

Abstract
BACKGROUND AND PURPOSE
High levels of SKP2 are a poor prognostic factor in multiple human cancers and mostly correlate with low p27KIP1 levels. Prodigiosin is a bacterial tripyrrole pigment with strong pro-apoptotic activity. Induction of cell cycle blockade underlies one of its anticancer actions but the mechanisms involved are unclear. The aim of this study was to explore the role of the SKP2–p27KIP1 axis in prodigiosin's cytostatic effect on human lung adenocarcinoma cells.
EXPERIMENTAL APPROACH
Prodigiosin's effects on cell cycle progression and long-term cell proliferation of human lung adenocarcinoma cells were characterized by flow cytometry and colony formation assay, respectively. Real-time RT-PCR and promoter activity analyses were performed for assessing transcriptional control, while cycloheximide chase analysis evaluated protein stability. Immunoblotting was employed for mechanistic study.
KEY RESULTS
Prodigiosin increased p27KIP1 expression mainly by stabilizing p27KIP1 through transcriptional repression of SKP2. Importantly, SKP2 overexpression or p27KIP1 depletion restored the colony forming capacity of prodigiosin-treated cells. Furthermore, prodigiosin induced PKB dephosphorylation, leading to PKB inhibition as revealed by decreased serine 9 phosphorylation of GSK-3β. Constitutive PKB activation reduced prodigiosin-induced SKP2 repression. Prodigiosin also down-regulated E2F1 (mediates PI3K/PKB-induced SKP2 transcription), but E2F1 overexpression failed to restore SKP2 expression in prodigiosin-treated cells.
CONCLUSIONS AND IMPLICATIONS
Transcriptional repression of SKP2 and the consequent accumulation of p27KIP1 are essential for prodigiosin's antiproliferative action. Mechanistically, prodigiosin induces PKB inhibition to down-regulate SKP2 in a GSK-3β- and E2F1-independent manner. Our findings further implicate the potential for developing prodigiosin as a novel class of SKP2-targeting anticancer agent.
Keywords: prodigiosin, SKP2, p27KIP1, AKT, PKB, antiproliferation, lung adenocarcinoma
Introduction
Lung cancer has been the global leading cause of cancer-related mortality (Jemal et al., 2011). Lung adenocarcinoma is the major component of non–small cell lung cancer (NSCLC), which represents about 85–90% of all cases of lung cancer (Cataldo et al., 2011). Around 50% of lung cancer patients are diagnosed with advanced-stage disease, the treatment of which primarily depends on systemic chemotherapy (Ramalingam et al., 2011). Platinum-based chemotherapy in combination with additional chemotherapeutics, including paclitaxel, is currently the standard therapy but it is often compromised by low response rate and rapid development of drug resistance (Yoshida et al., 2010). Molecularly targeted agents such as EGF receptor inhibitors geftinib and erlotinib confer significant efficacy advantage over combination chemotherapy (Maemondo et al., 2010). However, the application of EGF receptor inhibitors is limited to selected patient subpopulations with distinct mutations in the EGFR gene, and the efficacy of these drugs is often counteracted by mechanisms leading to acquired resistance (Xu et al., 2010). Identification of new effective chemotherapeutic agents and novel oncogenic drivers as molecular targets for therapy is therefore urgently needed.
Cancer cells are characterized by uncontrolled cell proliferation owing to cell cycle deregulation. The cyclin-dependent kinase (CDK) inhibitor p27KIP1 is an integral negative regulator of the cell cycle and functions by preventing S-phase entry, binding and inhibiting the activity of CDK2 (Borriello et al., 2011). As such, high levels of p27KIP1 are required for the maintenance of a quiescent state, while a progressive decline in p27KIP1 during G1 is needed to allow the G1–S transition (Chu et al., 2008). Consistent with the inhibitory role of p27KIP1 on cell cycle progression, treatments that augment p27KIP1 expression generally lead to G1 cell cycle arrest and reduced cell proliferation (Ling et al., 2007; Huang et al., 2008; Liu and Yamauchi, 2009). Clinically, low levels of p27KIP1 protein are observed in multiple human epithelial malignancies, including lung cancer, and are generally associated with poor prognosis (Chu et al., 2008). Of note, accelerated proteolysis of p27KIP1 largely accounts for the low p27KIP1 levels in human cancers (Frescas and Pagano, 2008).
The action of the E3 ubiquitin ligase complex SCFSKP2[S-phase kinase protein 1 (SKP1)/Cullin/F-box protein: S-phase kinase protein 2 (SKP2)] represents the most potent mechanism to initiate p27KIP1 polyubiquitination and subsequent proteasome-dependent proteolysis (Chu et al., 2008). Mechanistically, SKP2 functions as the receptor component of SCFSKP2 to recruit p27KIP1 for ubiquitination. In line with its ability to destabilize p27KIP1, Skp2−/− mouse embryonic fibroblasts exhibit defects in cell proliferation along with cellular accumulation of p27KIP1 (Nakayama et al., 2000). Given its promoting role in cell proliferation, SKP2 abundance or activity is positively regulated by multiple oncogenic signalling pathways, including Notch, IKK/NF-κB and the PI3K/PKB signalling pathway (Chan et al., 2010). Not surprisingly, high levels of SKP2 are found in a broad range of human cancers associated with poor prognosis and, notably, in most cases correlate with low p27KIP1 levels (Frescas and Pagano, 2008). The oncogenic potential of SKP2 is further substantiated by recent genetic evidence illustrating the indispensable role of SKP2 in tumourigenesis initiated by an array of oncogenic stimuli, including BCR-ABL overexpression, Pten loss and Rb1 deficiency (Agarwal et al., 2008; Lin et al., 2010; Wang et al., 2010). For that reason, SKP2 has been proposed as a potential target for therapy. In fact, small molecules targeting SCFSKP2 activity or SCFSKP2 complex assembly have been examined in leukaemia cells and in xenograft tumour model, respectively, and proved to be effective as anticancer agents (Chen et al., 2008; Lin et al., 2010).
Prodigiosin (2-methyl-3-pentyl-6-methoxyprodiginine) is an anticancer tripyrrole red pigment produced by micro-organisms such as Serratia marcescens (Chang et al., 2011). Prodigiosin is currently subjected to preclinical trials for the treatment of pancreatic cancer conducted by Aida Pharmaceuticals Inc. (http://www.crunchbase.com/company/aida-pharmaceuticals). In addition, obatoclax, a prodigiosin analogue and a BH3 mimetic agent, has been shown to display promising anticancer efficacy in preclinical studies and at present is in several clinical trials either as a single agent or in combination with other chemotherapeutic drugs (Pérez-Tomás and Viñas, 2010). These notions together highlight the potential of using prodigiosin as a lead compound for developing novel cancer therapeutics. Currently, four possible mechanisms of action involved in the anticancer effect of prodigiosin have been described. These include (i) induction of intracellular acidification, (ii) induction of DNA cleavage by acting as inhibitors of topoisomerase I and II, (iii) modulation of MAPK activity and (iv) inhibition of cell cycle progression (Pérez-Tomás and Viñas, 2010). It is noteworthy that the first three modes of action lead to induction of apoptosis. Intriguingly, prodigiosin at non-cytotoxic concentrations provokes cell cycle blockade, while higher levels of prodigiosin induce apoptosis (Williamson et al., 2006; Soto-Cerrato et al., 2007a). Prodigiosin is known to induce apoptosis in a variety of human cancer cell lines irrespective of p53 status and multidrug resistance but shows little toxicity to normal cells, conferring great advantages to prodigiosin as an anticancer agent (Pandey et al., 2009; Pérez-Tomás and Viñas, 2010). The molecular mediators whereby prodigiosin induces apoptosis are gradually being elucidated. Transcriptional up-regulation of pro-apoptotic NAG-1 and down-regulation of anti-apoptotic survivin have been documented to explain the pro-apoptotic action of prodigiosin (Soto-Cerrato et al., 2007b; Ho et al., 2009). Regarding the molecular basis of prodigiosin's cytostatic effect, so far, only the CDK inhibitor p21CIP1 is reported to be a transcriptional target of prodigiosin ostensibly mediating prodigiosin-induced cell cycle arrest in MCF-7 breast cancer cells (Soto-Cerrato et al., 2007a). Intriguingly, questions remain as to whether p21CIP1 up-regulation is a general mode of action of prodigiosin's cytostatic effect, and whether additional molecular targets exist that are responsible for the antiproliferative action of prodigiosin.
In this study, we aimed to elucidate the molecular basis underlying the antiproliferative effect of prodigiosin on human lung adenocarcinoma cells. We observed that prodigiosin at non-cytotoxic concentrations induces G1 cell cycle blockade along with increased expression of p27KIP1 in addition to p21CIP1. Importantly, SKP2 was identified as a novel molecular target of prodigiosin; the down-regulation of SKP2 by prodigiosin and the resulting accumulation of p27KIP1 were shown to be critical for the ability of prodigiosin to suppress cell proliferation. Mechanistically, prodigiosin was demonstrated to engage PKB-mediated signalling to induce transcriptional repression of SKP2 in an E2F1-independent manner. Overall, our results are the first to demonstrate the involvement of the SKP2–p27KIP1 axis in the antiproliferative action of prodigiosin. These findings not only provide new insights into the molecular understanding of prodigiosin's antiproliferative effect, but also highlight the potential of prodigiosin in the development of SKP2-targeted anticancer therapeutics.
