

Abstract
BACKGROUND AND PURPOSE
Resveratrol is a polyphenol abundantly found in grape skin and red wine. In the present study, we investigated whether resveratrol exerts protective effects against cardiac ischaemia/reperfusion and also explored its mechanisms.
EXPERIMENTAL APPROACH
Infarct size and functional recovery in rat isolated perfused hearts subjected to no-flow global ischaemia followed by reperfusion were measured. Cultured neonatal rat cardiomyocytes were exposed to H2O2 (100 µmol·L−1) to induce cell injury. Intracellular ion concentrations were measured using specific dyes. Western blotting was used to quantify protein expression levels.
KEY RESULTS
In rat isolated perfused hearts, treatment with resveratrol (20 and 100 µmol·L−1) 15 min before ischaemia considerably improved left ventricular functional recovery and infarct size. In cultured neonatal rat cardiomyocytes, resveratrol significantly attenuated the increase in reactive oxygen species (ROS) and loss of mitochondrial inner membrane potential. Resveratrol also suppressed the increase in intracellular concentrations of Na+ ([Na+]i) and Ca2+ ([Ca2+]i) after H2O2 application; however, it did not suppress the ouabain-induced [Ca2+]i increase. By measuring changes in intracellular pH recovery after acidification, we also confirmed that acid-induced activation of the Na+-H+ exchanger (NHE) was prevented by pretreatment with resveratrol. Furthermore, resveratrol inhibited the H2O2-induced translocation of PKC-α from the cytosol to the cell membrane; this translocation is believed to activate NHE.
CONCLUSION AND IMPLICATIONS
Resveratrol exerts cardioprotection by reducing ROS and preserving mitochondrial function. The PKC-α-dependent inhibition of NHE and subsequent attenuation of [Ca2+]i overload may be a cardioprotective mechanism.
Keywords: resveratrol, cardiomyocyte, Na+-H+ exchanger, cardioprotection, PKC-α
Introduction
Resveratrol, a polyphenol abundantly found in red wine and grapes, has been reported to exert beneficial effects such as anti-inflammation, inhibition of tumour cell growth, extension of lifespan and cardioprotection against ischaemia/reperfusion injury. Several mechanisms of these effects have been proposed, such as antioxidative action (Ray et al., 1999; Danz et al., 2009), up-regulation of NO synthesis (Hattori et al., 2002; Das et al., 2005) and activation of the sirtuin-1 (Sirt1) pathway (Chen et al., 2009). However, the precise mechanisms of resveratrol-induced cardioprotection have not yet been established.
The sarcolemmal Na+-H+ exchanger (NHE) plays a role in regulation of intracellular pH (pHi) by H+ extrusion driven by the transmembrane Na+ gradient in cardiomyocytes. NHE activity is increased during ischaemia in response to intracellular acidosis (Avkiran and Marber, 2002). Increases in the intracellular concentration of Na+ ([Na+]i) induced by increased Na+-H+ exchange facilitate Ca2+ influx through the Na+-Ca2+ exchanger (NCX) working in reverse mode and exacerbate cell injury (transporter nomenclature follows Alexander et al., 2011). Hence, the inhibition of NHE is expected to exert cardioprotection by indirectly limiting increases in intracellular concentration of Ca2+ ([Ca2+]i). It has also been reported that several NHE inhibitors protected cardiomyocytes subjected to oxidative stress and ischaemia/reperfusion with consistent improvement in functional recovery (Scholz et al., 1995; Knight et al., 2001; Teshima et al., 2003; Garciarena et al., 2011). Furthermore, a clinical study revealed that the inhibition of NHE during reperfusion therapy preserved cardiac function in patients with acute myocardial infarction (Rupprecht et al., 2000). In addition, the sub-analysis of a large clinical trial showed that the use of cariporide, a NHE inhibitor, in patients who underwent high-risk coronary artery bypass graft surgery reduced the incidence of myocardial infarction and mortality (Boyce et al., 2003). Recently, resveratrol has been reported to decrease the expression of the NHE isoform 1 (NHE-1; SLC9A1) by repressing its promoter in rat myoblasts (Jhumka et al., 2009). This result indicates a possible link between resveratrol and the inhibition of NHE. In the present study, we investigated whether the inhibition of NHE is a possible mechanism of the cardioprotective effects of resveratrol.
Methods
All animal care and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and the Oita University animal care guidelines.
