects of cigarette smoke on NO bioavailability: role of arginine metabolism and oxidative stress. Hypertension. 2006;48:278–285. doi: 10.1161/01.HYP.0000231509.27406.42. [DOI] [PubMed] [Google Scholar]