Methods
Drugs
Prodigiosin was isolated and purified from S. marcescens C3 and quantified by HPLC as previously described (Ho et al., 2009). Purified prodigiosin was stored in methanol at a concentration of 10 mM, in the dark at −20°C until used. Cycloheximide (Sigma-Aldrich, St. Louis, MO, USA) were prepared as 10 mM stock solutions in DMSO. MG132 was purchased from Tocris Bioscience (Bristol, UK) and was stored in DMSO at a concentration of 20 mM. All chemicals in stock solutions were stored in aliquots at −20°C before use.
Cell culture
Human lung adenocarcinoma cell lines A549 (ATCC no. CCL-185), CL1-5 (Chen et al., 2001) and H23 (ATCC no. CRL-5800) were grown in RPMI1640 medium (Invitrogen, Carlsbad, CA, USA), while 293T cells (ATCC no. CRL-11268) were grown in DMEM medium (Invitrogen). Both media were supplemented with 10% heat-inactivated fetal bovine serum (Gibco BRL®, Grand Island, NY, USA), 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin. Cells were allowed to grow at 37°C in a humidified, 5% CO2 atmosphere. Effective concentrations were determined.
Determination of effective concentrations of prodigiosin
The effect of prodigiosin on cell viability was determined using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS assay) kit (Promega, Madison, WI, USA) according to the protocol described in Ho et al. (2009). Cell viability was calculated as (Asample−Ablank) / (Acontrol−Ablank) × 100%, while cytotoxicity = 100% − cell viability (%). In this study, the concentrations of prodigiosin that caused 15% or 25% reduction in cell viability with no apparent pro-apoptotic effect after 24 h-treatment were considered to be non-cytotoxic concentrations and were used to study the effect on cell cycle progression (Soto-Cerrato et al., 2007a), while the dosage that lowered the viability to 50% (IC50) at 24 h was regarded as the cytotoxic concentration. For A549 cells, prodigiosin at 50 nM and 100 nM reduced cell viability by 15% and 25%, respectively, whereas the IC50 of prodigiosin was 420 nM.
Flow cytometry
A549, CL1-5 and H23 cells (5 × 105) grown on 60 mm Petri dishes were treated with various doses of prodigiosin for 24 h. Subsequently, cells were harvested, washed with 1X ice-cold PBS and resuspended in 1 mL of 1X ice-cold PBS. Next, cells were fixed by gradually adding 3 mL of ice-cold absolute ethanol and stored at −20°C for 24 h. Cell pellets were then collected by centrifugation, resuspended in 0.5 mL of 1X ice-cold PBS and mixed well with 0.5 mL of 0.1% Triton X-100 solution supplemented with 100 µg·mL−1 of ribonuclease and 20 µg·mL−1 of propidium iodide (PI), followed by incubation at 37°C for 1 h in the dark. Fluorescence emitted from the PI-DNA complexes was then quantified using Beckman Coulter Cytomics™ FC500 (Beckman-Coulter, Brea, CA, USA).
Immunoblotting
Immunoblotting was performed as previously described (Ho et al., 2009). When MG132 treatment was required, 20 µM of MG132 was added 2 h before cell harvesting. Antibodies against human p21CIP1 and p27KIP1 were purchased from BD Biosciences (Franklin Lakes, NJ, USA). Human SKP2 antibody was obtained from Invitrogen, while the primary antibodies against PKB, phosphor-Ser473-PKB, phosphor-Ser9-GSK-3β and E2F1 were all purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-HA and anti-Flag antibodies were obtained from GeneMark (Taichung, Taiwan). Antibodies against human cyclin E and GSK-3β were purchased from Epitomics (Burlingame, CA, USA). Anti-GAPDH and anti-β-tubulin, both serving as controls for equal loading, were purchased from Abcam (Cambridge, UK) and Sigma-Aldrich, respectively.
Reverse transcription PCR (RT-PCR)
Total RNA extraction, first-strand cDNA preparation and SYBR green-based real-time PCR were performed as reported previously (Ho et al., 2009). The primer pairs used for semiquantitative PCR are listed as follows: p21CIP1 forward, 5′-CCTCTTCGGCCCAGTGGAC-3′ and p21CIP1 reverse, 5′-CCGTTTTCGACCCTGAGAG-3′; p27KIP1 forward 5′-TGCAACCGACGATTCTTCTAC-3′ and p27KIP1 reverse, 5′-TCCTTGCTTCATCAAGCAGTG-3′; SKP2 forward, 5′-GCTGCTAAAGGTCTCTGGTGT-3′ and SKP2 reverse, 5′-AGGCTTAGATTCTGCAACTTG-3′ (Sonoda et al., 2006). For quantitative real-time RT-PCR, we employed the following primer pairs: p21CIP1 forward, 5′-AGACTCTCAGGGTCGAAAAC-3′ and p21CIP1 reverse, 5′-TGGAGTGGTAGAAATCTGTCATG-3′; p27KIP1 forward, 5′-TGCAACCGACGATTCTTCTAC-3′ and p27KIP1 reverse, 5′-CTTCTGTTCTGTTGGCTCTTTTG-3′; SKP2 forward, 5′-CTGTCTCAAGGGGTGATTGC-3′ and SKP2 reverse, 5′-TTCGATAGGTCCATGTGCTG-3′ (Chen et al., 2009). The levels of mRNA expression of genes of interest were normalized to that of TATA-binding protein (TBP). Final results are expressed as the ratio of copy numbers of the mRNA of genes of interest to TBP mRNA copy numbers and presented as mean ± SEM of three independent experiments.
Luciferase reporter assay
The human p27KIP1 (encoded by CDKN1B) promoter containing the region from −1358 to +132 (Pang et al., 2008) and the human SKP2 promoter covering the region between −1148 and +20 from the translational start site (Huang and Hung, 2006) were PCR-amplified as described in the respective reports. The PCR-amplified fragments were then cloned into the luciferase reporter plasmid pGL4.18 vector (Promega) to generate the promoter reporter plasmids of human p27KIP1 (pCDKN1B-Luc) and SKP2 (pSKP2-Luc), respectively. Luciferase activity assay was performed using Dual-Luciferase® Reporter assay kit (Promega). In brief, cells (5 × 104) were seeded onto six-well plates and allowed to grow overnight. Cells were then transiently transfected with pCDKN1B-Luc, pSKP2-Luc or p4XE2F1-Luc in combination with a plasmid expressing Renilla luciferase by jetPEI™ transfection reagent (Polyplus, New York, NY, USA). Twenty-four hours later, cells were treated with prodigiosin for 24 h, and the drug-treated cell lysates were prepared and subjected to Dual-Luciferase® Reporter assay following manufacturer's recommendation. The intensity of luminescence was determined on GloMax® 20/20 luminometer (Promega). Luciferase activity was normalized to Renilla luciferase activity, and final data were presented as the fold change of the luciferase activity compared with that of control vectors.
Cycloheximide chase analysis
A549, CL1-5 and H23 cells (5 × 105) were seeded onto 60 mm Petri dishes and treated without or with 100 nM of prodigiosin. Eighteen hours later, cells were treated with cycloheximide (60 µg·mL−1) to stop de novo protein synthesis. Cell lysates were then harvested at 0, 1, 3 and 6 h following cycloheximide treatment and then subjected to immunoblotting for the levels of p27KIP1.
RNA interference
Endogenous levels of p27KIP1 and GSK-3β were depleted through RNA interference-mediated suppression mechanism by targeting the sequence 5′-GCGCAAGTGGAATTTCGATTT-3′ of human p27KIP1 mRNA (GenBank accession no. NM_004064) and 5′-AGCAAATCAGAGAAATGAAC-3′ of human GSK-3β mRNA (GenBank accession no. NM_002093), respectively. The lentiviral vectors pLKO.1 expressing a short hairpin interfering RNA (shRNA) that targets the aforementioned sequence of human p27KIP1 (clone ID: TRCN0000039930) and human GSK-3β (clone ID: TRCN0000039565) were purchased from the National RNAi Core Facility located at Academic Sinica, Taiwan. Additionally, the pLKO.1-shLuc plasmid carrying a shRNA targeting luciferase (clone ID: TRCN0000072243; target sequence: 5′-CTTCGAAATGTCCGTTCGGTT-3′) was used as a negative control.
Construction of pBabe.puro-based expression plasmids
pBabe.puro-Myr-Flag-PKB1, a constitutively active mutant of PKB1 cloned in the retroviral expression vector pBabe.puro, was purchased from Addgene (Addgene plasmid 15294, Cambridge, MA, USA). To construct the SKP2-expressing vector pBabe-SKP2, the open reading frame (ORF) of human SKP2 was PCR-amplified from the first-strand cDNA pools of A549 cells and then cloned into the pBabe.puro vector at the EcoRI site, followed by validation for accurate orientation and DNA sequence. The primers used for cloning the human SKP2 ORF were 5′-ACGCTATGCACAGGAAGCAC-3′ (forward) and 5′-CTTCATAGACAACTGGGCTTTTG-3′ (reverse). pBabe-HA-E2F1 was constructed by retrieving the HA-E2F1 fragment from pSG5L-HA-E2F1 (Addgene plasmid 10736) by BamHI–EcoRI double digestion, followed by insertion between the BamHI and EcoRI sites of the pBabe.puro vector.
pLKO.1-derived lentiviral and pBabe.puro-derived retroviral particle production and infection
HEK-293T cells (7 × 105) were transiently transfected for 24 h by jetPEI™ transfection reagent (Polyplus) with 2.5 µg of pLKO.1-shLuc, pLKO.1-shp27 or pBabe.puro-based plasmids along with the plasmids expressing gag-pol and VSV-G proteins required for the package of viral particles. Viral particles released into the fresh culture media replaced at 24 and 48 h following initial transfection were harvested by centrifugation and the supernatant containing viral particles was collected. For performing viral infection, A549 cells (5 × 105) were incubated for 48 h with viral particle-enriched media supplemented with 8 µg·mL−1 of polybrene (Sigma-Aldrich) to promote infection efficiency. Subsequently, cells with stable infection were selected following treatment with puromycin (3 µg·mL−1) for 48–72 h and the efficiency of p27KIP1 depletion or ectopic expression of genes of interest was validated by immunoblotting.