Isolated perfused heart experiments
Isolated perfused heart experiments and infarct size measurements were performed using the Langendorff apparatus. Sprague-Dawley rats (250–300 g) were anaesthetized with an intraperitoneal injection of a mixture of ketamine (60 mg·kg−1) and xylazine (10 mg·kg−1). After the depth of anaesthesia was confirmed by stable heart rate and a lack of flexor responses to a paw pinch, rat hearts were isolated and perfused in retrograde mode at 37.0°C under a constant pressure of 75 mmHg. After stabilization for 20 min, the hearts were subjected to no-flow global ischaemia for 20 min, followed by reperfusion for 30 min. In the resveratrol-treated group, the hearts were perfused with 20 µmol·L−1 resveratrol in Krebs-Henseleit buffer (composition (in mmol L−1) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 glucose, 10 HEPES) 15 min before ischaemia. Left ventricular (LV) pressure was monitored using a pressure transducer to record the peak positive and negative first derivatives of LV pressure (dP/dtmax and dP/dtmin respectively). LV developed pressure (LVDP) was defined as the difference between LV systolic and diastolic pressures. LV pressure, coronary perfusion pressure and electrocardiograms were continuously recorded using a polygraph recorder (Nihon Kohden, Tokyo, Japan). Data were analysed using Lab Chart software (ADInstruments, Colorado Springs, CO, USA). At the end of reperfusion, the hearts were rapidly removed from the Langendorff apparatus and sliced across the long axis of their left ventricles into 2-mm-thick transverse sections. These slices were incubated in 1% triphenyltetrazolium chloride in a phosphate buffer (0.1 mol L−1: pH 7.4) at 37°C for 20 min. Infarct areas were enhanced by storage in 10% formaldehyde solution for 24 h and determined by planimetry of each slice. The volumes were calculated using image analysis software (Image J) by multiplying each area by the slice thickness and summing them for each heart.
Primary culture of neonatal rat cardiomyocytes
Cardiac ventricular myocytes were prepared from Sprague-Dawley rats aged 1–3 days and cultured as described elsewhere (Teshima et al., 2003). In brief, rat pups were killed by decapitation and the cardiac ventricles were removed and digested with trypsin (2 mg·mL−1) at 37°C. The cardiomyocytes were then isolated and cultured in Dulbecco's modified Eagle's minimal essential medium (DMEM) supplemented with 5% fetal bovine serum, penicillin (100 U·mL−1), streptomycin (100 mg·mL−1), vitamin B12 (2 µg·mL−1) and bromodeoxyuridine (0.1 mmol·L−1). These cells were placed in serum-free DMEM for 24 h before the experiments. In each isolation, we harvested cardiomyocytes from 11 neonatal rats, which were divided into appropriate groups. An experiment from one isolation was counted as n= 1.
Assay for cell death
For quantification of cellular viability, the cardiomyocytes were stimulated with hydrogen peroxide (H2O2) (100 µmol·L−1) for 2 h with or without 20 µmol·L−1 resveratrol (Sigma, St Louis, MO, USA), which was administered 1 h before the application of H2O2. The cardiomyocytes were stained with 50 µg·mL−1 propidium iodide (PI) (Roche, Basel, Switzerland), and images were captured by confocal microscopy (543 nm He/Ne laser line) using a ×20 objective lens (Carl Zeiss, Oberkochen, Germany). After scanning, the cells were permeabilized with saponin (50 µmol·L−1) (Sigma) to calculate the percentage of PI-positive cells in each group.
Assay for mitochondrial inner membrane potential
To assay mitochondrial inner membrane potential (ΔΨm), FACS analysis was performed. The cardiomyocytes were stimulated by exposure to H2O2 (100 µmol·L−1) for 30 min with or without incubation with 20 µmol·L−1 resveratrol (except for the experiments using various concentrations of resveratrol), which was added 1 h before H2O2 (except for the experiments in various pre-incubating durations with resveratrol). Other chemicals were added 30 min before resveratrol. Tetramethylrhodamine ethyl ester (TMRE; 100 nmol·L−1) (Invitrogen, Carlsbad, CA, USA) was loaded into the cells at 37°C for 30 min. The cells were harvested by trypsinization and analysed using the FACSCalibur system (Becton Dickinson, Bedford, MA, USA). Fluorescence intensity was monitored at 585 nm (FL-2 channel).
Assay for reactive oxygen species
We also investigated the changes in levels of reactive oxygen species (ROS) by FACS analysis. Resveratrol (20 µmol·L−1) was added 1 h before H2O2 application. The cardiomyocytes were loaded with 2 µmol·L−1 CM-H2DCFDA (Invitrogen) in the absence or presence of 100 µmol·L−1 H2O2 and incubated at 37°C for 20 min. The cells were harvested by trypsinization and fluorescence intensity was monitored at 530 nm (FL-1 channel). Data were analysed using the WinMDI analysis software.
Assays for [Na+]i and [Ca2+]i
CoroNa Green (1 µmol·L−1) and Fluo-4 (1 µmol·L−1) (Invitrogen) were used to examine [Na+]i and [Ca2+]i levels respectively. The cardiomyocytes were loaded with dyes at 37°C for 30 min. The scanning began immediately after the application of 100 µmol·L−1 H2O2, and fluorescence intensity was sequentially monitored every 5 min by confocal microscopy (488 nm argon laser line; ×40 objective lens) (Carl Zeiss).
