

Abstract
BACKGROUND AND PURPOSE
The lung adenocarcinoma cell line, A549, undergoes epithelial-mesenchymal cell transition (EMT) in response to TGF-β. Glucocorticoids do not prevent the EMT response, but TGF-β induced resistance to the cytokine-regulatory action of glucocorticoids. We sought to characterize the impairment of glucocorticoid response in A549 cells.
EXPERIMENTAL APPROACH
A549 cells were exposed to TGF-β for up to 96 h before glucocorticoid treatment and challenge with IL-1α to assess glucocorticoid regulation of IL-6 and CXCL8 production. Nuclear localization of the glucocorticoid receptor α (GRα) was ascertained by immunofluorescence and Western blotting. Transactivation of the glucocorticoid response element (GRE) was measured with a transfected GRE-secreted human placental alkaline phosphatase reporter.
KEY RESULTS
TGF-β (40–400 pM) reduced the maximum inhibitory effect of dexamethasone on IL-1α-induced IL-6 and CXCL8 production. The impaired glucocorticoid response was detected with 4 h of TGF-β (40 pM) exposure (and 4 h IL-1α to induce CXCL8 expression) and therefore was not secondary to EMT, a process that requires longer incubation periods and higher concentrations of TGF-β. TGF-β also impaired dexamethasone regulation of granulocyte-macrophage colony-stimulating factor in thrombin-stimulated BEAS-2B epithelial cells. Impaired regulation of CXCL8 was associated with markedly reduced GRE transactivation and reduced induction of mRNA for IκBα, the glucocorticoid-inducible leucine zipper and the epithelial sodium channel (SCNN1A). The expression, cellular levels and nuclear localization of GRα were reduced by TGF-β.
CONCLUSIONS AND IMPLICATIONS
We have identified mechanisms underlying the impairment of responses to glucocorticoids by TGF-β in the A549 and BEAS-2B cell lines.
Keywords: steroid resistance, TGF-β, epithelial cells, lung cancer, ERK, p38MAPK, epithelial mesenchymal transition (EMT), inflammation, cytokines
Introduction
Glucocorticoids are among the most commonly used of all prescription medicines (Gross and Cidlowski, 2008). The treatment with glucocorticoids of many chronic inflammatory conditions is remarkably successful. However, in a number of chronic inflammatory conditions, and notably in severe respiratory disease, there are patient phenotypes in which glucocorticoids are not sufficient to control disease (Adcock and Barnes, 2008). Glucocorticoids are also widely used in the treatment of solid tumours. In most circumstances, glucocorticoid treatment has palliative effects. However, in several conditions, there is an established or intended adjuvant chemotherapy effect, shown both in vitro studies and by observations of synergy with conventional chemotherapy [see Keith (2008)]. For example, the use of glucocorticoids has a well-established adjuvant chemotherapy effect in the treatment of lymphoma, although it is still uncertain whether the mechanism is explained by glucocorticoid-induced apoptosis. Emergent glucocorticoid resistance remains a limitation of glucocorticoid use in lymphoma (Smith and Cidlowski, 2010).
There is increasing evidence that chronic inflammatory diseases and certain types of tumour are accompanied by a pathology that includes the process of epithelial mesenchymal cell transition (EMT). This differentiation process is a normal feature of development in the lung and elsewhere. The formation of tubes by epithelium is followed by the generation of supporting mesenchymal cells, smooth muscle and fibroblasts through the EMT process (Lee et al., 2006). In epithelial tumours, EMT is thought to play a key role in invasiveness and metastasis, as the transition between epithelial and fibroblast-like phenotypes is accompanied by a loss of cell polarity, increased production and release of matrix metalloproteases and acquisition of a migratory phenotype (Radisky et al., 2007). Thus, EMT has been implicated in breast and lung cancers and has been modelled using cell lines such as the lung adenocarcinoma A549 cell line (Kasai et al., 2005).
Given the importance of EMT to tumour growth and spread, we set out to examine the glucocorticoid sensitivity of the EMT process itself. It has recently been shown that EMT responses in A549 cells are not prevented by glucocorticoid treatment (Doerner and Zuraw, 2009). In the course of studies examining the EMT sensitivity to glucocorticoids, we unexpectedly found that TGF-β, a prototypical stimulus for induction of EMT, was able to induce resistance to the cytokine-regulatory actions of glucocorticoids. Here, we describe the characteristics of TGF-β-induced glucocorticoid resistance, showing that it involves impaired nuclear translocation of the glucocorticoid receptor α (GRα), which is the transactivationally active and more abundant splice variant of GR and a failure to activate the glucocorticoid-mediated glucocorticoid response element (GRE), with consequent loss of glucocorticoid-dependent gene regulation.
Methods
Cell culture and treatments
Human A549 lung adenocarcinoma cells (ATCC, Manassas, VA, USA) were grown in phenol red-free Dulbecco's modified Eagle's media (DMEM) containing 5% vv−1 heat-inactivated fetal calf serum (FCS), 15 mM HEPES, 0.2% vv−1 sodium bicarbonate, 2 mM L-glutamine, 1% vv−1 non-essential amino acids, 1% vv−1 sodium pyruvate, 5 IU·mL−1 penicillin and 50 µg·mL−1 streptomycin and maintained at 37°C in a humidified 5% CO2/95% air atmosphere. Unless otherwise stated, A549 cells were seeded in tissue culture plates or chamber slides in growth medium as described above and allowed to adhere overnight. BEAS-2B cells (from ATCC, Manassas, VA, USA) were cultured in LHC-9 media containing 2% vv−1 heat-inactivated FCS, 15 mM HEPES, 0.2% vv−1 sodium bicarbonate, 2 mM L-glutamine, 50 IU·mL−1 penicillin and 50 µg·mL−1 streptomycin and maintained at 37°C in a humidified 5% CO2/95% air atmosphere. For experiments, the cells were seeded at 25 000 cells/well in 24 well plates and incubated in serum-free medium (phenol red-free Roswell Park Memorial Institute (RPMI) containing 15 mM HEPES, 0.2% vv−1 sodium bicarbonate, 50 IU·mL−1 penicillin, 50 µg·mL−1 streptomycin, 0.25% wv−1 BSA, 2 mM L-glutamine, 1% v/v sodium pyruvate and 1% v/v non-essential amino acids) for 24 h prior to treatment with TGF-β (40 pM) for 24 h. The cells were then treated with dexamethasone. IL-1α (1 ng·mL−1) was added 30 min after the addition of dexamethasone to stimulate the production of cytokines when required. Cells were supplemented with Monomed A (1% vv−1) 24 h after culture in serum-free medium.
Assessment of cell morphology
A549 seeded at 50 000–60 000 cells per well in a 6-well culture plate were allowed to attach and grow for 48 h. The medium was changed to a serum-free DMEM, containing 0.25% wv−1 BSA for 24 h. Cells were then treated with the glucocorticoid, Dexamethasone (100 nM or 1 µM), for 30 min prior to addition of TGF-β (4–400 pM). Cells were exposed to TGF-β for between 16 and 96 h. The cell morphology was then assessed by phase microscopy (IATIA system, IATIA Vision Sciences, Melbourne, VIC, Australia) (Curl et al., 2004) or by live cell imaging (Leica DMI6000B, Leica Microsystems GmbH, Wetzlar, Germany).