Colony formation assay
The capacity for long-term cell proliferation was evaluated by colony formation assay. The assay was carried out according to the previously reported procedure (Ho et al., 2009), except that A549 cells were plated at a density of 1000 cells per dish. The same procedure was repeated at least three times to calculate the numbers of colonies for statistical analysis.
Statistical analysis
All data are expressed as a means ± SEM from at least three independent experiments. Differences between groups were examined for statistical significance using Student's t-test. A P-value lower than 0.05 was used as the minimum criteria for statistical significance.
Results
Prodigiosin induces cell cycle arrest and increased expression of p27KIP1 in addition to p21CIP1 in multiple human lung adenocarcinoma cell lines
To explore the effect of prodigiosin on cell cycle progression in human lung adenocarcinoma cells, A549 cells were treated with 0, 50 and 100 nM of prodigiosin for 24 h, and cell cycle distribution was determined by flow cytometry analysis thereafter. Treatment with 50 and 100 nM prodigiosin, which, respectively, accounted for 15% and 25% reduction in cell viability (data not shown), had only a small effect on the number of hypodiploid (apoptotic) cells (sub-G1 phase) (Figure 1A, upper panel). Conversely, a marked accumulation of cell population in the G1 phase was observed after prodigiosin treatment (from 71.8 ± 3.4% at 0 nM to 85.2 ± 2.8% and 86.4 ± 3.6% at 50 and 100 nM, respectively; P < 0.01), with a concomitant decrease of cells in the S phase (from 13.2 ± 1.9% at 0 nM to 5.7 ± 2.2% at 50 nM, P < 0.05 and 4.8 ± 2.1% at 100 nM, P < 0.01) (Figure 1A, upper panel). At the molecular level, it is noteworthy that the same concentration range of prodigiosin evidently up-regulated p21CIP1 and p27KIP1 (Figure 1B, left panel). Taken together, these results indicated prodigiosin at non-cytotoxic doses induces G1 arrest of the cell cycle as well as increased expression of p21CIP1 and p27KIP1 in A549 cells. Importantly, the effect of prodigiosin on cell cycle progression and the expression of p21CIP1 and p27KIP1 is not restricted to A549 cells only, as similar results were observed in additional lung adenocarcinoma cell lines CL1-5 and H23 following prodigiosin treatment (Figure 1). Overall, it appears to be a general mode of action of prodigiosin at non-cytotoxic concentrations to induce G1 cell cycle arrest, along with the up-regulation of p21CIP1 and p27KIP1 in human lung adenocarcinoma cells.
Figure 1.
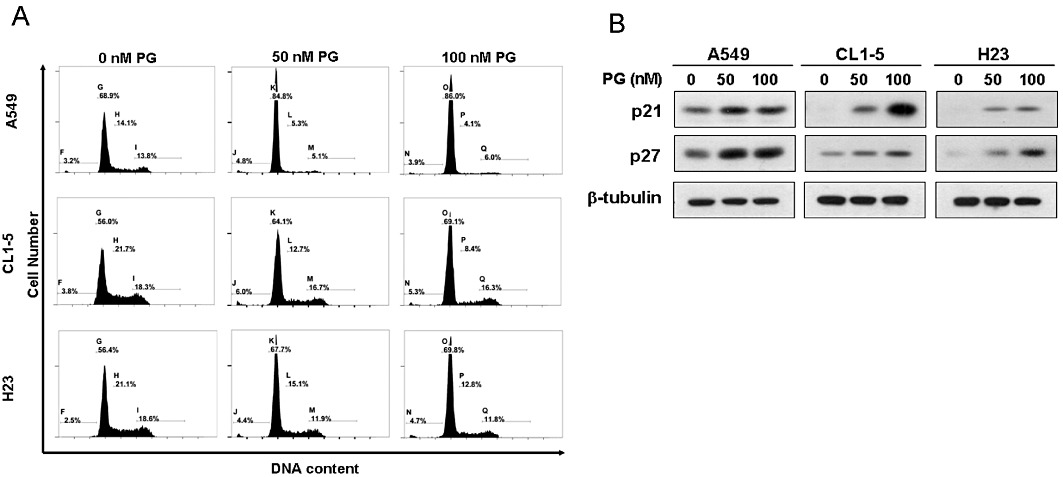
Prodigiosin induces G1 cell cycle arrest and increased expression of p21CIP1 and 27KIP1 in multiple human lung adenocarcinoma cell lines. (A) Prodigiosin at non-cytotoxic concentrations arrests cells in the G1 phase. A549, CL1-5 and H23 cells were treated with prodigiosin (0–100 nM) for 24 h and then subjected to flow cytometry analysis for cell cycle distribution. It is clear that apoptoptic cells (sub-G1 phases) were barely elicited by prodigiosin at the concentrations used. An evident increase in the % of cells in the G1 phase was observed, along with a concomitant decrease in cells in the S phase. (B) Increase in the levels of p21CIP1 and p27KIP1 in prodigiosin-treated cells. A549, CL1-5 and H23 cells were treated with prodigiosin (0∼100 nM) for 24 h and then subjected to immunoblotting analysis for the protein levels of p21CIP1 and p27KIP1. Human β-tubulin was used as the loading control. Shown here is a representative blot from at least three independent experiments. PG, prodigiosin.
Prodigiosin up-regulates p27KIP1 largely through protein stabilization
Next we investigated the mechanism whereby prodigiosin up-regulates p21CIP1 and p27KIP1. We first determined whether prodigiosin modulates the mRNA levels of p21CIP1 and p27KIP1 using RT-PCR analysis. It is apparent that in A549, CL1-5 and H23 cells, treatment with 50 or 100 nM prodigiosin clearly increased the level of p21CIP1 mRNA (Figure 2A, upper panel), in accordance with the mechanism operated in MCF-7 breast cancer cells (Soto-Cerrato et al., 2007a). Quantitative real-time RT-PCR analysis further revealed a two- to sixfold increase in p21CIP1 mRNA levels after treatment with 100 nM prodigiosin among the lung adenocarcinoma cells examined (Figure 2B). In sharp contrast, p27KIP1 mRNA levels were barely modulated by prodigiosin, as illustrated by both RT-PCR assay (Figure 2A, middle panel) and quantitative real-time RT-PCR analysis (Figure 2C). In line with its effect on mRNA expression, the promoter activity of CDKN1B (the gene encoding p27KIP1) was not significantly affected following prodigiosin treatment (Figure 2D). Together these results exclude the transcriptional control of p27KIP1 expression by prodigiosin but instead highlight the possibility that prodigiosin up-regulates p27KIP1 at the post-transcriptional level. Since the decline of p27KIP1 abundance during cell cycle progression is mainly a result of increased proteolysis (Chu et al., 2008), we explored the effect of prodigiosin on the protein stability of p27KIP1 using cycloheximide chase analysis. As shown in Figure 2E, it is evident that prodigiosin treatment effectively increased p27KIP1 protein stability in all cell lines examined. Particularly, the half-life of p27KIP1 protein was increased from 5.6 h to far longer than 6 h in A549 cells, while that in CL1-5 and H23 cells were enhanced from 1.7 to 4.2 h and from 1.4 to 4.8 h, respectively (Figure 2F). Taken together, these results demonstrate that prodigiosin-induced p27KIP1 up-regulation is less likely to be caused by transcriptional activation but rather is a result of p27KIP1 stabilization.
Figure 2.