Measurement of pHi
pHi was measured by monitoring the fluorescence of a pH indicator, BCECF-AM (Invitrogen). Cardiomyocytes were plated on glass-bottomed dishes and loaded with BCECF-AM (2 µmol·L−1) for 15 min at room temperature in Krebs-HEPES solution (in mmol L−1: 135 NaCl, 5.9 KCl, 1.5 CaCl2, 1.2 MgCl2, 11.6 HEPES, 11.5 d-glucose). The cells were dually excited at wavelengths of 488 and 458 nm with a fixed emission at 540 nm, and images were sequentially acquired at 0.5 s intervals using a confocal microscope system (LSM 710, Carl Zeiss). At the end of the experiments, pHi was calculated from the ratio of the two fluorescence intensities (488 nm/458 nm) using the nigericin high-K+ protocol (Thomas et al., 1979). In this protocol, the cells were superfused with a high-K+ solution (in mmol L−1: 145 KCl, 5 NaCl, 0.01 EDTA, 5 HEPES) at varying pH levels ranging from 5.86 to 8.5 in the presence of 5 mg·L−1 nigericin. We confirmed that the ratios of fluorescence intensity linearly correlated with the pHi levels in a preliminary experiment.
Assays for NHE activity
NHE activity was measured by monitoring the recovery rate of pHi from rapid acidification using the NH4Cl pre-pulse technique as described elsewhere (Sabri et al., 1998). The cells were superfused with Krebs-HEPES solution for 5 min, followed by NH4Cl solution (25 mmol·L−1) for 6 min; thereafter, the solution was replaced with Na+-free Krebs-HEPES solution. After 10 min of perfusion, the perfusate was switched back to Na+-containing Krebs-HEPES solution and the cells were superfused for 5 min. The solution in the dishes was continuously changed during the experiment by perfusing fresh solution while aspirating the old solution at the same rate (5 mL·min−1) using a Harvard syringe system (Harvard Apparatus, Holliston, MA, USA). The change in pHi was sequentially analysed. During superfusion with Na+-free Krebs-HEPES solution, pHi was stable at an acidic level and did not recover until the perfusate was switched back to Krebs-HEPES solution. The rate of pHi recovery was operationally defined as NHE activity. We quantified this rate by measuring the slope of a straight line fitted to the initial 1 min from the onset of recovery. Resveratrol was added 1 h before the experiment. H2O2 and other reagents were added from the beginning of perfusion with NH4Cl and continued until the end of the experiment.
Western immunoblot analysis
The cells were incubated with resveratrol for 1 h. H2O2 and phorbol 12-myristate 13-acetate (PMA) were loaded 10 min before cell harvesting. Membranous and cytosolic proteins were extracted using the Subcellular Proteome Extraction Kit (Merck, Darmstadt, Germany) according to the manufacturer's instructions. Western immunoblot analyses were performed as previously described (Thuc et al., 2010). The primary antibody for PKC-α was purchased from Cell Signaling Technology (Danvers, MA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. Equal amounts of protein were fractionated on 8.5% SDS-polyacrylamide gels and transferred electrophoretically onto a polyvinylidene difluoride membrane. The membranes were blocked with 5% non-fat milk in Tris-buffered saline containing 0.05% Tween 20 for 1 h at room temperature, and incubated with primary antibodies at a 1:1000 dilution for 1 h at room temperature. After washing, the membranes were incubated with a 1:4000 dilution of horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The blots were visualized by enhanced chemiluminescence and exposure to Hyper-film. Band signals were quantified densitometrically using Image J software. All the band signals were normalized by the GAPDH protein levels and the values were expressed as percentage of the mean value of the control.
Statistical analysis
All data are expressed as mean ± SE. Multiple comparisons among groups were performed by one-way anova with Bonferroni post hoc test. Sequential changes were analysed by two-way anova. A value of P < 0.05 was considered statistically significant.
Results
LV functional recovery and infarct size
At baseline, all haemodynamic parameters, such as LVDP, coronary perfusion pressure and heart rate, were the same between each group. The hearts subjected to global ischaemia exhibited considerable decrease in LVDP during reperfusion. Treatment with 20 or 100 µmol·L−1 resveratrol 15 min before global ischaemia showed a higher recovery of LVDP; however, treatment with 10 µmol·L−1 resveratrol did not improve LVDP recovery (Figure 1A). The infarct size was significantly decreased in the 20 and 100 µmol·L−1 resveratrol-treated groups compared with that in the control group. Treatment with 10 µmol·L−1 resveratrol did not reduce infarct size as compared with control (Figure 1B).
Figure 1.