Subcellular fractionation
Cells were lysed in lysis buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA and 1 mM EGTA) containing phosphatase inhibitor cocktail, protease inhibitor cocktail and 1 mM dithiothreitol (DTT). The lysates were passed 10 times through a 25 gauge needle and then centrifuged at 720×g for 5 min. The nuclear pellet was then washed with lysis buffer, passed 10 times through a 25 gauge needle and centrifuged again at 720×g for 10 min. The nuclear pellet was then suspended in lysis buffer containing 10% vv−1 glycerol and 0.1% wv−1 sodium dodecyl sulphate and sonicated for 3 s. To obtain the cytoplasmic fraction, the supernatants from the first centrifugation were re-centrifuged at 10 000×g for 10 min and the supernatants were transferred into fresh tubes and centrifuged again at 100 000×g for 1 h.
Western blot analysis
Cells were lysed in lysis buffer (100 mM NaCl; 10 mM Tris–HCL (pH 7.4); 1 mM EDTA; 1% vv−1 TritonX-100) containing phosphatase inhibitor cocktail (#P5726 Sigma-Aldrich, Castle Hill, Australia) and protease inhibitor cocktail (#P1860 Sigma-Aldrich) for 20 min on ice. Extracts were collected by centrifugation at 10 000×g for 10 min, and protein concentration was determined by Bradford assay. Samples were resolved by SDS-PAGE on 12.5% polyacrylamide gel and transferred onto nitrocellulose membranes. Several markers of TGF-β receptor activation were examined, including the phosphorylation of the MAPK, ERK (anti-phospho-ERK1/2, ERK 1/2 rabbit polyclonal: Santa Cruz Biotechnology, Santa Cruz, CA, USA) and p38 MAPK (phospho-p38 MAPK rabbit polyclonal: Cell Signalling, Danvers, MA, USA; p38 MAPK rabbit polyclonal: Santa Cruz Biotechnology), and the phosphorylation of Smad2 (phospho-smad2 rabbit polyclonal, smad2/3 rabbit polyclonal: Cell Signalling). α-Smooth muscle actin (α-SMA) (α-SMA mouse monoclonal: Sigma-Aldrich) protein levels and total cellular levels of GRα rabbit polyclonal: Santa Cruz Biotechnology] were also assessed in separate experiments. Signals were detected using horseradish peroxidase-conjugated secondary antibodies and Amersham™ ECL chemiluminescence substrate (GE Healthcare, NSW, Australia). The levels of total protein and β-actin (β-actin mouse monoclonal: Abcam, Cambridge, UK) were used as housekeeping controls for protein loading. Images of Western blots were captured using an Image Quant 350 Digital Imaging System (GE Healthcare) and densitometry was carried out using ImageJ (v1.44, National Institute of Health, Bethesda, MD, USA).
Immunofluorescence
Cells seeded at 30 000 cells per chamber in an eight-chamber cell culture slide (Nunc, Roskilde, the Netherlands) were allowed to attach for 24 h then starved in DMEM containing 0.25% BSA for a further 24 h. Cells were then treated with TGF-β (40 pM) for 24 h before the addition of dexamethasone (10 nM) for 2 h. Cells were fixed in 10% vv−1 neutral buffered formalin for 5 min, washed in PBS and incubated with the primary antibody against GRα (GR rabbit polyclonal #sc-1003: Santa Cruz Biotechnology) overnight at 4°C. Slides were washed twice with PBS and incubated with FITC-conjugated secondary antibody for 1 h at room temperature, prior to nuclear staining using 4′-6-diamidino-2-phenylindole (DAPI) (Santa Cruz Biotechnology) and coverslipping using fluorescence anti-fade mounting medium (DAKO, VIC, Australia). Fluorescence microscopy (Leica DM6000B) and confocal microscopy (Zeiss Meta, North Ryde, Australia) were used to confirm co-localization of DAPI and GRα immunoreactivity.
Cell viability
At the conclusion of different incubation periods, as described in the text, cell viability was assessed using flow cytometry and Trypan blue exclusion methodologies. Briefly, wells were inspected for the presence of detached cells, and cell monolayers were washed with 1 mL PBS, then harvested with trypsin prior to staining for apoptotic cells (annexin-V-FITC) or cells having undergone cytolysis (propidium iodide) and analysed by flow cytometry as described in our previous studies (Khau et al., 2011), or incubated with Trypan blue (0.2% wv−1) for 5 min before counting stained and unstained cells.
Cytokine detection by elisa
Supernatants were assayed for IL-8 (CXCL8), granulocyte macrophage colony-stimulating factor (GM-CSF) and IL-6 according to manufacturer's instructions (Pierce Endogen, Rockford, IL, USA, for CXCL8; Becton Dickinson, Heidelberg, Germany, for IL-6 and GM-CSF). Briefly, plates were coated with capture antibody overnight, washed several times in PBS (containing 0.1% vv−1 Tween20) and blocked with 4% FCS in PBS for 1 h. Appropriate dilutions of supernatants and standards were incubated with biotin-labelled detecting antibody at ambient temperature for 3 h with detection by horseradish peroxidase-conjugated streptavidin complex and visualization using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. The reaction was stopped by addition of 0.18 M sulphuric acid and the absorbance was measured at 450 nm.
RNA extraction and RT-qPCR
Total RNA was purified from cells using Qiagen RNeasy miniprep kit (Qiagen, Doncaster, VIC, Australia), following manufacturer's instructions. The culture medium from the 24-well plate was removed and the cells were washed once with PBS and lysed with 0.3 mL RLT buffer containing 1% vv−1β-mercaptoethanol. The RLT lysates containing 0.3 mL of 70% vv−1 ethanol in diethylpyrocarbonate water were stored at −80°C until purification. Total RNA was eluted into 50 µL RNase-free water. RNA (2.5 µL) was reverse transcribed with 0.25 µL of 0.15 g·L−1 random primer, 0.25 µL of 10 mM dNTP mix, 0.125 µL of RNase inhibitor (RNaseOUT), 0.125 µL Superscript III (SSIIIRT), 0.25 µL of 0.1 M DTT, 1 µL first strand buffer (250 mM, Tris-HCL; 375 mM KCL; 15 mM MgCl2 pH 8.3) (5x) (Applied Biosystems, Scoresby, VIC, Australia) in a final volume of 5 µL. Reverse transcription was carried out using the following thermal protocol: 0–5 min, 25°C; 5–60 min, 50°C; 60–75 min, 75°C. The resulting cDNA was diluted with 195 µL Milli-Q water (Merck, Millipore, Kilsythe, VIC, Australia) and stored at −20°C until used. Real-time PCR was performed in triplicate for each gene of interest in a 384-well plate using ABI Prism 7900HT sequence detection system (Applied Biosystems). Each 10 µL reaction consisted of 3 µL diluted cDNA, 5 µL Platinum SYBR Green qPCR Supermix-UDG (Invitrogen, Mulgrave, VIC, Australia) and 1.0 µM of each of the relevant forward and reverse primers (Table 1). Primer sequences were obtained either from published references, or designed using Primer Express software (Applied Biosystems) with mRNA sequences from the National Centre for Biotechnology Information (http://www.ncbi.nlm.nih.gov). The conditions for PCR amplification were: 50°C for 2 min then 95°C for 10 min followed by 40 cycles of 95°C for 0.15 min, and 60°C for 1 min. The threshold cycle determined for each gene was normalized against that obtained for 18S ribosomal RNA, which was measured as internal control.
Table 1.