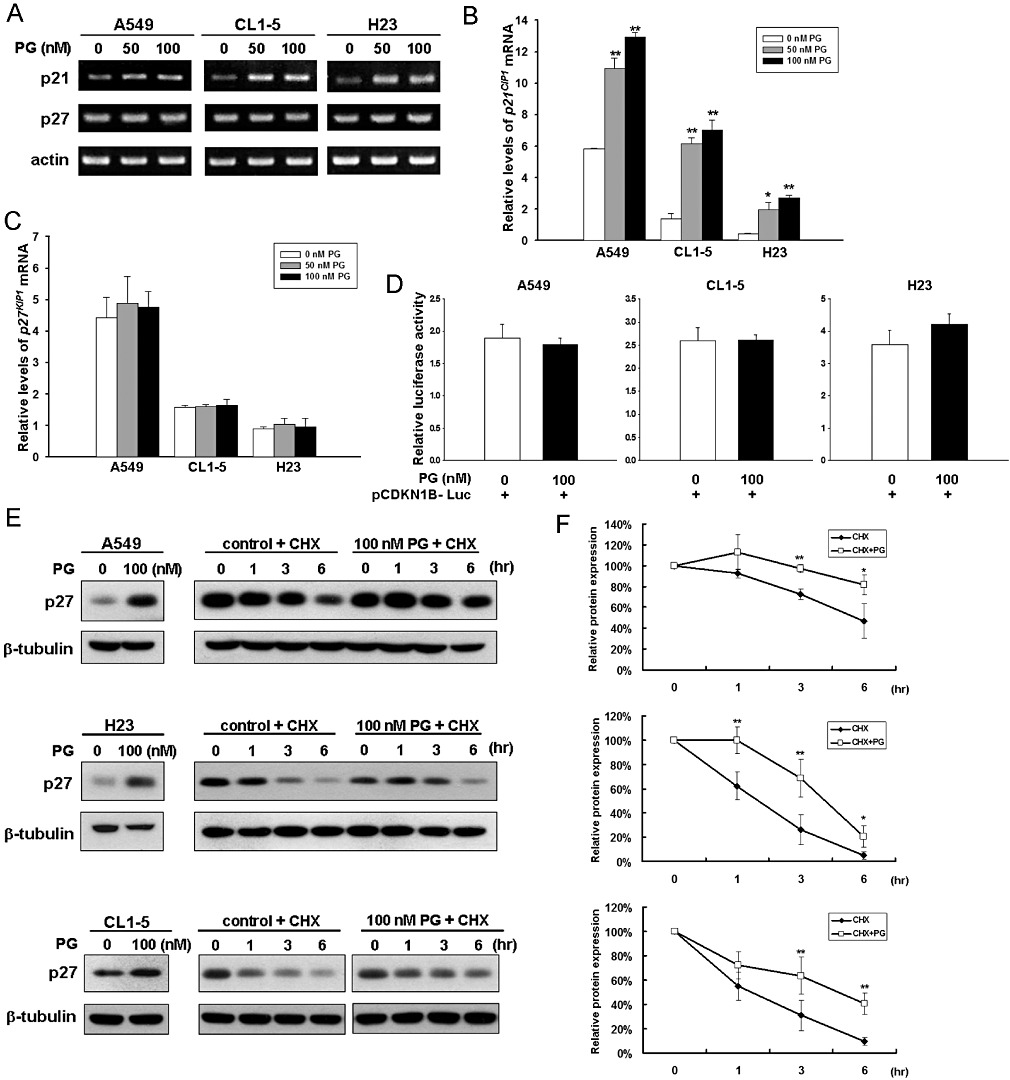
Prodigiosin up-regulates p27KIP1 predominantly by increasing protein stability. A549, CL1-5 and H23 cells were treated with prodigiosin (0–100 nM) for 24 h. Total RNA was extracted thereafter and then subjected to (A) semi-quantitative RT-PCR and quantitative real-time RT-PCR analyses for mRNA expression of p21CIP1 (B) and p27KIP1 (C). Equal loading was confirmed by the levels of human actin mRNA. An evident increase in p21CIP1 mRNA expression was observed, while the level of p27KIP1 mRNA was barely changed. All experiments were repeated at least three times with triplicate samples in each experiment. Data are expressed as means ± SEM. *P < 0.05; **P < 0.01. (D) Prodigiosin shows limited effect on the CDKN1B promoter activity. A549, CL1-5 and H23 cells were transiently transfected with the p27KIP1 promoter reporter plasmid pCDKN1B-Luc 24 h before prodigiosin treatment (100 nM). The extent of the p27KIP1 promoter activity was assessed 24 h later by determining luciferase activity. (E) Prodigiosin stabilizes p27KIP1. A549, CL1-5 and H23 cells were treated without or with prodigiosin (100 nM) in the presence of cycloheximide (60 µg·mL−1) added 18 h after prodigiosin treatment. The protein levels of p27KIP1 at 0, 1, 3 and 6 h following addition of cycloheximide were determined by immunoblotting analysis. Human β-tubulin was used as the loading control. (F) Prodigiosin extends the half-life of p27KIP1 protein. ImageJ software was used to determine the intensity of p27KIP1 protein signals on three independent immunoblots from cycloheximide experiments shown in (E). Upper panel: A549; middle panel: H23; lower panel: CL1-5. CHX, cycloheximide.
Prodigiosin transcriptionally down-regulates SKP2 to stabilize p27KIP1
We next investigated the mechanism responsible for prodigiosin-induced p27KIP1 stabilization. It is well known that the major mechanism controlling p27KIP1 stability depends on the SKP2-containing SCF ubiquitin ligase, where SKP2 recruits p27KIP1 to the SCF complex for ubiquitination and subsequent proteasomal degradation (Chan et al., 2010). Accordingly, we were interested to test whether prodigiosin down-regulates SKP2 to promote p27KIP1 stabilization. To this end, A549 cells were treated with prodigiosin (0–100 nM) for 24 h, and the level of SKP2 in drug-treated cells was evaluated by immunoblotting. It is clear that prodigiosin effectively down-regulated SKP2, accompanied by an increase in p27KIP1 levels (Figure 3A, left panel). Similarly, an evident reduction of SKP2 and a concomitant increase of p27KIP1 were also observed in CL1-5 and H23 cells following prodigiosin treatment (Figure 3A, centre and right panel respectively). Altogether, these results supported the notion that prodigiosin induces p27KIP1 stabilization through SKP2 down-regulation.
Figure 3.
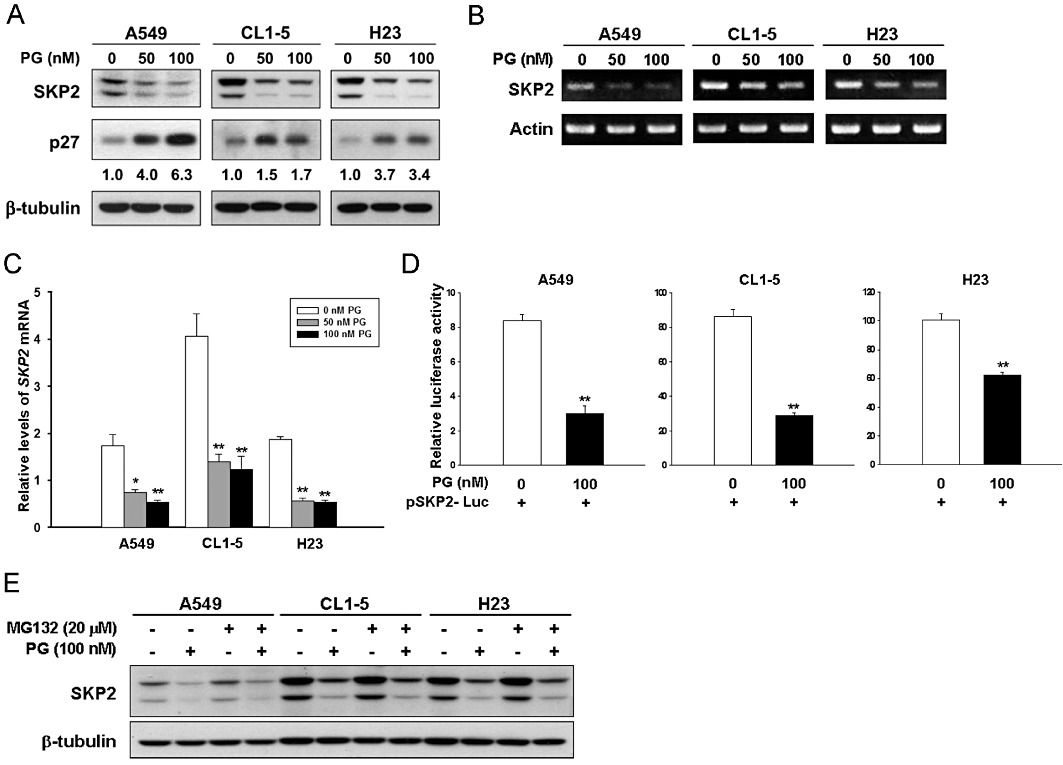
Prodigiosin transcriptionally down-regulates SKP2. (A) Prodigiosin induces a reduction in SKP2 protein levels. A549, CL1-5 and H23 cells were treated with prodigiosin (0–100 nM) for 24 h and then subjected to immunoblotting analysis for the protein levels of SKP2 and p27KIP1. Human β-tubulin was used as the loading control. (B, C) Prodigiosin decreases SKP2 mRNA expression. A549, CL1-5 and H23 cells were treated with prodigiosin (0–100 nM) for 24 h and then subjected to semi-quantitative RT-PCR (B) and quantitative real-time RT-PCR (C) analyses for the mRNA levels of SKP2. Human actin mRNA was used as the loading control. (D) Prodigiosin lowers the activity of the SKP2 promoter. A549, CL1-5 and H23 cells were transiently transfected with the SKP2 promoter reporter plasmid pSKP2-Luc 24 h before treatment with prodigiosin (100 nM). The level of the SKP2 promoter activity was determined 24 h later by luciferase activity assay. All experiments were repeated at least three times with triplicate samples in each experiment. Data are expressed as means ± SEM. *P < 0.05; **P < 0.01. (E) Prodigiosin shows limited effect on SKP2 protein stability. A549, CL1-5 and H23 cells were treated with prodigiosin (100 nM) for 24 h in the absence or presence of MG132 (20 µM) added 2 h before lysate preparation. SKP2 protein levels were evaluated by immunoblotting thereafter. Human β-tubulin was used as the loading control.