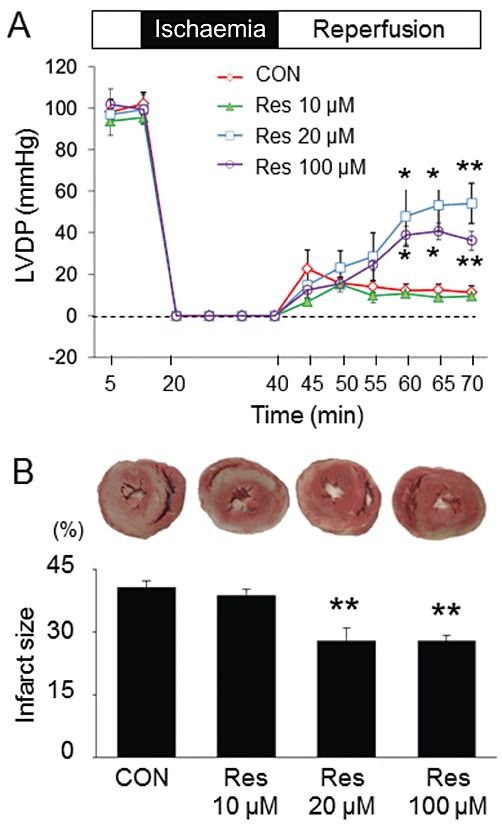
Effects of resveratrol on LV functional recovery and infarct size. (A) Treatment with 20 or 100 µmol·L−1 resveratrol (Res) 15 min before ischaemia improved the recovery of LVDP in rat isolated perfused hearts subjected to 20 min of global ischaemia, followed by 30 min of reperfusion. (B) Treatment with 20 or 100 µmol·L−1 resveratrol reduced the infarct size compared with the control group. Treatment with 10 µmol·L−1 resveratrol neither improved LV functional recovery nor reduced the infarct size in rat hearts. Data are means ± SE (n= 5 for each group). *P < 0.05, **P < 0.01 significantly different from control (CON).
Cell death
Cell death was induced by incubation with H2O2 for 2 h. Figure 2A shows the representative images and quantitative results of PI staining in the cardiomyocytes. Incubation with H2O2 significantly increased the number of PI-positive cells, which was suppressed by pretreatment with resveratrol (20 µmol·L−1).
Figure 2.
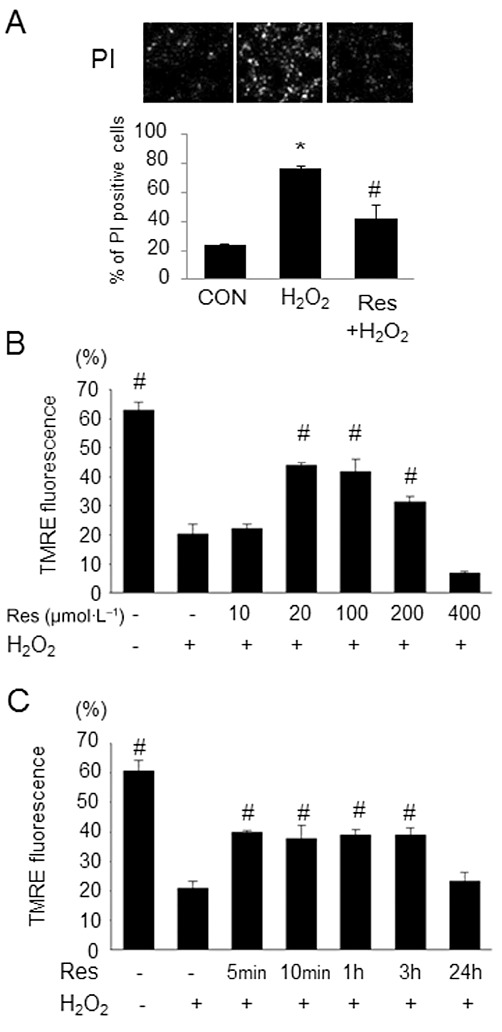
Effects of resveratrol on cell death and the dissipation of ΔΨm induced by H2O2. (A) Representative images of PI staining of cardiomyocytes stimulated by H2O2 (100 µmol·L−1) in the presence or absence of resveratrol (Res; 20 µmol·L−1) and quantitative percentages of PI-positive cells. Resveratrol suppressed the increase of PI positivity induced by H2O2. (B) Summarized data of FACS analysis with various concentrations of resveratrol. Resveratrol preserved ΔΨm in a dose-dependent manner. (C) Quantitative results of FACS analysis with various durations of pre-incubation with resveratrol. Data are means ± SE (n= 7 for each group). *P < 0.05 significantly different from control (CON); #P < 0.05 significantly different from H2O2.
Mitochondrial inner membrane potential
Dissipation of ΔΨm is a critical event in the process of cell death (Kroemer et al., 1998). We examined whether the cardioprotective effect of resveratrol is associated with the preservation of ΔΨm by assessing the change in TMRE fluorescence by FACS analysis. Incubation with H2O2 decreased TMRE fluorescence, indicating the depolarization of ΔΨm. The percentage of cells exhibiting higher TMRE fluorescence was evaluated, as described elsewhere (Thuc et al., 2010). As shown in Figure 2B, ΔΨm is preserved at resveratrol concentrations of 20–200 µmol·L−1, but not at concentrations as low as 10 µmol·L−1 or as high as 400 µmol·L−1. The protective effect of pre-incubation with resveratrol (20 µmol·L−1) occurred as early as 5 min and continued up to 3 h. Cardioprotection was not observed with pre-incubation for 24 h (Figure 2C).