Primer sequences for RT-PCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| 18s | CGCCGCTAGAGGTGAAAT | TCTTGGCAAATGCTTTCGCTC |
| CXCL8 | CTGGCCGTGGCTCTCTTG | CCTTGGCAAAACTGCACCTT |
| IL-6 | AGCTCTATCTCGCCTCCAGGA | CGCTTGTGGAGAAGGAGTTCA |
| IκBα | TACCAACTACAATGGCCACACG | TAGCCATGGATAGAGGCTAAGTGTAGA |
| GILZ | TCCTGTCTGAGCCCTGAAGAG | AGCCACTTACACCGCAGAAC |
| SCNN1a | AGCACAACCGCATGAAGAC | TGAGGTTGATGTTGAGGCTG |
| MKP1 | CCACAAGGCAGACATCAGCTC | TCTATGAAGTCAATGGCCTCGTT |
A549 cell transfection
A549 cells for transfection were seeded at a density of 90 000 cells/well in a 24 well plate in growth medium devoid of penicillin and streptomycin. After 24 h, the medium was replaced with 0.5 mL DMEM containing only HEPES and sodium bicarbonate, and cells were equilibrated for 1 h. The GRE-secreted human placental alkaline phosphatase (SEAP) reporter construct (Clontech, Mountain View, CA, USA) was incubated with pmax-green fluorescent protein (GFP) internal control reporter (all 150 ng·per well) and 1.0 µL Lipofectamine 2000 reagent in 100 µL·well−1 of OptiMEM for 20 min at room temperature. Cells were then transfected for 6 h before replacing transfection media with 1 mL DMEM containing 0.25% BSA and all other supplements. After 1 h, the cells were treated with 40 pM TGF-β and Monomed A (1% vv−1) and after 24 h were incubated in dexamethasone (30 nM)/vehicle following the usual protocol.
Supernatants were collected before treatment with dexamethasone, 24 and 48 h after incubation with IL-1α, and stored at −20°C for the assessment of gene activity by determining the amount of SEAP using a chemiluminescence kit (Roche Applied Science, NSW, Australia) with chemiluminescence measured using Topcount (Perkin Elmer, Glen Waverley, VIC, Australia). At the conclusion of each experiment, several fields of the cells in each well were imaged using Leica DMI6000B to assess transfection efficiency by GFP expression.
Statistical analyses
Data are presented as the mean ± SEM for n individual experiments. Each experiment was repeated on a minimum of three separate occasions. All data were statistically analysed using GraphPad Prism 5.0 (Graphpad, San Diego, CA, USA). In most cases, either one or two-way anova with repeated measures were used to analyse the data, and treatment groups were compared with Bonferroni's post hoc tests. A P-value of <0.05 was considered to be statistically significant.
Materials
The materials used in these experiments were from the following sources: DMEM, LHC-9, RPMI (Gibco/Invitrogen, Mulgrave, VIC, Australia); FCS, HEPES, sodium bicarbonate, L-glutamine, non-essential amino acids, sodium pyruvate, penicillin-streptomycin solution, bovine serum albumin, dexamethasone, IL-1α, Monomed A, trypsin, EDTA, EGTA, SDS, Tris-HCL, Annexin-V-FITC, PI, SB431542, budesonide, FITC-conjugated secondary anti-body (Sigma-Aldrich, Castle Hill, NSW, Australia); TGFβ (R&D systems, Minneapolis, MN, USA); IL-6, TMB substrate (BD biosciences, North Ryde, NSW, Australia); SB202190, SB203580, bFGF (Calbiochem/Merck, Kilsyth, VIC, Australia); U0126 (Tocris, Bristol, UK); glycerol (Ajax Finechem/Thermo Fisher Scientific, Scoresby, VIC, Australia); NaCl, MgCl2, Trypan blue, sucrose, KCl (BDH/Merck, Kilsyth, VIC, Australia); nitrocellulose membranes (Amersham/GE health care, Rydalmere, NSW, Australia); HRP-conjugated streptavidin (Thermo Scientific, Scoresby, VIC, Australia); 24-well cell culture plate (BD Biosciences, North Ryde, NSW, Australia); 6-well cell culture plate, 8-chamber cell culture slides (Nunc, Roskilde, the Netherlands).
Results
TGF-β induces glucocorticoid resistance
Cells incubated in the presence of TGF-β (4–400 pM) for 96 h acquired a mesenchymal phenotype, as shown by changes in morphology (Supporting Information Figure S1A), the acquisition of a proliferative response to basic fibroblast growth factor (bFGF (Supporting Information Table S1) and a number of gene expression changes characteristic of mesenchymal cells, including increased expression of smooth muscle α-actin (fold control: 1.46 ± 0.14, n = 5) and fibronectin (2.54 ± 0.68, n = 5). Dexamethasone (100 nM), added before TGF-β, had little effect on these gene changes (smooth muscle α-actin, 1.60 ± 0.23, n = 7 and fibronectin 2.80 ± 0.84, n = 5, respectively), consistent with recent reports indicating that EMT in A549 cells is not glucocorticoid-responsive (Doerner and Zuraw, 2009). The proliferative response to bFGF acquired following TGF-β exposure was not inhibited by dexamethasone, whereas the proliferative effect of FCS in cells naïve to TGF-β was reduced by dexamethasone (Supporting Information Table S1).
The effects of prolonged TGF-β exposure on CXCL8 levels were further examined using IL-1α as a stimulus, as bFGF only marginally increased (∼30%) levels of CXCL8. Dexamethasone (1–100 nM) concentration-dependently reduced the effects of IL-1α on CXCL8 generation with a maximum effect of approximately 80% inhibition. However, in cells exposed to TGF-β (400 pM) for 96 h, the maximum effect of dexamethasone was greatly reduced, although the location of the concentration–effect curve was not changed (Figure 1A). An examination of TGF-β (400 pM) exposure for durations of between 4 and 96 h revealed that the suppression was evident within 4 h and maximal at 24 h (Figure 1B), noting however that the cytokine response to IL-1α stimulation was measured after 24 h making the total incubation time with TGF-β 28 h when it was added 4 h before dexamethasone. TGF-β (24 h) also reduced the effect of dexamethasone (10 nM) on IL-6 levels (Figure 1C). These effects of TGF-β were concentration dependent (4–400 pM) (Figure 1C) and were greater when the TGF-β was present throughout the incubation period than when it was removed before dexamethasone/IL-1α exposure (data not shown). In order to establish whether the impairment of dexamethasone actions induced by TGF-β was a class effect of glucocorticoids, budesonide (0.1–100 nM) was tested in cells incubated in the presence or absence of TGF-β (40 pM) for 96 h. The TGF-β-induced reduction in the response to budesonide was similar to that observed for dexamethasone (Figure 1A cf. D).
Figure 1.
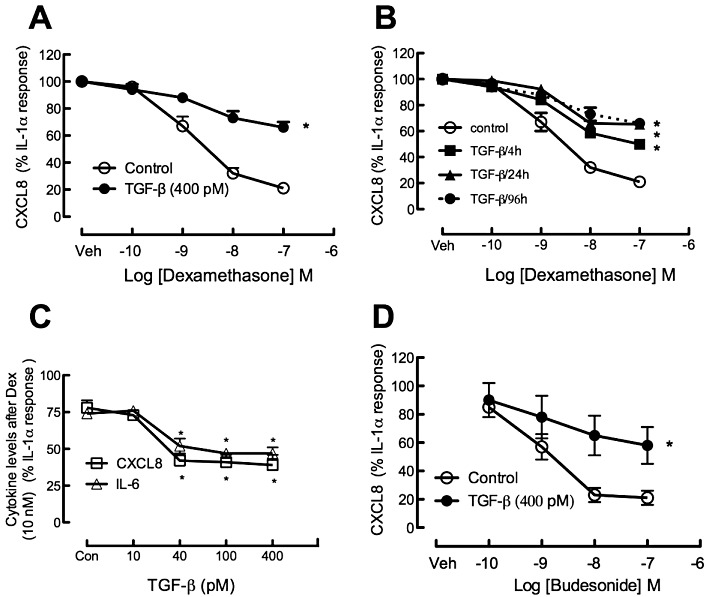
(A) Effect of TGF-β (400 pM) on dexamethasone (0.1–100 nM) regulation of IL-1α (1 ng·mL−1)-induced CXCL8 production by A549 cells. Cells were incubated with vehicle or TGF-β for 96 h, then incubated with dexamethasone (0.1–100 nM) or vehicle (Veh) for 30 min prior to addition of IL-1α. CXCL8 levels in the supernatant were determined after a further 48 h incubation. The data are presented as a percentage of the CXCL8 level in vehicle-treated cells incubated with IL-1α (80 ± 5 ng·mL−1) or the level in cells pre-incubated with TGF-β before IL-1α stimulation (31 ± 3 ng·mL−1) and are presented as the mean and SEM of seven independent experiments. (B) The effect of TGF-β (400 pM) over pre-incubation times from 4 h up to 96 h was investigated in experiments of otherwise identical design to those in A. (C) Dexamethasone (Dex; 10 nM) inhibition of IL-1α-induced CXCL8 and IL-6 under control conditions (Con) and in increasing concentrations of TGF-β (10–400 pM). (D) Effect of TGF-β on budesonide regulation of IL-1α-induced CXCL8 levels. *P < 0.05, control vs. TGF-β; anova.