As SKP2 down-regulation by prodigiosin was clearly established, we went on to explore the underlying mechanism. To test whether prodigiosin regulates SKP2 at the level of transcription, total RNA was extracted from A549, CL1-5 and H23 cells treated with prodigiosin (0–100 nM) for 24 h and then subjected to RT-PCR analysis. We found that prodigiosin treatment clearly decreased SKP2 mRNA levels in all cell lines tested (Figure 3B). Additionally, quantitative real-time RT-PCR analysis indicated that 100 nM prodigiosin lowered the level of SKP2 mRNA to 31%, 30% and 28% of that in untreated controls in A549, CL1-5 and H23 cells respectively (Figure 3C). To further substantiate the transcriptional control of SKP2 expression by prodigiosin, a human SKP2 promoter reporter vector was constructed to examine the effect of prodigiosin on SKP2 transcription. As shown in Figure 3D, prodigiosin at 100 nM evidently inhibited the SKP2 promoter activity in all cell lines examined, indicating that prodigiosin induces transcriptional repression of SKP2 to lower the SKP2 mRNA expression. Lastly, as the main mechanism that regulate SKP2 levels is through the ubiquitin–proteasome system (Bashir et al., 2004; Wei et al., 2004), we further examined the effect of prodigiosin on SKP2 protein stability. To this end, A549, CL1-5 and H23 cells were treated with prodigiosin (100 nM) for 24 h in the absence or presence of the proteasome inhibitor MG132 to block proteasome-mediated degradation. We found that, although MG132 alone preserved SKP2 from degradation, it failed to restore the level of SKP2 reduced by prodigiosin, illustrating the limited effect of prodigiosin on SKP2 stability (Figure 3E). Cycloheximide chase analyses further confirmed that prodigiosin treatment did not significantly alter the half-life of SKP2 protein (Supplementary Figure S1). In conclusion, it is apparent that prodigiosin does not down-regulate SKP2 by the classical mechanism (i.e. protein stability) but rather controls its level of transcription.
Essential roles of SKP2 down-regulation and p27KIP1 up-regulation in the antiproliferative effect of prodigiosin
With prodigiosin's effect on p27KIP1 and SKP2 unravelled, we were anxious to define the functional significance of SKP2 down-regulation and the ensuing p27KIP1 stabilization in the antiproliferative effect of prodigiosin. To address this issue, we tested the consequence of p27KIP1 depletion or SKP2 overexpression on cell proliferation following prodigiosin treatment. To this end, A549 cells were infected with a lentiviral vector expressing p27KIP1-targeting shRNA (shp27) to suppress the level of endogenous p27KIP1, thus antagonizing prodigiosin-induced increase in p27KIP1 (Figure 4A). These p27KIP1-depleted stable clones were then subjected to colony formation assay to evaluate the effect of prodigiosin on their capacity for long-term cell proliferation. It is apparent that prodigiosin (100 nM) strongly inhibited the formation of colonies from cells carrying the luciferase-targeting shRNA (shLuc); in contrast, p27KIP1 depletion effectively protected cells from prodigiosin-induced repression of cell proliferation (Figure 4B, C). Similarly, ectopic SKP2 expression in A549 cells was achieved by infection with a retroviral vector pBabe.puro (pBabe) carrying the SKP2 open reading frame (SKP2) in order to counteract prodigiosin-induced SKP2 down-regulation and the ensuing p27KIP1 stabilization (Figure 4D). Although the colony forming ability of pBabe vector-infected cells was potently suppressed by prodigiosin, SKP2 overexpression made the cells resistant to the antiproliferative effects of prodigiosin (Figure 4E, F). Taken together, these results indicate that SKP2 down-regulation and the consequent p27KIP1 accumulation represents a fundamental mechanism underlying the antiproliferative effect of prodigiosin.
Figure 4.
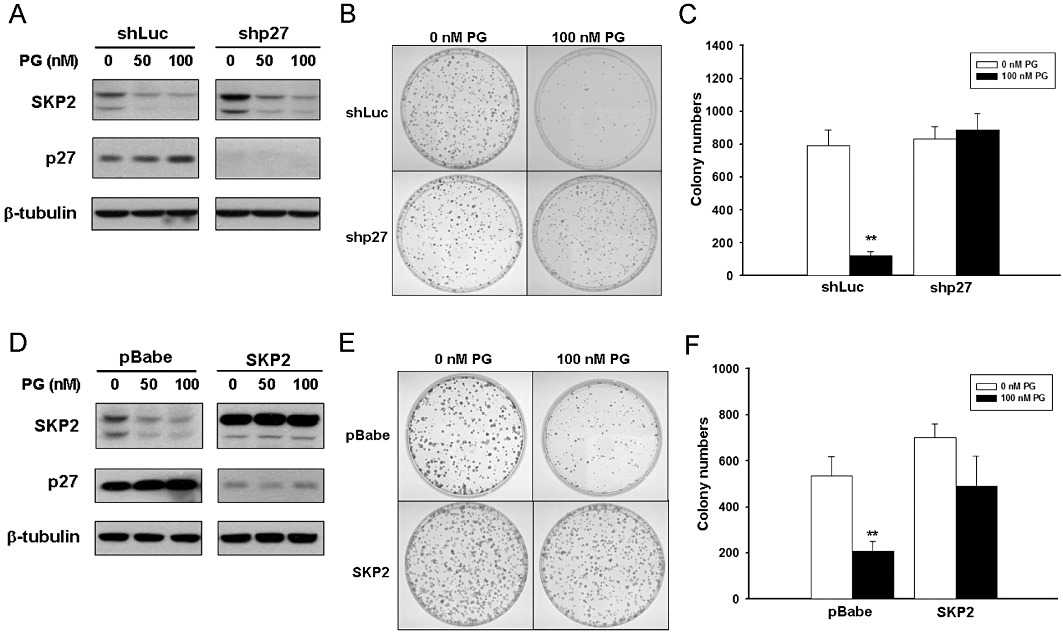
Essential roles of p27KIP1 up-regulation and SKP2 down-regulation in the antiproliferative effect of prodigiosin. (A) Depletion of endogenous p27KIP1 by RNA interference. A549 cells were infected with lentiviral vectors expressing luciferase shRNA (shLuc) or p27KIP1 shRNA (shp27), followed by puromycin selection for stable infectants. p27KIP1 depletion was validated by immunoblotting analysis. Human β-tubulin was used as the loading control. (B) p27KIP1 depletion confers resistance to prodigiosin-induced antiproliferation. Shown here are representative images of the colony forming capacity of shLuc- or shp27-infected cells treated without or with 100 nM of prodigiosin. (C) Quantitative analysis of the colony forming capacity of prodigiosin-treated A549 cells without or with p27KIP1 depletion. Colony numbers were counted in three independent experiments shown in (C). Data are expressed as means ± SEM. **P < 0.01. (D) Ectopic expression of SKP2. A549 cells were infected with the retroviral vector pBabe.puro alone (pBabe) or with the pBabe.puro vector carrying the SKP2 open reading frame (SKP2). SKP2 overexpression and the resulting decrease in p27KIP1 levels were verified by immunoblotting analysis. Human β-tubulin was used as the loading control. (E) Ectopic expression of SKP2 restores the capacity of colony formation in prodigiosin-treated cells. Shown here are representative images of the colony forming capacity in control or SKP2-overexpressing cells following treatment without or with prodigiosin (100 nM). (F) Quantitative analysis for the colony forming capacity of prodigiosin-treated A549 cells without or with ectopic expression of SKP2. Colony numbers were counted in three independent experiments shown in (E). Data are expressed as means ± SEM. **P < 0.01.
Prodigiosin engages PKB-mediated signalling to induce SKP2 down-regulation
We next investigated the upstream signalling that mediates prodigiosin-induced SKP2 down-regulation. It is noteworthy that previous studies have implicated PI3K/PKB signalling in promoting SKP2 expression (Chan et al., 2010). We therefore investigated whether prodigiosin modulates the PI3K/PKB signalling to down-regulate SKP2. To determine the effect of prodigiosin on PKB, A549 cells were treated with increasing doses of prodigiosin for 24 h, and the level of PKB phosphorylation at serine 473 (Ser473) was determined by immunoblotting analysis. As shown in Figure 5A, prodigiosin treatment caused a dose-dependent reduction in the level of Ser473-phosphorylated PKB. To further validate the consequence of PKB dephosphorylation at Ser473, we evaluated the phosphorylation status of GSK-3β, a well-known downstream substrate of PKB whose activity is inhibited by PKB-mediated phosphorylation at serine 9 (Ser9). We found that prodigiosin dose-dependently lowered the level of GSK-3β with Ser9 phosphorylation, illustrating the activation of GSK-3β as a consequence of PKB dephosphorylation/inhibition following prodigiosin treatment (Figure 5A). Additionally, time course studies revealed that prodigiosin (100 nM) induced a time-dependent Ser473 dephosphorylation of PKB. In particular, the level of Ser473-phosphrylated PKB was clearly reduced by prodigiosin at 8 h and sharply declined after 12 h of treatment. Intriguingly, the kinetic change of Ser473-phosphorylated PKB levels was seemingly paralleled by a decrease in the levels of Ser9-phosphorylated GSK-3β and SKP2 along with an increase in p27KIP1 expression (Figure 5B).
Figure 5.