Levels of ROS
Oxidative stress is one of the key determinants of myocardial ischaemia/reperfusion injury (Zweier and Talukder, 2006). We also examined the role of ROS in the cardioprotective effect of resveratrol. Figure 3 shows the representative histograms and quantitative results from FACS analysis of ROS levels in cardiomyocytes. Treatment with H2O2 shifted the peak of the histogram to the right, indicating an increase in ROS level (Figure 3A). The summarized data showed a significant increase in DCF fluorescence in H2O2-treated cells, which was suppressed by pretreatment with resveratrol (Figure 3B).
Figure 3.
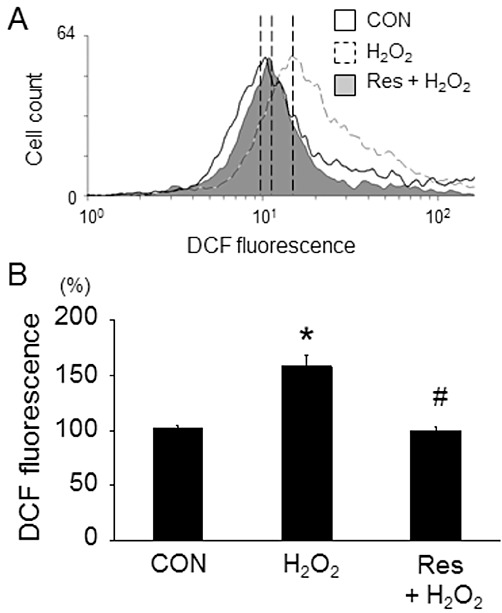
Effects of resveratrol on ROS level. (A) Representative FACS histograms obtained from cardiomyocytes loaded with CM-H2DCFDA (2 µmol·L−1) in the presence or absence of H2O2 (100 µmol·L−1). The area under black line represents control, the grey area represents resveratrol-treated cells and the area under dotted line represents the cells stimulated by H2O2. Incubation with H2O2 for 20 min shifted the histogram to the right, indicating an increase in ROS level. Treatment with resveratrol (Res; 20 µmol·L−1) for 1 h before H2O2 application almost totally suppressed the increase in ROS level induced by H2O2. (B) Quantitative results analysed the intensity of fluorescence in the peak of the histograms. Data are means ± SE (n= 6 for each group). *P < 0.05 significantly different from control, #P < 0.05 significantly different from H2O2.
[Na+]i and [Ca2+]i
[Ca2+]i overload following the accumulation of [Na+]i leads to the dissipation of ΔΨm. Therefore, we examined the effects of resveratrol on H2O2-induced [Na+]i and [Ca2+]i increases in cardiomyocytes. Figure 4A shows the representative images and sequential changes of CoroNa Green fluorescence in six randomly selected cells incubated with or without resveratrol. [Na+]i gradually increased during the first 30 min after H2O2 application and thereafter the rate of increase was augmented to reach its peak at approximately 45 min. Incubation with resveratrol diminished the increase in [Na+]i. Figure 4B shows the representative images and quantitative results of Fluo-4 fluorescence in six randomly selected cardiomyocytes. [Ca2+]i was rapidly and considerably increased at approximately 30 min after H2O2 application and reached a plateau at approximately 40 min. Incubation with resveratrol almost completely inhibited the increase in [Ca2+]i due to H2O2 application. The fluorescence of CoroNa Green and Fluo-4 were steeply decreased 50 min after the incubation with H2O2 because of the rupture of the cardiomyocytes. To examine a possible effect of resveratrol on NCX, we investigated the changes in [Ca2+]i by ouabain, a Na+/K+ ATPase inhibitor. [Ca2+]i continuously increased during the 60 min scanning after ouabain application. Incubation with resveratrol did not affect the increase in [Ca2+]i due to ouabain application (Figure 4C). Taken together, these results indicate that resveratrol may affect NHE, but not NCX, to suppress [Ca2+]i overload.
Figure 4.