Incubation with TGF-β for 96 h caused a substantial reduction in the amount of CXCL8 generated in response to IL-1α (IL-1α, 80 ± 5 ng·mL−1, vs. IL-1α + TGF-β (400 pM), 31 ± 3 ng·mL−1; n = 7 P < 0.05), but increased the amount of IL-6 [IL-1α, 3.1 ± 1.1, IL-1α + TGF-β (400 pM) 11.1 ± 1.5 ng·mL−1]. The results with CXCL8 raised the possibility that the decreased response to dexamethasone was simply a consequence of an already diminished CXCL8 production. To address this possibility, comparisons were made of the inhibitory effects of dexamethasone on concentrations of IL-1α (1.0 ng·mL−1) in the presence of TGF-β that were equi-effective at increasing CXCL8 levels, with those of IL-1α (0.1–0.3 ng·mL−1) in the absence of TGF-β. These experiments established that the diminished response to the glucocorticoid was not related to the lowered CXCL8 generation (Supporting Information Table S2). Furthermore, with 24 h exposure to TGF-β, the CXCL8 responses and, to a greater extent, the IL-6 responses to IL-1α were increased rather than decreased, but dexamethasone regulation was also reduced by TGF-β under these conditions (Supporting Information Table S3).
Analysis of CXCL8 mRNA levels indicated a similar pattern of response to combinations of TGF-β and IL-1α in the presence and absence of dexamethasone 10 nM as observed for CXCL8 protein levels (Table 2), suggesting that the interaction occurred at regulation of CXCL8 transcription, but the mechanism of mRNA regulation was not further investigated.
Table 2.
Effect of dexamethasone (100 nM) on CXCL8 mRNA in A549 cells with or without prior treatment with TGF-β after 4 h IL-1α incubation
| Control | TGF-β | |||
|---|---|---|---|---|
| Control | IL-1α | Control | IL-1α | |
| Control | ||||
| CXCL8 | 1.001 | 64.1 ± 23.2 | 0.9 ± 0.2 | 75.2 ± 9.9 |
| IL-6 | 1.00 | 19.4 ± 6.1 | 3.0 ± 1.1 | 119.8 ± 36.5 |
| Dexamethasone | ||||
| CXCL8 | 0.3 ± 0.0 | 16.9 ± 3.4* | 0.3 ± 0.1 | 47.2 ± 4.9* |
| IL-6 | 2.2 ± 0.5 | 8.8 ± 1.8 | 3.9 ± 0.9 | 81.0 ± 19.3 |
Table 1mRNA levels are normalized to 18sRNA and then related to the level under control conditions.
P < 0.05 dexamethasone vs. control.
We sought to establish whether the impairment in glucocorticoid actions was evident in the BEAS-2B cell line, a line that represents some of the characteristics of central airway epithelium. In experiments using thrombin (0.03 U·mL−1) as a stimulus and measuring GM-CSF as a cytokine response, a concentration-dependent decrease in GM-CSF levels was established in response to 3–100 nM dexamethasone (Figure 2). Incubation of BEAS-2B cells for 24 h before dexamethasone with TGF-β (40 pM) greatly reduced the regulatory effect of this steroid on GM-CSF production in response to thrombin. Further experiments in BEAS-2B cells indicated that IL-1α-induced GM-CSF regulation by dexamethasone was also impaired in cells pre-incubated in TGF-β (data not shown).
Figure 2.
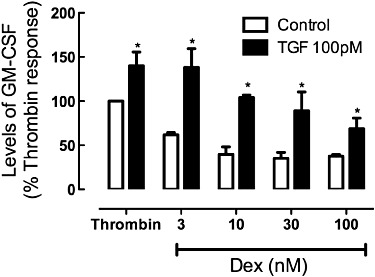
Effect of TGF-β on regulation of thrombin-induced GM-CSF levels by dexamethasone (Dex; 3–100 nM) in BEAS-2B cells. The cells were incubated in serum-free Roswell Park Memorial Institute for 24 h before addition of TGF-β (100 pM) and after a further 24 h dexamethasone was added 30 min before thrombin (0.03U mL−1). Levels of GM-CSF in the supernatant were measured by elisa and are presented as mean and SEM of the percentage of the level under control thrombin stimulation (0.96 ± 0.44 ng·mL−1, n = 3). *P < 0.05, TGF-β vs. control.
Potential relationship of TGF-β-induced glucocorticoid impairment to EMT
TGF-β-induced glucocorticoid insensitivity could be the result of an inherent resistance of the mesenchymal phenotype to regulation by glucocorticoids, or it may relate to phenotype-independent actions of TGF-β. The time course and the concentration dependence of TGF-β-induced EMT was compared with that of induction of dexamethasone-induced resistance in A549 cells. Using morphological criteria for EMT, it was established that the shape changes occurred after a delay of 24–36 h and that they required concentrations of more than 40 pM (Supporting Information Figure S1A,B). In contrast, TGF-β induced dexamethasone impairment with a 4 h pre-exposure to TGF-β (Figure 1B) and concentrations as low as 40 pM (Figure 1C), as compared to the 400 pM required for EMT-associated morphological changes (Supporting Information Figure S1B).
TGF-β-induced glucocorticoid impairment is associated with activin-like kinase 5 (ALK5)-induced impairment of GRE activation
The mechanism underlying the effect of TGF-β was examined using the selective ALK5 (TGFβRI kinase) inhibitor SB431542 (1 µM), previously established to reduce signalling induced through TGF-β receptor activation in a variety of cell types (Inman et al., 2002; Walsh et al., 2008; Ehnert et al., 2010). We established that TGF-β activated the canonical Smad2 pathway by monitoring the phosphorylation of this proximal target of TGF-β signalling (Figure 3). Over a 4 h TGF-β exposure, the phosphorylation of Smad2 was increased in a manner markedly reduced by co-incubation with the ALK5 inhibitor, SB431542 (1 µM). TGF-β had no detectable effects on the phosphorylation of ERK1/2 or p38MAPK (Supporting Information Figure S2). Glucocorticoid impairment induced by TGF-β (40 pM) was completely prevented by continuous incubation with SB431542 (1 µM) for 30 min prior to TGF-β. The addition of SB431542 (1 µM) 30 min prior to IL-1α (24 h after TGF-β addition) partially attenuated the TGF-β-induced glucocorticoid impairment (Figure 4). None of SB203580, SB202190 (p38MAPK inhibitors) or U0126 (MEK inhibitor) reversed the glucocorticoid impairment (Supporting Information Figure S3).