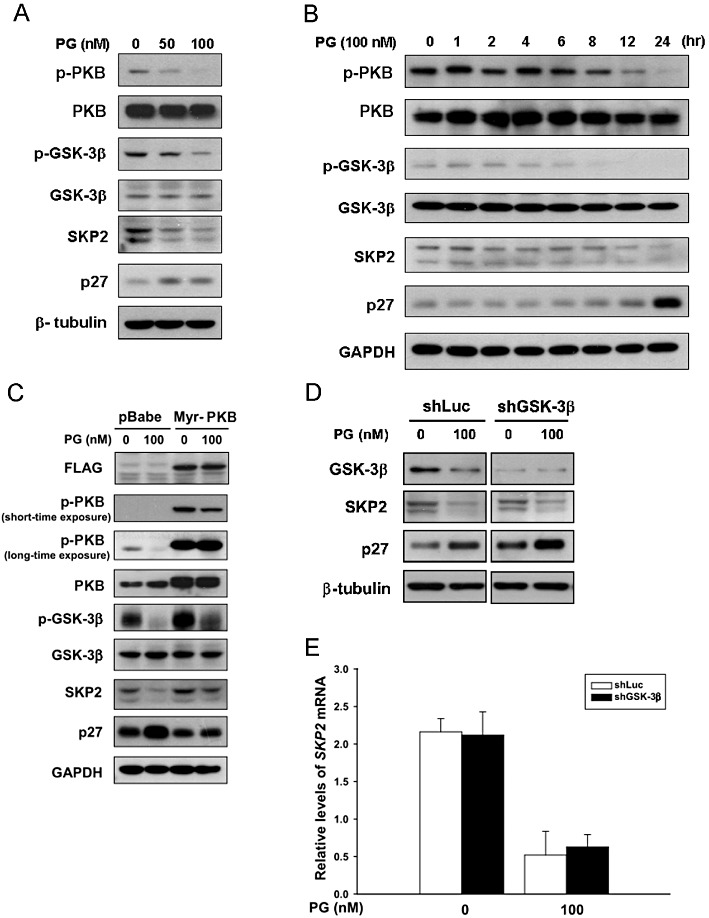
Prodigiosin engages PKB-mediated signalling to induce SKP2 down-regulation. (A) Prodigiosin induces PKB inhibition. A549 cells were treated with graded doses of prodigiosin (0–100 nM) for 24 h, followed by immunoblotting analysis for the levels of Ser473-phosphorylated PKB (p-PKB), total PKB (PKB), Ser9-phosphorylated (inactivated) GSK-3β (p-GSK-3β), total GSK-3β, SKP2 and p27KIP1. Human β-tubulin was used as the loading control. (B) Time-dependent Ser473 dephosphorylation of PKB, activation of GSK-3β, down-regulation of SKP2 and the ensuing accumulation of p27KIP1 following prodigiosin treatment. A549 cells were treated with 100 nM of prodigiosin for 24 h, and the levels of proteins shown in (A) were evaluated by immunoblotting at the time points indicated. Human GAPDH was used as the loading control. (C) Constitutive PKB activation restores SKP2 expression in prodigiosin-treated cells. A549 cells were infected with pBabe.puro vector alone (pBabe) or the pBabe vector expressing the constitutively active mutant of PKB (Myr-PKB). Stably infected A549 cells were then treated without or with prodigiosin (100 nM) for 24 h and the levels of protein shown in (A) were determined thereafter. Constitutive PKB activation was validated by the high levels of Ser473-phosphorylated PKB and Ser9-phosphorylated GSK-3β. Human GAPDH was used as the loading control. (D) GSK-3β is not essential for prodigiosin-induced reduction of SKP2. A549 cells were infected with lentiviral vectors expressing luciferase-targeting shRNA (shLuc) or GSK-3β-targeting shRNA (shGSK-3β). The stable infectants were then subjected to 24 h-treatment with 100 nM prodigiosin, followed by immunoblotting analyses for GSK-3β, SKP2 and p27KIP1. It is evident that GSK-3β was largely depleted by shGSK-3β, but SKP2 levels were still reduced by prodigiosin regardless of GSK-3β depletion. Human β-tubulin was used as the loading control. (E) GSK-3β depletion fails to restore SKP2 transcription in prodigiosin-treated cells. Control or GSK-3β-depleted A549 cells were subjected to real-time RT-PCR analysis to evaluate SKP2 mRNA expression after prodigiosin treatment for 24 h. In agreement with the protein expression pattern shown in (D), the level of SKP2 mRNA was lowered by prodigiosin irrespective of GSK-3β depletion. All experiments were repeated at least three times with triplicate samples in each experiment. Data are expressed as means ± SEM. **P < 0.01.
Since PKB activity promotes SKP2 expression and is inhibited by prodigiosin, we investigated whether prodigiosin down-regulates SKP2 through inhibition of PKB. To test this, A549 cells were infected with a pBabe.puro vector expressing an N-terminally myristoylated PKB (Myr-PKB), a constitutively active mutant of PKB. These Myr-PKB-expressing A549 cells were subjected to treatment with prodigiosin (100 nM) for 24 h and immunoblotting analysis thereafter. It is noteworthy that expression of Myr-PKB indeed neutralized the inhibitory effect of prodigiosin on PKB activity, as verified by the high level of Ser473-phosphorylated PKB and the restoration of Ser9-phosphorylated GSK-3β that was nearly absent in control cells treated with prodigiosin (Figure 5C). Importantly, prodigiosin failed to lower SKP2 when PKB activity was preserved by Myr-PKB, confirming the essential role of PKB inhibition in prodigiosin-induced SKP2 down-regulation.
As PKB inhibition leads to GSK-3β activation and GSK-3β has been reported to lower SKP2 expression in colon cancer cells (Wang et al., 2008), we further examined the role of GSK-3β in prodigiosin-induced SKP2 down-regulation. To this end, endogenous GSK-3β expression in A549 cells was suppressed by a lentiviral vector-expressing GSK-3β-targeting shRNA (shGSK-3beta), followed by 24 h treatment with prodigiosin (0 or 100 nM) and immunoblotting or quantitative real-time RT-PCR analyses for SKP2 expression thereafter. As shown in Figure 5D, the level of endogenous GSK-3β protein was nearly depleted by GSK-3β shRNA; nevertheless, GSK-3β depletion failed to restore SKP2 expression at both the protein and mRNA levels (Figure 5D, E, respectively). Altogether, we concluded that GSK-3β is not essential for prodigiosin-induced SKP2 down-regulation, which further indicates that other effectors downstream of PKB inhibition are responsible for prodigiosin's inhibitory effect on SKP2 expression.
E2F1 is not responsible for prodigiosin-induced SKP2 down-regulation
Given that SKP2 is transcriptionally repressed by prodigiosin, we were interested to identify the transcription factor(s) involved in this effect. Previous studies have identified SKP2 as a transcriptional target of E2F1 (Zhang and Wang, 2006), and, intriguingly, E2F1 has been shown to mediate PI3K/PKB signalling-induced transcription of SKP2 (Reichert et al., 2007). We therefore investigated whether E2F1 is the transcription factor mediating prodigiosin-induced transcriptional repression of SKP2. As shown in Figure 6A, a dose-dependent reduction in E2F1 protein levels was observed following prodigiosin treatment for 24 h. We next examined whether prodigiosin down-regulates E2F1 to lower SKP2. To this end, A549 cells were infected with a pBabe.puro vector encoding HA-tagged E2F1 (HA-E2F1) for ectopic expression of E2F1. Indeed, the inhibitory effect of prodigiosin on E2F1 expression and activity was compromised in A549 cells overexpressing HA-E2F1, as illustrated by the high levels of E2F1 and its bona fide target gene cyclin E (Ohtani et al., 1995), respectively (Figure 6B). However, SKP2 levels were still effectively reduced by prodigiosin regardless of enforced E2F1 expression (Figure 6B). Together these results exclude the involvement of E2F1 in prodigiosin-induced SKP2 down-regulation.
Figure 6.
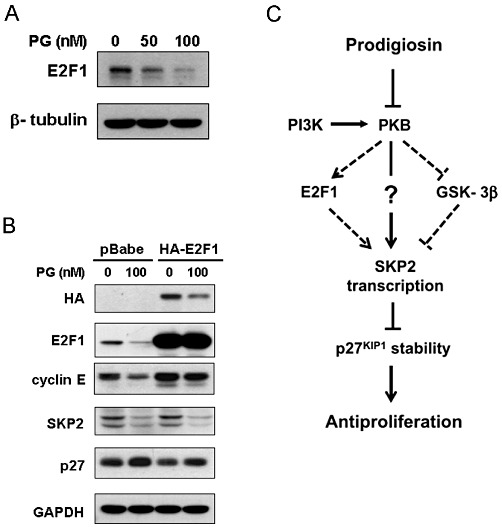
E2F1 is not responsible for prodigiosin-induced transcriptional repression of SKP2. (A) Dose-dependent reduction in E2F1 by prodigiosin. A549 cells were treated with increasing doses of prodigiosin (0–100 nM) for 24 h, and the protein levels of E2F1 were determined thereafter using immunoblotting analysis. Human β-tubulin was used as the loading control. (B) Ectopic expression of E2F1 fails to restore SKP2 expression in prodigiosin-treated cells. A549 cells were infected with pBabe.puro vector alone (pBabe) or with pBabe vector expressing HA-tagged E2F1 (HA-E2F1). The effect of E2F1 overexpression on SKP2 levels in prodigiosin-treated cells was examined thereafter. E2F1 activity was preserved in E2F1-overexpressing cells treated with prodigiosin, as shown by the high levels of cyclin E expression, a bona fide transcriptional target of E2F1. (C) A proposed model for the mechanistic understanding of prodigiosin's antiproliferative action. In this study, we established that prodigiosin induces PKB inhibition for transcriptional repression of SKP2, leading to cellular accumulation of p27KIP1 to suppress cell proliferation. Neither GSK-3β nor E2F1 is involved in this process. The effectors downstream of PKB responsible for prodigiosin-induced repression of SKP2 transcription remain to be identified.
Discussion and conclusions
In this study, we have presented evidence demonstrating that prodigiosin targets the SKP2–p27KIP1 axis to induce antiproliferation in human lung adenocarcinoma cells. In particular, we showed that prodigiosin treatment leads to cell cycle arrest in the G1 phase, along with increased expression of CDK inhibitors p21CIP1 and p27KIP1. Subsequent analyses revealed that prodigiosin up-regulates p27KIP1 largely by inducing p27KIP1 stabilization as a result of transcriptional repression of SKP2. Furthermore, we proved that prodigiosin engages PKB-mediating signalling to down-regulate SKP2. Notably, ectopic expression of SKP2 or depletion of p27KIP1 prevented the inhibitory effect of prodigiosin on long-term cell proliferation, illustrating the essential role of SKP2 down-regulation and the resulting p27KIP1 accumulation in prodigiosin's antiproliferative action. To the best of our knowledge, this is the first report indicating that the SKP2–p27KIP1 axis is involved in the antiproliferative effect of prodigiosin.