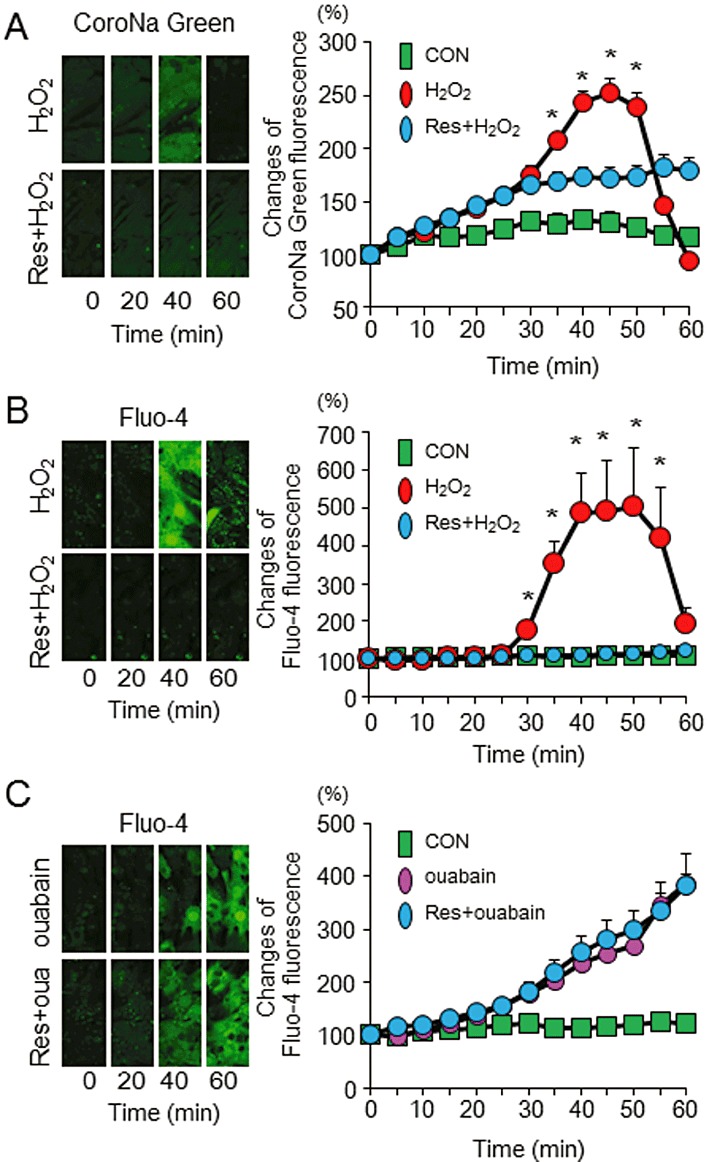
Effects of resveratrol on [Na+]i and [Ca2+]i. (A) Representative sequential images of CoroNa Green fluorescence obtained from the cardiomyocytes loaded with H2O2 (100 µmol·L−1) in the presence or absence of resveratrol (Res; 20 µmol·L−1) and quantitative data from each group. [Na+]i started to increase after the H2O2 application and peaked at approximately 45 min. This increase was attenuated by pretreatment with resveratrol. (B) Representative sequential images of Fluo-4 fluorescence obtained from the cardiomyocytes loaded with H2O2 in the presence or absence of resveratrol and quantitative data from each group. An increase in [Ca2+]i by the H2O2 application was suppressed by pretreatment with resveratrol. (C) Representative sequential images of Fluo-4 fluorescence obtained from the cardiomyocytes loaded with ouabain (1 mmol·L−1) in the presence or absence of resveratrol (20 µmol·L−1) and quantitative data from each group. Pretreatment with resveratrol did not affect the increase in [Ca2+]i by ouabain. Data are means ± SE (n= 6 for each group). *P < 0.05 significantly different from Res + H2O2.
NHE activity
Next, we examined the effect of resveratrol on NHE activity. Figure 5A shows the representative alteration in the pHi of cardiomyocytes. Perfusion with NH4Cl solution increased pHi, which rapidly decreased after the perfusate was switched to a Na+-free solution. During 10 min of Na+-free perfusion, pHi was stable at approximately 6.5. The recovery of pHi began after the cells were superfused with Na+-containing Krebs solution. A bold line indicates the slope of the initial 1 min of pHi recovery in each experiment (Figure 5B). Figure 5C shows the summarized data of the pHi recovery rates from five independent experiments. Exposure to H2O2 for 10 min increased NHE activity, which was attenuated by treatment with resveratrol and chelerythrine, a PKC inhibitor. Furthermore, PMA, a PKC activator, increased NHE activity, which was also attenuated by treatment with resveratrol. Taken together, these results showed that resveratrol inhibits NHE activity in a PKC-dependent manner.
Figure 5.