Figure 3.

(A) Effect of TGF-β (100 pM) on Smad2 phosphorylation after a 30 min incubation, and its susceptibility to inhibition by the ALK5 inhibitor, SB431542 (1 µM), the MEK inhibitor, U0126 (1 µM), and the p38MAPK inhibitors, SB2029190 (1 µM) and SB203580 (1 µM). (B) Grouped data for 3 independent experiments, with 5, 30, 120 and 240 min exposure to TGF-β. *P < 0.05 compared with TGF-β effect in the absence of inhibitor.
Figure 4.
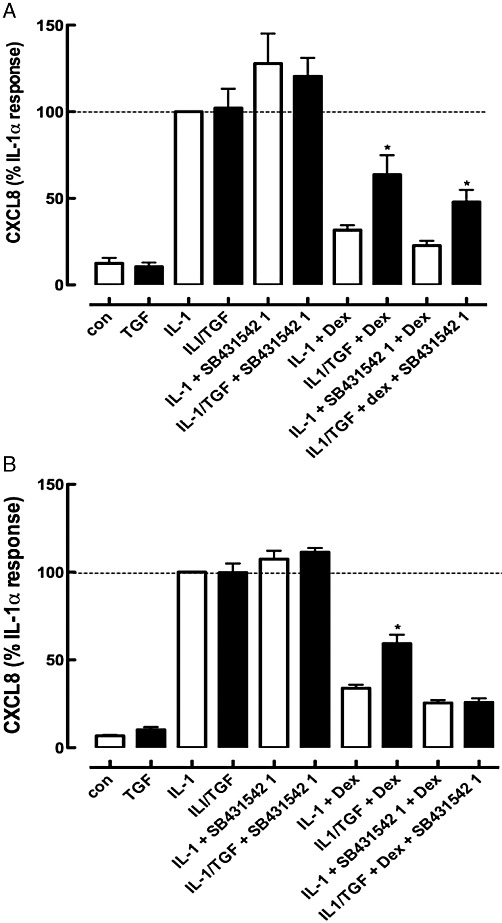
Effect of the ALK5 inhibitor (SB431542, 1 µM) on TGF-β-induced glucocorticoid impairment when added 30 min before TGF-β (A) or 30 min prior to IL-1α (B). n = 9 independent experiments. *P < 0.05, significantly different from IL-1α/TGF-β/Dex.
In A549 cells transfected with a plasmid bearing the GRE-controlled SEAP expression vector, dexamethasone (0.1–100 nM) induced a concentration-dependent increase in GRE activity that peaked at 30 nM. Incubation with 40 pM TGF-β for 24 h before dexamethasone treatment caused a very marked suppression of the GRE activity (Figure 5A); this effect of TGF-β was concentration-dependent and detectable at 10 pM, reaching a maximum at 40–100 pM (Figure 5B). In a further series of experiments examining the effects of SB431542 (1 µM), it was established that the ALK5 inhibitor could prevent the TGF-β impairment of the GRE response to dexamethasone (30 nM), whereas inhibition of ERK or p38MAPK had no effect (Figure 5C). The artificial GRE construct may respond differently to TGF-β than endogenous GRE-regulated genes. Thus, measurement of the GRE-regulated gene for IκBα was used to ascertain its susceptibility to TGF-β impairment of the ability of dexamethasone to stimulate IκBα mRNA. Dexamethasone induced a threefold increase in IκBα mRNA at 4 to 8 h, but in the presence of TGF-β (40 pM), this was reduced to a 1.3-fold increase (Figure 6). Similar patterns of gene regulation were observed for two other genes containing a GRE in the promoter region, namely those for the α sub-unit of the epithelial sodium channel, SCNN1A, and the glucocorticoid-inducible leucine zipper (GILZ). Expression of several other GRE-containing genes was measured. However, dexamethasone did not increase levels of annexin-1, secretory leukocyte protease inhibitor and the β2-adrenoceptor (data not shown). In addition, while MAP kinase phosphatase-1 (MKP-1) levels were increased 5.5 ± 0.4-fold by dexamethasone, MKP-1 levels were also increased by TGF-β 3.3 ± 0.6-fold, precluding use of this gene expression response to analyse the TGF-β impairment.
Figure 5.
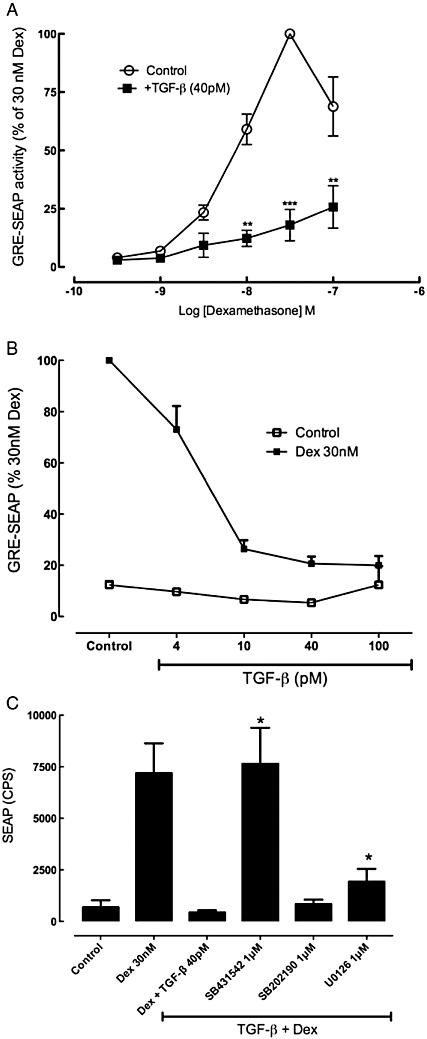
(A) Effect of TGF-β on glucocorticoid-induced GRE activity in A549 cells that were incubated with TGF-β (40 pM) for 24 h before stimulation by dexamethasone (0.3–100 nM) for a further 24 h. The level of SEAP in the supernatants was expressed as a percentage of the level induced in response to 30 nM dexamethasone and the data are mean and SEM for n = 5 independent experiments. (B) Effect of TGF-β (4–100 pM) on GRE activation by dexamethasone (Dex; 30 nM). (C) A fixed optimal dexamethasone (Dex) concentration of 30 nM was used to examine the effects of potential TGF-β signalling pathway inhibitors, including the ALK5 kinase inhibitor, SB431542 (1 µM), the p38MAPK inhibitor, SB202190 (1 µM) and the MEK inhibitor, U0126 (1 µM). *P < 0.05, compared with dexamethasone + TGF-β (representative of three independent experiments, each conducted in triplicate).
Figure 6.
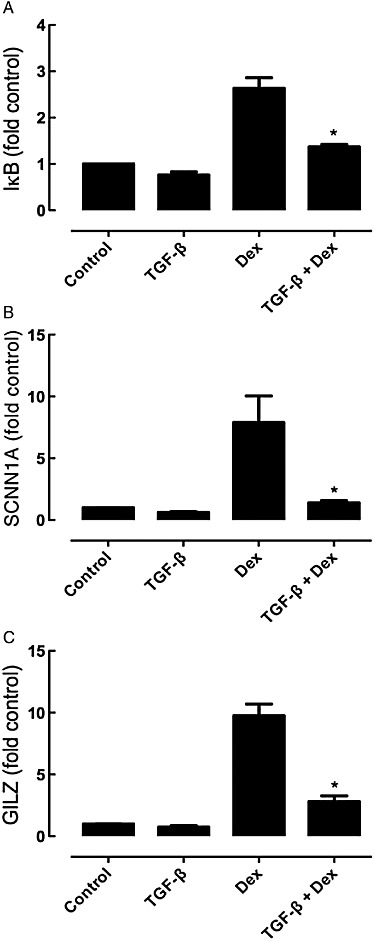
Effect of TGF-β on dexamethasone (Dex, 10 nM)-induced increases in (A) IκBα, (B) SCNN1A and (C) GILZ mRNA levels after 4 h incubation with IL-1α (1 ng·mL−1). Data are presented as the mean and SEM of n = 3 experiments (each determined in triplicate) and the gene expression is normalized to concurrently measured 18 s RNA levels and referenced to levels in control/IL-1α treated cells (1.0). *P < 0.05 compared to corresponding response in the absence of TGF-β pretreatment.