Using human breast adenocarcinoma cell line MCF-7 as the cell model, Soto-Cerrato et al. (2007a) have shown that non-cytotoxic doses of prodigiosin trigger G1 phase arrest of the cell cycle and an increase in p21CIP1 levels, which is attributed to increased p21CIP1 mRNA abundance induced by the TGF-β signalling pathway. Likewise, we observed that prodigiosin induced G1 cell cycle arrest and increased the expression of p21CIP1 mRNA and protein in multiple human lung adenocarcinoma cell lines. Importantly, we further identified p27KIP1 and SKP2 as additional molecular targets of prodigiosin. Given that p21CIP1 and p27KIP1 are crucial for restraining the G1–S phase transition (Niculescu III et al., 1998; Chu et al., 2008), the up-regulation of p21CIP1 and p27KIP1 by prodigiosin in human lung adenocarcinoma cells would seem to be an effective mode of action for it to induce antiproliferation.
In contrast to its promoting effect on p21CIP1 mRNA expression, prodigiosin barely modulates CDKN1B transcription but rather up-regulates p27KIP1 by inducing p27KIP1 stabilization (Figure 2). This finding led us to discover that one mechanism whereby prodigiosin stabilizes p27KIP1 is through down-regulation of SKP2, an F-box protein responsible for targeting the proteolysis of p27KIP1 (Frescas and Pagano, 2008) (Figure 3). In fact, an inverse pattern of expression between SKP2 and p27KIP1 was consistently observed in prodigiosin-treated cells throughout this study. The notion that prodigiosin down-regulates SKP2 to stabilize p27KIP1 was further substantiated by our observations that the increase in p27KIP1 levels following prodigiosin treatment was nearly abolished by manipulations that counteract prodigiosin-induced SKP2 down-regulation, either by enforced expression of SKP2 (Figure 4) or by inducing constitutive activation of PKB (Figure 5). Our results therefore are consistent with the notion that p27KIP1 is the primary physiological target of SKP2 with respect to cell proliferation, as supported by the fact that the generalized hypoplasia phenotypes of the Skp2−/− mice is associated with cellular accumulation of p27Kip1 and, notably, can be rescued by concomitant deletion of Cdkn1b (Nakayama et al., 2000; 2004). In line with this, we found that cells with ectopic expression of SKP2 or depletion of p27KIP1 were resistant to prodigiosin-induced antiproliferation (Figure 4). It is worth noting that the induction of SKP2 down-regulation and the ensuing p27KIP1 accumulation represents a major mode of action underlying the antiproliferative effect of a broad range of anticancer agents, including bortezomib (Uddin et al., 2008; 2009) and imatinib (Liu et al., 2008). Our findings thus add prodigiosin to the growing list of chemotherapeutic agents that target the SKP2–p27KIP1 axis to suppress cell proliferation.
A number of reports have indicated that prodigiosin induces transcriptional up-regulation of some genes while inhibition of others. For example, prodigiosin is known to increase the mRNA levels of p21CIP1, NAG-1, DR4 and DR5 (Soto-Cerrato et al., 2007a, b). Similarly, in this study, we showed an increase in p21CIP1 mRNA levels following prodigiosin treatment. In contrast, both the mRNA levels and the promoter activity of survivin (Ho et al., 2009) and SKP2 (in this study) were down-regulated by prodigiosin. It is also noteworthy that the expression of certain genes is not subjected to transcriptional control by prodigiosin. For instance, in this study, we demonstrated that neither the mRNA expression nor the promoter activity of the gene encoding p27KIP1 was altered by prodigiosin. Together, these findings rule out the possibility that induction of transcriptional inhibition is a general effect of prodigiosin.
The data presented here indicate that prodigiosin induces Ser473 dephosphorylation and thus inhibition of PKB to facilitate SKP2 down-regulation (Figure 5A, B). This notion was substantiated by the finding that prodigiosin-induced SKP2 down-regulation was nearly abolished when PKB activity was preserved through enforced expression of a constitutively active mutant of PKB (Figure 5C). In fact, recent studies have unravelled the role of PI3K/PKB signalling as a positive regulator of SKP2 at multiple levels, including transcription, stability, activity and subcellular localization (Chan et al., 2010). In this context, PI3K/PKB signalling is required for SKP2 transcription, because inhibition of PI3K activity by LY294002 or depletion of PKB1 by siRNA lowers the levels of SKP2 mRNA and SKP2 promoter activity (Reichert et al., 2007). At the post-transcriptional level, PKB1-mediated phosphorylation of SKP2 at Ser72 results in cytoplasmic localization and stabilization of SKP2, and also facilitates the assembly of the SKP2–SCF complex to enhance the ubiquitination and subsequent proteolysis of p27KIP1 (Ecker and Hengst, 2009; Gao et al., 2009; Lin et al., 2009). Accordingly, it is plausible that inhibition of PKB-mediated signalling is a fundamental mechanism whereby prodigiosin down-regulates the expression of SKP2 and, possibly, the assembly and activity of the SKP2–SCF complex as well.
Signalling effectors acting downstream of PKB inhibition to induce SKP2 down-regulation were also examined in this study. Prodigiosin was found to promote GSK-3β activation, as demonstrated by the decrease in PKB-mediated Ser9 phosphorylation (Figure 5A, B). Interestingly, GSK-3β is responsible for sodium butyrate (NaBT)-induced transcriptional repression of SKP2 to promote nuclear p27KIP1 accumulation, leading to intestinal cell differentiation (Wang et al., 2008). For that reason, GSK-3β appears to be a potential candidate mediating prodigiosin's inhibitory effect on SKP2 expression. To our surprise, GSK-3β depletion failed to restore SKP2 expression following prodigiosin treatment (Figure 5D, E), illustrating that GSK-3β is not essential for prodigiosin-induced SKP2 down-regulation.
In this study, we demonstrated that prodigiosin down-regulates SKP2 mainly at the level of mRNA expression. Specifically, it is the level of SKP2 transcription that is regulated by prodigiosin, as demonstrated by prodigiosin-induced inhibition of SKP2 promoter activity (Figure 3D) in addition to the limited effect of prodigiosin on SKP2 mRNA stability (Supplementary Figure S2). Accumulating evidence has identified SKP2 as a transcriptional target of multiple transcription factors including E2F1, NF-κB, CBF1, GABP, FOXM1 and MYC (Chan et al., 2010 and references therein; Bretones et al., 2011), while SP1 and FOXP3 act as SKP2 transcriptional repressors (Huang and Hung, 2006; Zuo et al., 2007). Intriguingly, E2F1 was proved to mediate PI3K/PKB signalling-induced SKP2 transcription in colon cancer cells (Reichert et al., 2007). We found that prodigiosin down-regulated E2F1 (Figure 6A), but the involvement of E2F1 in prodigiosin-induced SKP2 repression was ruled out later because ectopic E2F1 expression failed to restore the expression of SKP2 (Figure 6B). Therefore, the effectors other than GSK-3β and E2F1 that act downstream of PKB to mediate prodigiosin-induced transcriptional repression of SKP2 still need to be identified.
In conclusion, our study identified SKP2 and p27KIP1 as novel molecular targets responsible for the antiproliferative action of prodigiosin in human lung adenocarcinoma cells. Mechanistically, prodigiosin induces PKB inhibition to transcriptionally down-regulate SKP2 in a GSK-3β- and E2F1-independent manner, consequently leading to p27KIP1 accumulation to suppress cell proliferation (Figure 6C). Importantly, this mode of action provides new insights into the molecular basis underlying the antiproliferative action of prodigiosin. Moreover, considering the oncogenic potential of SKP2 in multiple human cancers and the potent senescence-inducing effect of SKP2 inactivation on cancer cells (Lin et al., 2010), SKP2 represents a highly promising drug target for cancer treatment. In view of that, prodigiosin holds great potential for chemotherapy or to serve as a lead compound for the development of novel anticancer therapeutics targeting SKP2.
Acknowledgments
This work was supported in part by grants from the Taichung Veterans General Hospital and National Chung Hsing University, Taichung, Taiwan (TCVGH-NCHU997606 to C-CC/C-HL), from the National Science Council, Taiwan, ROC (NSC 99-2320-B-005- 004-MY3 to C-CC) and from the Ministry of Education, Taiwan, ROC under the ATU plan. The authors are indebted to Prof Yun-Wei Lin (National Chiayi University), Prof Jeremy JW Chen (National Chung Hsing University) and Prof Jiunn-Liang Ko (Chung Shan Medical University) for providing human lung adenocarcinoma cell lines A549, CL1-5 and H23, respectively, as well as Cong-Hao Lai and Ya-Ting Yang for their technical assistance. The authors are also grateful to Dr Wan-Lai Chang for scientific editing.
Glossary
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GSK-3β
glycogen synthase kinase-3β
- MG-132
carbobenzoxy-L-leucyl-L-leucyl-L-leucinal
- NAG
non-steroidal anti-inflammatory drug-activated gene-1
- PI3K
phosphoinositide 3-kinases
- SCFSKP2
SKP1/Cullin/F-box protein: SKP2
- SKP1
S-phase kinase protein 1
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Cycloheximide chase assay reveals the limited effect of prodigiosin on SKP2 protein stability. (A) A549, CL1-5 and H23 cells were treated without or with prodigiosin (100 nM) in the presence of cycloheximide (60 μg·mL−1) added 18 h after prodigiosin treatment. The protein levels of SKP2 at 0, 1, 3 and 6 h following addition of cycloheximide were determined by immunoblotting analysis. Human β-tubulin was used as the loading control. (B) ImageJ software was used to determine the intensity of SKP2 proteinsignals on three independent immunoblots from cycloheximide chase experiments shown in (A). Upper panel: A549; middle panel: CL1-5; lower panel: H23. ♦: cells with cycloheximide treatmentalone; □: cells co-treated with prodigiosin andcycloheximide. PG, prodigiosin; CHX, cycloheximide.