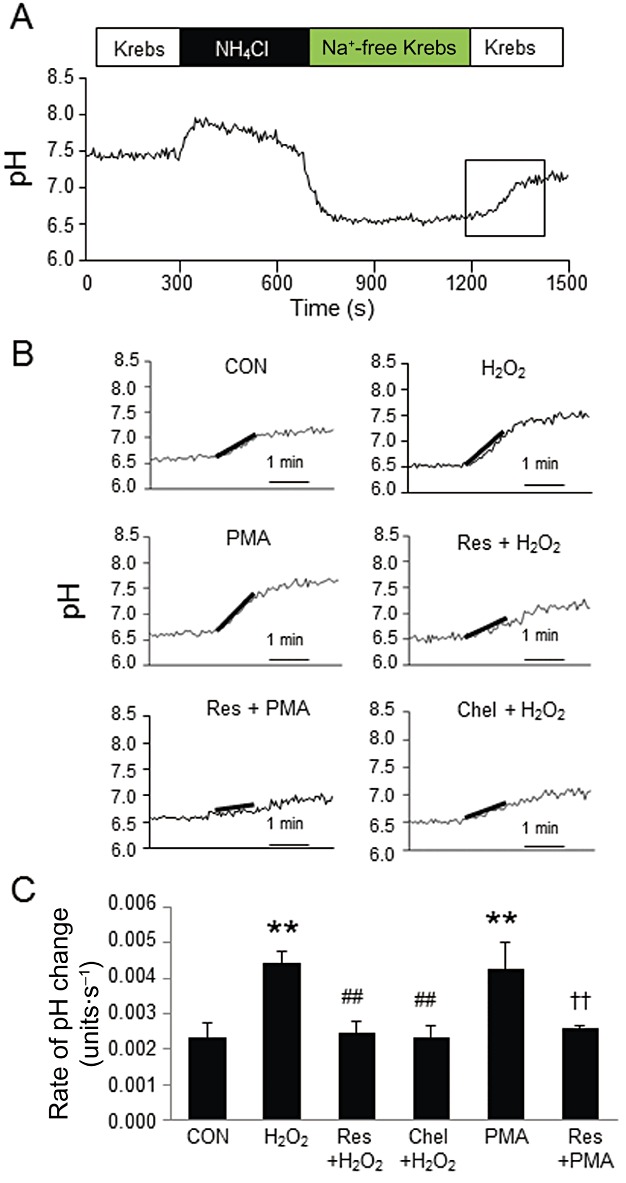
Effect of resveratrol on NHE activity. (A) pHi was continuously measured using the pH sensitive dye, BCECF. The rate of pHi recovery during the initial 60 s was defined as NHE activity (outlined by rectangle). (B) Representative pHi recovery responses from rapid acidification in each group. A line was fitted to pHi trace during the first minute of recovery. The slopes were measured and compared. (C) Summarized quantitative data from independent experiments in each group. Exposure to H2O2 (100 µmol·L−1) increased NHE activity in the cardiomyocytes, which was suppressed by pretreatment with resveratrol (Res; 20 µmol·L−1) or chelerythrine (Chel; 1 µmol·L−1). PMA (5 µmol·L−1) also activated NHE, which was attenuated by pretreatment with resveratrol. Data are mean ± SE (n= 5 for each group). **P < 0.01 significantly different from control, ##P < 0.01 significantly different from H2O2, ††P < 0.01 significantly different from PMA.
Translocation of PKC
PKC isozymes are reported to translocate to different parts of the cell and exert divergent effects in ischaemia/reperfusion injury. During ischaemia, PKC-α is translocated from the cytosol to the cell membrane (Yoshida et al., 1996). Therefore, we examined the effect of resveratrol on the translocation of PKC-α. Incubation with H2O2 for 10 min increased the expression of PKC-α in the sarcolemmal membrane and decreased that in the cytosol, which was prevented by pretreatment with resveratrol (Figure 6A,B). The PMA-induced increase in PKC-α expression in the sarcolemmal membrane was also considerably reduced by treatment with resveratrol (Figure 6C). These results indicate that resveratrol inhibited NHE activity by inhibiting the translocation of PKC-α.
Figure 6.
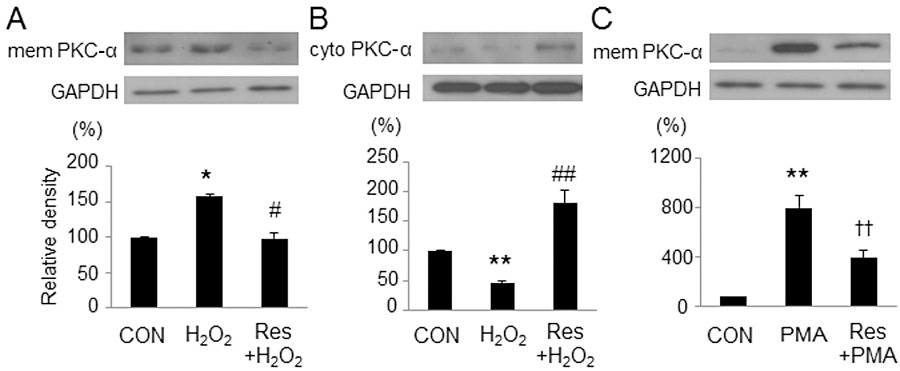
Effects of resveratrol on the translocation of PKC-α. Representative images and quantitative results of immunoblots for PKC-α. All band signals were normalized by GAPDH and compared with control. (A, B) The H2O2-induced translocation of PKC-α from the cytosol (cyto) to the sarcolemmal membrane (mem) was reversed by pretreatment with resveratrol (Res). (C) The PMA-induced translocation of PKC-α to the sarcolemmal membrane was attenuated by resveratrol. Data are mean ± SE (n= 5 for each group). *P < 0.05, **P < 0.01 significantly different from control; #P < 0.05, ##P < 0.01 significantly different from H2O2; ††P < 0.01 significantly different from PMA.