We next investigated the mechanism of impairment of GRE activation in A549 cells by establishing the levels and location of GRα in cells exposed to TGF-β. Examination of GRα immunofluorescence indicated that TGF-β reduced the GRα level (Figure 7A,B). The localization to the nucleus in response to dexamethasone (10 nM) was reduced by TGF-β, as was the baseline nuclear localization of GRα (Figure 7). The impact of TGF-β on nuclear localization was confirmed by immunoblot of nuclear extracts of cells incubated with dexamethasone (30 nM), in the presence and absence of TGF-β (40 pM). Upon exposure to dexamethasone (30 nM), immunoreactive-GRα levels increased in the nuclear compartment in vehicle-pretreated cells, but not in those incubated with 40 pM TGF-β (Figure 7C,D). Furthermore, experiments measuring GR-α expression at 4 h after dexamethasone showed that GR-α levels were reduced by TGF-β and the combination of dexamethasone and TGF-β (Figure 7E).
Figure 7.
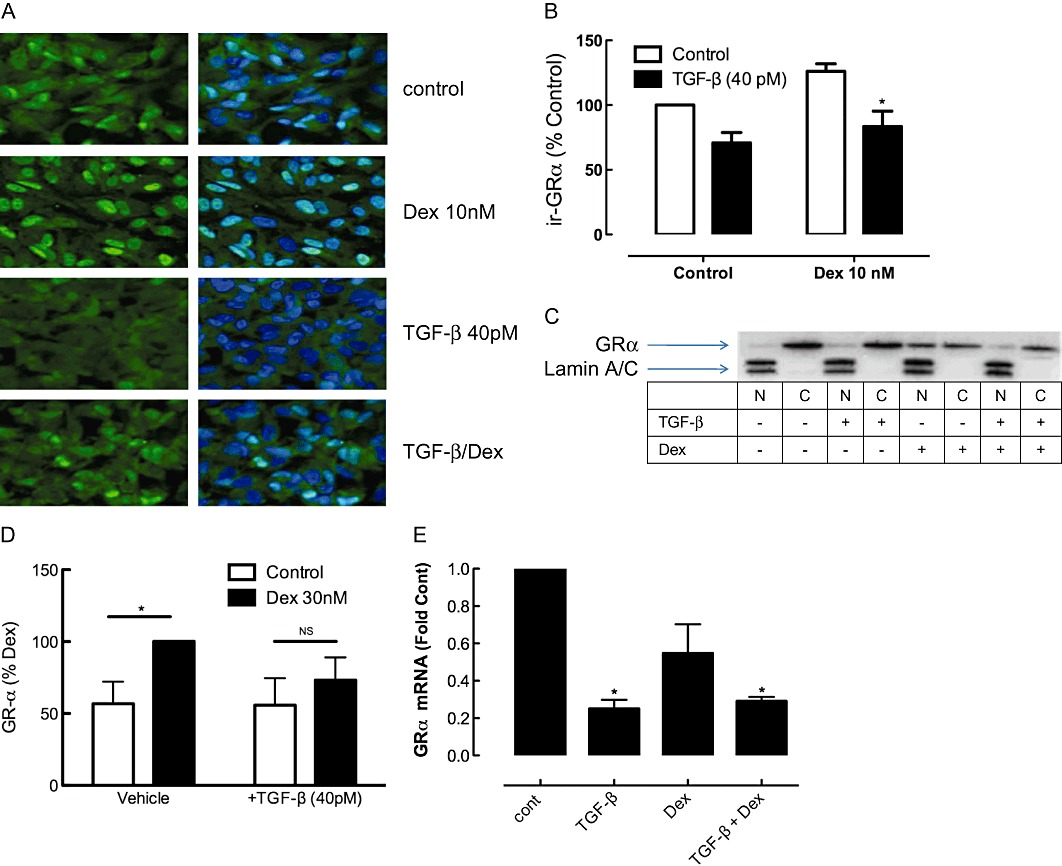
(A) Immunofluorescence of GRα in A549 cells treated with TGF-β for 24 h and dexamethasone (Dex; 10 nM) for 2 h before fixation. The images were obtained on Leica DMI6000B live cell imaging system at a magnification of 400×. Nuclei are stained blue with DAPI, or GRα is detected with a FITC-labelled secondary antibody. The image in Figure 7A is representative of five similar experiments, grouped data for which is presented in (B). (C) Cells were incubated using an identical protocol to that in Figure 7A except that 30 nM dexamethasone (Dex) was used. At the end of the 2 h dexamethasone incubation, cells were separated into cytosolic (C) and nuclear (N) extracts that were subjected to SDS-PAGE and immunoblotted with anti-GRα. Equal proportions of the whole cell extract representing the nuclear and cytosolic fractions were loaded. The nuclear fraction is identified by enrichment of the nuclear proteins, lamin A and C. Equivalence of cytosolic fraction loading was established by β-actin levels (not shown). (D) Grouped data for GR-α levels in the nuclear fraction, corrected for lamin A and C and expressed as a percentage of the level in vehicle/Dex 30 nM group (n = 4). (E) GRα gene expression is shown 4 h after exposure to dexamethasone (Dex; 30 nM), TGF-β 100 pM or the combination. Data are presented as mean and SEM of 3 experiments (each determined in triplicate). *P < 0.05, significantly different from control gene expression.
Discussion and conclusions
We sought to establish the glucocorticoid sensitivity of EMT in the A549 lung adenocarcinoma cell line. Our findings suggest that glucocorticoids have little detectable impact on TGF-β-induced EMT, consistent with a recent report in A549 cells (Doerner and Zuraw, 2009), but at variance with observations in a mink lung epithelial cell line (Zhang et al., 2010). However, we made the novel observation that exposure to TGF-β was associated with a reduced capacity of glucocorticoids to regulate CXCL8 generation in response to IL-1α. Further investigation revealed that TGF-β induced a broad impairment of glucocorticoid actions. While our study focussed on IL-1α as a stimulus of the A549 cell line, TGF-β impairment of glucocorticoid responses was also evident in the BEAS-2B cell line (continuous line generated from human central airways) with thrombin as the stimulus and measuring GM-CSF as the glucocorticoid-sensitive signal.
Several key questions regarding the glucocorticoid insensitivity were addressed to exclude trivial explanations. During prolonged incubation (96 h) with TGF-β, the IL-1α-induced CXCL8 generation was diminished, raising the question of whether diminished response to dexamethasone was secondary to the lower level of CXCL8. However, when CXCL8 levels in the absence of TGF-β were matched to those in its presence by lowering the concentration of the stimulus (IL-1α), the diminished maximum effect of dexamethasone was still evident. Moreover, in a 24 h TGF-β pretreatment protocol, the absolute level of CXCL8 generated in response to IL-1α was unaffected, whereas dexamethasone effects were still impaired. In the case of IL-6, TGF-β synergized with IL-1α to produce larger amounts of IL-6, yet dexamethasone responses were impaired. Thus, the impairment in dexamethasone response has no relationship to any effect of TGF-β on the magnitude of the IL-1α-induced cytokine response. The effect of TGF-β on cell viability was examined by flow cytometry that revealed that treatments, including TGF-β, dexamethasone and the combination did not affect either cell viability or the proportion of apoptotic cells.