Figure S2 Prodigiosin shows limited effect on SKP2 mRNA stability. A549 cells were treated without or with 100 nM of PG for 24 h. Actinomycin D (Act.D) (2 μg·mL−1) were added during the final 6 h, and total RNAs of cells collected at the indicated time points were prepared, reverse transcribed and then subjected to quantitative real-time PCR analysis. The levels of SKP2 mRNA expression were normalized to that of TBP. Final results were expressed as the ratio of copy numbers of SKP2 mRNA to TBP mRNA copy numbers and presented as mean ± SEM of three independent experiments. *P < 0.05.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Agarwal A, Bumm TG, Corbin AS, O'Hare T, Loriaux M, VanDyke J, et al. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood. 2008;112:1960–1970. doi: 10.1182/blood-2007-09-113860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Borriello A, Bencivenga D, Criscuolo M, Caldarelli I, Cucciolla V, Tramontano A, et al. Targeting p27Kip1 protein: its relevance in the therapy of human cancer. Expert Opin Ther Targets. 2011;15:677–693. doi: 10.1517/14728222.2011.561318. [DOI] [PubMed] [Google Scholar]
- Bretones G, Acosta JC, Caraballo JM, Ferrándiz N, Gómez-Casares MT, Albajar M, et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27KIP1 through SKP2 in human leukemia cells. J Biol Chem. 2011;286:9815–9825. doi: 10.1074/jbc.M110.165977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo VD, Gibbons DL, Pérez-Soler R, Quintás-Cardama A. Treatment of non–small-cell lung cancer with erlotinib or gefitinib. N Engl J Med. 2011;364:947–955. doi: 10.1056/NEJMct0807960. [DOI] [PubMed] [Google Scholar]
- Chan CH, Lee SW, Wang J, Lin HK. Regulation of Skp2 expression and activity and its role in cancer progression. Scientificworldjournal. 2010;10:1001–1015. doi: 10.1100/tsw.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chen WC, Ho TF, Wu HS, Wei YH. Development of natural anti-tumor drugs by microorganisms. J Biosci Bioeng. 2011;111:501–511. doi: 10.1016/j.jbiosc.2010.12.026. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Peck K, Hong TM, Yang SC, Sher YP, Shih JY, et al. Global analysis of gene expression in invasion by a lung cancer model. Cancer Res. 2001;61:5223–5230. [PubMed] [Google Scholar]
- Chen JY, Wang MC, Hung W. Transcriptional activation of Skp2 by BCR-ABL in K562 chronic myeloid leukemia cells. Leuk Res. 2009;33:1520–1524. doi: 10.1016/j.leukres.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the p27 E3 ligase SCFSkp2 results in p27- and Skp2-mediated cell cycle arrest and activation of autophagy. Blood. 2008;111:4690–4699. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- Ecker K, Hengst L. Skp2: caught in the Akt. Nat Cell Biol. 2009;11:377–379. doi: 10.1038/ncb0409-377. [DOI] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TF, Peng YT, Chuang SM, Lin SC, Feng BL, Lu CH, et al. Prodigiosin down-regulates survivin to facilitate paclitaxel sensitization in human breast carcinoma cell lines. Toxicol Appl Pharmacol. 2009;235:253–260. doi: 10.1016/j.taap.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Huang HC, Way TD, Lin CL, Lin JK. EGCG stabilizes p27kip1 in E2-stimulated MCF-7 cells through down-regulation of the Skp2 protein. Endocrinology. 2008;149:5972–5983. doi: 10.1210/en.2008-0408. [DOI] [PubMed] [Google Scholar]
- Huang YC, Hung WC. 1,25-dihydroxyvitamin D3 transcriptionally represses p45Skp2 expression via the Sp1 sites in human prostate cancer cells. J Cell Physiol. 2006;209:363–369. doi: 10.1002/jcp.20741. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YH, Li T, Yuan Z, Haigentz JM, Weber TK, Pérez-Soler R. Erlotinib, an effective epidermal growth factor receptor tyrosine kinase inhibitor, induces p27KIP1 up-regulation and nuclear translocation in association with cell growth inhibition and G1/S phase arrest in human non-small-cell lung cancer cell lines. Mol Pharmacol. 2007;72:248–258. doi: 10.1124/mol.107.034827. [DOI] [PubMed] [Google Scholar]
- Liu S, Yamauchi H. p27-associated G1 arrest induced by hinokitiol in human malignant melanoma cells is mediated via down-regulation of pRb, Skp2 ubiquitin ligase, and impairment of cdk2 function. Cancer Lett. 2009;286:240–249. doi: 10.1016/j.canlet.2009.05.038. [DOI] [PubMed] [Google Scholar]
- Liu Y, Perdreau SA, Chatterjee P, Wang L, Kuan SF, Duensing A. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res. 2008;68:9015–9023. doi: 10.1158/0008-5472.CAN-08-1935. [DOI] [PubMed] [Google Scholar]
- Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Niculescu AB, III, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Chander R, Sainis KB. Prodigiosins as anti-cancer agents: living upto their name. Curr Pharm Des. 2009;15:732–741. doi: 10.2174/138161209787582192. [DOI] [PubMed] [Google Scholar]
- Pang PH, Lin YH, LeeYH HHH, Hsu SP, Juan SH. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol. 2008;215:161–171. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- Pérez-Tomás R, Viñas M. New insights on the antitumoral properties of prodiginines. Curr Med Chem. 2010;17:2222–2231. doi: 10.2174/092986710791331103. [DOI] [PubMed] [Google Scholar]
- Ramalingam SS, Owonikoko TK, Khuri FR. Lung Cancer: new biological insights and recent therapeutic advances. CA Cancer J Clin. 2011;61:91–112. doi: 10.3322/caac.20102. [DOI] [PubMed] [Google Scholar]
- Reichert M, Saur D, Hamacher R, Schmid RM, Schneider G. Phosphoinositide-3-kinase signaling controls S-phase kinase-associated protein 2 transcription via E2F1 in pancreatic ductal adenocarcinoma cells. Cancer Res. 2007;67:4149–4156. doi: 10.1158/0008-5472.CAN-06-4484. [DOI] [PubMed] [Google Scholar]
- Sonoda H, Inoue H, Ogawa K, Utsunomiya T, Masuda TA, Mori M. Significance of skp2 expression in primary breast cancer. Clin Cancer Res. 2006;12:1215–1220. doi: 10.1158/1078-0432.CCR-05-1709. [DOI] [PubMed] [Google Scholar]
- Soto-Cerrato V, Viñals F, Lambert JR, Pérez-Tomás R. The anticancer agent prodigiosin induces p21WAF1/CIP1 expression via transforming growth factor-beta receptor pathway. Biochem Pharmacol. 2007a;4:1340–1349. doi: 10.1016/j.bcp.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Soto-Cerrato V, Viñals F, Lambert JR, Kelly JA, Pérez-Tomás R. Prodigiosin induces the proapoptotic gene NAG-1 via glycogen synthase kinase-3β activity in human breast cancer cells. Mol Cancer Ther. 2007b;6:362–369. doi: 10.1158/1535-7163.MCT-06-0266. [DOI] [PubMed] [Google Scholar]
- Uddin S, Ahmed M, Bavi P, El-Sayed R, Al-Sanea N, AbdulJabbar A, et al. Bortezomib (Velcade) induces p27Kip1 expression through S-phase kinase protein 2 degradation in colorectal cancer. Cancer Res. 2008;68:3379–3388. doi: 10.1158/0008-5472.CAN-07-6109. [DOI] [PubMed] [Google Scholar]
- Uddin S, Ahmed M, Hussain AR, Jehan Z, Al-Dayel F, Munkarah A, et al. Bortezomib-mediated expression of p27Kip1 through S-phase kinase protein 2 degradation in epithelial ovarian cancer. Lab Invest. 2009;89:1115–1127. doi: 10.1038/labinvest.2009.75. [DOI] [PubMed] [Google Scholar]
- Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat Genet. 2010;42:83–88. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhou Y, Wang X, Evers BM. p27Kip1 nuclear localization and cyclin-dependent kinase inhibitory activity are regulated by glycogen synthase kinase-3 in human colon cancer cells. Cell Death Differ. 2008;15:908–919. doi: 10.1038/cdd.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Williamson NR, Fineran PC, Leeper FJ, Salmond GP. The biosynthesis and regulation of bacterial prodiginines. Nat Rev Microbiol. 2006;4:887–899. doi: 10.1038/nrmicro1531. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu H, Chen J, Zhou Q. Acquired resistance of lung adenocarcinoma to EGFR-tyrosine kinase inhibitors gefitinib and erlotinib. Cancer Biol Ther. 2010;9:572–582. doi: 10.4161/cbt.9.8.11881. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Zhang G, Haura EB. Targeting epidermal growth factor receptor: central signaling kinase in lung cancer. Biochem Pharmacol. 2010;80:613–623. doi: 10.1016/j.bcp.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang C. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 2006;25:2615–2627. doi: 10.1038/sj.onc.1209286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo T, Liu R, Zhang H, Chang X, Liu Y, Wang L, et al. FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J Clin Invest. 2007;117:3765–3773. doi: 10.1172/JCI32538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.