The initial observations were made with A549 cells treated for 96 h during which time the cells undergo an EMT process, characterized in the present study by acquisition of mesenchymal cell contractile proteins, collagen synthesis capacity and acquisition of proliferative responsiveness to bFGF. We established that TGF-β-induced glucocorticoid insensitivity was not secondary to the phenotypic impact of prolonged incubation in TGF-β, as the insensitivity to glucocorticoids could be induced within 4 h of exposure to TGF-β, as was detectable in the CXCL8 mRNA response 4 h after IL-1α, and in protein levels after a further 20 h incubation. Changes in A549 morphology to the fibroblast phenotype, comprising isolated spindle-shaped cells only become pronounced after 72 to 96 h TGF-β exposure. EMT-associated morphological change required concentrations of 100 pM TGF-β, whereas the induction of glucocorticoid insensitivity had a threshold concentration of 4 to 40 pM TGF-β. Primary human parenchymal fibroblast cultures showed a high level of sensitivity to the glucocorticoid regulation of cytokine production (data not shown) as do airway fibroblasts (Ward et al., 2008), suggesting that the mesenchymal phenotype per se is not associated with glucocorticoid insensitivity.
The magnitude of the TGF-β effect in the presence of dexamethasone, being a two- to threefold increase in IL-1α-induced CXCL8 levels, would be expected to affect neutrophil recruitment and angiogenic responses. These CXCL8 functions are of potential significance in the lung cancer context. There is evidence of an association between TGF-β and pulmonary metastases in non-small cell lung carcinoma (NSCLC) (Saji et al., 2003). Conversely, other studies indicate that NSCLC progression is accelerated in patients with lowered TGFβRII expression (Borczuk et al., 2005), and higher TGF-β expression is associated with better chemotherapy responses (Irigoyen et al., 2010). The net impact of TGF-β remains unclear. The use of dexamethasone as an adjunct to chemotherapy in the management of lung cancer, albeit of equivocal efficacy (Keith, 2008), supports the relevance of understanding interactions between TGF-β, IL-1α and dexamethasone in the lung cancer microenvironment.
Several mechanisms for glucocorticoid resistance in the context of airway disease have been explored in considerable detail, including a role for up-regulation of GRβ (Hamid et al., 1999) or MKP-1 (Bhavsar et al., 2008b), inactivation by oxidative stress of histone deacetylase-2 (HDAC2) (Bhavsar et al., 2008a) and/or an excess of pro-inflammatory transcription factors (Adcock and Caramori, 2001). However, there has been little investigation of glucocorticoid resistance in structural cell types, as opposed to inflammatory cell types, so the extent to which these previously described mechanisms are pertinent to epithelial cells is not clear. One study showed that hypoxia induces glucocorticoid resistance in A549 cells by reducing HDAC2 activity (Charron et al., 2009). Previous work has established a number of links between TGF-β and GR signalling processes, but to our knowledge, this is the first report providing evidence that TGF-β has the potential to contribute to glucocorticoid resistance.
Given that CXCL8 and IL-6 may be regulated by both transrepressional and transactivational activities of glucocorticoids, we undertook a broad approach to the investigation of the potential mechanisms. The mechanism of the impairment of glucocorticoid regulation of cytokines was initially investigated using a GRE-SEAP reporter construct. TGF-β concentration dependently and profoundly reduced GRE activity. The activity of dexamethasone was suppressed across the range of concentrations having effects on IL-1α-induced cytokine production. This impairment of GRE transactivation action was shown to have pathophysiological relevance, as it was paralleled by a reduction in dexamethasone-stimulation of IκBα mRNA levels, a well-characterized, positive GRE-containing glucocorticoid-inducible gene in A549 cells (Newton et al., 1998). Although the IκBα may contribute to the regulation of cytokine production in response to IL-1α, we used this response to verify that the reduced transactivation of the reporter gene was evident with a physiological GRE-regulated gene product. The similarity in the pattern of inhibition of dexamethasone-induced expression of GILZ and SCNN1A is consistent with a broad impact of TGF-β on glucocorticoid-inducible genes. A specific defect in transactivation may have occurred as a result of either excess recruitment of co-repressors such as NCoR, and/or impaired recruitment of co-activators such as SRC1 (Waters et al., 2004). Alternatively, or additionally, the impairment in transactivation could have resulted from reduced nuclear GRα as a result of lowered levels or reduced nuclear entry. We sought evidence for the latter possibility, given that it was likely to be associated with impairment of transrepression. It is generally accepted that CXCL8 is regulated by transrepressional mechanisms. The corollary of TGF-β profoundly compromising the ability of glucocorticoids to regulate CXCL8 is that the mechanisms underlying this effect of TGF-β should show the potential to affect transrepression.
The reduction of about 30% in cellular immunoreactive-GRα was quantitatively less impressive than the extent of reduction in GRE activation or GRα nuclear localization. Thus, our results suggest involvement of a more specific impairment of nuclear localization, in addition to the impact of reduced cellular levels of GRα, which were accompanied by markedly reduced GRα mRNA levels. The quantitative nature of the relationship between GRα nuclear localization and the activation of different GRE-regulated genes is not known and may be non-linear. The level of reduction in nuclear localization of GRα would be expected to impair GR transrepression and may be sufficient to explain the marked reductions in GRE transactivation that we observed. However, our results do not exclude additional influences of TGF-β on the recruitment or de-recruitment of GR co-repressors or co-activators, respectively.
The mechanisms regulating nuclear localization of GRα and its transactivational activity are complex and incompletely understood. The impairment in nuclear localization may be due to reduced nuclear entry, accelerated nuclear export or a combination of these effects. Moreover, synthesizing a composite understanding of these mechanisms from literature on studies in different species, cell types and contexts may prove misleading. Notwithstanding these reservations, several regulatory phosphorylations of the GR have been identified in previous studies that may explain the current observations. Phosphorylation by cyclin-dependent kinase 5 at two distinct residues, GR203 and GR211, reduces GR transactivational activity in neuronal cell lines (Kino et al., 2007). The phosphorylation of GR203 reduces nuclear accumulation in rat hepatoma cells (Blind and Garabedian, 2008). Recently, it has been suggested that phosphorylation at one or more of residues 203, 211 and 226 is required for GRE-transactivation, based on work in COS-1 cells. Recruitment of the co-activator GRIP1 is the feature of transactivation that requires this phosphorylation pattern (Avenant et al., 2010). Our initial survey of a range of kinases that have been implicated in phosphorylation of the above-described residues indicates that inhibition of ERK1/2, p38MAPK or JNK has no capacity to restore the impaired GRE activation.
Our unexpected finding of induction of glucocorticoid insensitivity is of potential relevance to the broad use of glucocorticoids in the treatment of allergy, inflammation and cancer. Associations between TGF-β expression and glucocorticoid resistance have been identified in allergic airways disease (Chakir et al., 2003). We are particularly interested in the possible role of TGF-β in chronic tissue remodelling associated with incessant cycles of tissue injury and repair in which TGF-β mobilization is well-established (Margadant and Sonnenberg, 2010). Further defining the pathways by which such resistance is induced, and determining the extent of overlap with or independence of the pathways used by other cytokines, such as TNF-α and IFN-γ that induce resistance to glucocorticoid actions, could lead to the identification of targets for a novel class of anti-inflammatory, steroid-sensitizing agents.
Acknowledgments
These studies were supported by a project grant from NHMRC 628691.
Glossary
- EMT
epithelial-mesenchymal transition
- GRα
glucocorticoid receptor α
- GRE
glucocorticoid response element
Conflicts of interest
The authors state no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 A. Effect of TGF-β (400 pM) on morphology of A549 cells pretreated with vehicle (control) or with TGF-β (4–400 pM). Images were taken using Iatia phase contrast microscopy at a magnification of 150 times and are representative of 8 similar experiments with 96 h TGF-βincubation. B. Time-course of response to TGF-β (400 pM) as measured in the Leica DMI6000B live cell imaging system (mag 100×), representative of 3 similar experiments.
Figure S2 A. Effect of TGF-β on ERK1/2 and p38MAPK phosphorylation after a 30 min incubation, and its susceptibility to inhibition by the ALK5 inhibitor, SB431542 (1 μM), the MEK inhibitor, U0126 (1 μM), and the p38MAPK inhibitors, SB2029190 (1 μM) and SB203580 (5 μM). B. Grouped data for n = 3 independent experiments conducted with 5, 30, 120 and 240 min exposure to TGF-β that induced an increase in Smad-2 phosphorylation (P < 0.05) that was unaffected by each of the inhibitors, except SB341542.
Figure S3 A. Effect of the MEK inhibitor, U0126 (1 μM) on TGF-β-induced glucocorticoid-resistance when added 30 min prior to TGF-β. B. Effect of the p38MAPK inhibitor, SB203580 (1 μM) on TGF-β-induced glucocorticoid-resistance when added 30 min prior to TGF-β. C. Effect of the p38MAPK inhibitor, SB202190 (1 μM) on TGF-β-induced glucocorticoid resistance when added 30 min prior to TGF-β. n = 5 independent experiments. *P < 0.05 cf IL-1α/ dexamethasone.
Table S1 A549 cell number changes in response to bFGF with and without 96 h pre-incubation with TGF-β
Table S2 Regulation of ‘matched’ CXCL8 (ng mL−1) levels by dexamethasone in the presence and absence of TGF-β (100 pM)
Table S3 This data is from an experiment of identical design to that outlined in figure 1A, except that the period of pre-incubation with TGF-β was 24 h rather than 96 h
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394–401. doi: 10.1378/chest.08-0440. [DOI] [PubMed] [Google Scholar]
- Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol. 2001;79:376–384. doi: 10.1046/j.1440-1711.2001.01025.x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avenant C, Kotitschke A, Hapgood JP. Glucocorticoid receptor phosphorylation modulates transcription efficacy through GRIP-1 recruitment. Biochemistry. 2010;49:972–985. doi: 10.1021/bi901956s. [DOI] [PubMed] [Google Scholar]
- Bhavsar P, Ahmad T, Adcock IM. The role of histone deacetylases in asthma and allergic diseases. J Allergy Clin Immunol. 2008a;121:580–584. doi: 10.1016/j.jaci.2007.12.1156. [DOI] [PubMed] [Google Scholar]
- Bhavsar P, Hew M, Khorasani N, Torrego A, Barnes PJ, Adcock I, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared with non-severe asthma. Thorax. 2008b;63:784–790. doi: 10.1136/thx.2007.090027. [DOI] [PubMed] [Google Scholar]
- Blind RD, Garabedian MJ. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J Steroid Biochem Mol Biol. 2008;109:150–157. doi: 10.1016/j.jsbmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borczuk AC, Kim HK, Yegen HA, Friedman RA, Powell CA. Lung adenocarcinoma global profiling identifies type II transforming growth factor-beta receptor as a repressor of invasiveness. Am J Respir Crit Care Med. 2005;172:729–737. doi: 10.1164/rccm.200504-615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- Charron CE, Chou PC, Coutts DJ, Kumar V, To M, Akashi K, et al. Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J Biol Chem. 2009;284:36047–36054. doi: 10.1074/jbc.M109.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curl CL, Harris T, Harris PJ, Allman BE, Bellair CJ, Stewart AG, et al. Quantitative phase microscopy: a new tool for measurement of cell culture growth and confluency in situ. Pflugers Arch. 2004;448:462–468. doi: 10.1007/s00424-004-1248-7. [DOI] [PubMed] [Google Scholar]
- Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009 doi: 10.1186/1465-9921-10-100. DOI: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehnert S, Baur J, Schmitt A, Neumaier M, Lucke M, Dooley S, et al. TGF-beta1 as possible link between loss of bone mineral density and chronic inflammation. PLoS ONE. 2010;5:e14073. doi: 10.1371/journal.pone.0014073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross KL, Cidlowski JA. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab. 2008;19:331–339. doi: 10.1016/j.tem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte JJ, et al. Increased glucocorticoid receptor beta in airway cells of glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 1999;159:1600–1604. doi: 10.1164/ajrccm.159.5.9804131. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Irigoyen M, Pajares MJ, Agorreta J, Ponz-Sarvise M, Salvo E, Lozano MD, et al. TGFBI expression is associated with a better response to chemotherapy in NSCLC. Mol Cancer. 2010;9:130. doi: 10.1186/1476-4598-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith BD. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer. 2008;8:84. doi: 10.1186/1471-2407-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khau T, Langenbach SY, Schuliga M, Harris T, Johnstone CN, Anderson RL, et al. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J. 2011;25:483–496. doi: 10.1096/fj.09-154096. [DOI] [PubMed] [Google Scholar]
- Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, et al. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21:1552–1568. doi: 10.1210/me.2006-0345. [DOI] [PubMed] [Google Scholar]
- Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010;11:97–105. doi: 10.1038/embor.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R, Hart LA, Stevens DA, Bergmann M, Donnelly LE, Adcock IM, et al. Effect of dexamethasone on interleukin-1beta-(IL-1β)-induced nuclear factor-kappaB (NF-kappaB) and κB-dependent transcription in epithelial cells. Eur J Biochem. 1998;254:81–89. doi: 10.1046/j.1432-1327.1998.2540081.x. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101:830–839. doi: 10.1002/jcb.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saji H, Nakamura H, Awut I, Kawasaki N, Hagiwara M, Ogata A, et al. Significance of expression of TGF-beta in pulmonary metastasis in non-small cell lung cancer tissues. Ann Thorac Cardiovasc Surg. 2003;9:295–300. [PubMed] [Google Scholar]
- Smith LK, Cidlowski JA. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog Brain Res. 2010;182:1–30. doi: 10.1016/S0079-6123(10)82001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MF, Ampasala DR, Hatfield J, Vander Heide R, Suer S, Rishi AK, et al. Transforming growth factor-beta stimulates intestinal epithelial focal adhesion kinase synthesis via Smad- and p38-dependent mechanisms. Am J Pathol. 2008;173:385–399. doi: 10.2353/ajpath.2008.070729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JE, Harris T, Bamford T, Mast A, Pain MC, Robertson C, et al. Proliferation is not increased in airway myofibroblasts isolated from asthmatics. Eur Respir J. 2008;32:362–371. doi: 10.1183/09031936.00119307. [DOI] [PubMed] [Google Scholar]
- Waters CE, Stevens A, White A, Ray DW. Analysis of co-factor function in a glucocorticoid-resistant small cell carcinoma cell line. J Endocrinol. 2004;183:375–383. doi: 10.1677/joe.1.05804. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lei W, Wang X, Tang Y, Song J. Glucocorticoid induces mesenchymal-to-epithelial transition and inhibits TGF-beta1-induced epithelial-to-mesenchymal transition and cell migration. FEBS Lett. 2010;584:4646–4654. doi: 10.1016/j.febslet.2010.10.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.