Discussion
The main findings of the present study are as follows: (i) resveratrol reduced infarct size and improved LV functional recovery in isolated hearts that were subjected to ischaemia/reperfusion; (ii) resveratrol attenuated cardiomyocyte death by reducing ROS and preserving mitochondrial function; and (iii) resveratrol prevented NHE activation by inhibiting the translocation of PKC-α from the cytosol to the sarcolemmal membrane in cardiomyocytes. This is the first report to demonstrate the PKC-dependent inhibition of NHE as a mechanism of rapid cardioprotection by resveratrol.
On the basis of our results, we have proposed a possible protective pathway for resveratrol in which resveratrol inhibits the translocation of PKC-α from the cytosol to the cell membrane and prevents the subsequent events leading to cell injury (Figure 7). PKC plays a critical role in ischaemia/reperfusion injury. PKC-α is translocated from the cytosol to the cell membrane during ischaemia/reperfusion (Yoshida et al., 1996), and membrane-associated PKC-α has been reported to activate NHE (Maly et al., 2002). In this report, the PKC inhibitor and genetic ablation of PKC-α suppressed the activation of NHE. These results indicate that PKC-α may contribute importantly to the activation of NHE. Furthermore, a study by Slater et al., (2003) showed that resveratrol inhibited the activity of conventional PKC-α in an in vitro assay system. In this report, resveratrol inhibited activation of PKC-α by competitive binding to the activator-binding sites of this kinase which prevented PKC-α from binding to plasma membrane and becoming active. These results strongly support our present finding that the inhibitory effect of resveratrol on NHE was PKC-α dependent. Activation of ERK1/2 is another pathway leading to NHE activation by oxidative stress (Snabaitis et al., 2002; Cingolani et al., 2011). Although our data clearly demonstrated a PKC-α-dependent NHE inhibition by resveratrol, the involvement of ERK1/2 is an interesting issue for further study. In a previous study, PKC activated NADPH oxidase (Dekker et al., 2000) which will increase ROS production, suggesting that PKC may work upstream of ROS. However, the present result that activation of NHE by H2O2 was inhibited by chelerythrine strongly supports the ROS-PKC-NHE activation sequence as shown in Figure 7. Considering that NHE is activated in response to intracellular acidosis, the link between acidosis and the translocation of PKC-α remains unclear.
Figure 7.
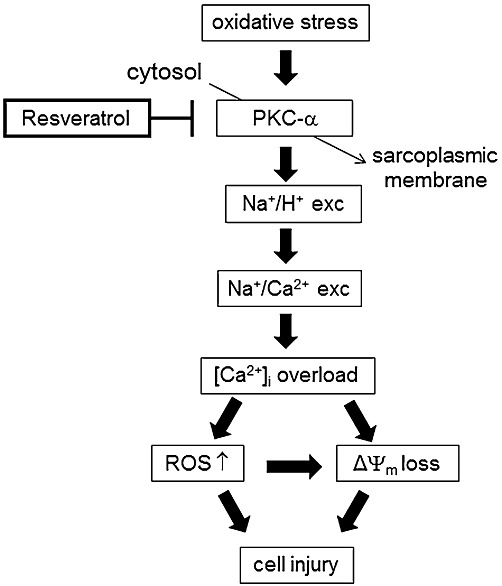
Proposed pathways of cardioprotective effects of resveratrol in ischaemia/reperfusion.
An increase in [Ca2+]i precedes irreversible myocardial injury. Much of the increase in [Ca2+]i during ischaemia is due to Ca2+ entry because of the reverse mode activity of the NCX. During ischaemia/reperfusion, pHi becomes acidic, which facilitates the extrusion of H+ from the cytosol in exchange for Na+ by NHE. Increased [Na+]i can be extruded by NCX in exchange for Ca2+, thereby increasing [Ca2+]i. Thus, the inhibition of NCX and NHE should attenuate [Ca2+]i overload and is a possible target for cardioprotection. Previous studies showed that NCX inhibitors such as KB-R7943 and SEA0400 exerted cardioprotective effects against ischaemia/reperfusion injury by attenuating [Ca2+]i overload (Yoshitomi et al., 2005; Motegi et al., 2007). In the present study, resveratrol prevented the increase not only in [Ca2+]i but also in [Na+]i by H2O2. Furthermore, resveratrol did not affect the increase in [Ca2+]i by ouabain, which facilitates Ca2+ influx through NCX working in reverse mode by inhibiting the Na+/K+ ATPase pump. These results indicate that resveratrol may affect NHE, but not NCX. We also assayed NHE activity by monitoring pHi recovery from acid loading and confirmed that resveratrol suppressed H2O2-induced NHE activation.
Mitochondria play a key role in the process of cell injury, and loss of the mitochondrial potential, ΔΨm, because of the opening of mitochondrial permeability transition pore (mPTP) is a critical step towards cell death. Opening of the mPTP collapses ΔΨm and halts oxidative phosphorylation, resulting in ATP depletion and cell death. The association of mitochondrial ATP-sensitive potassium (mitoKATP) channels with cardioprotection by the inhibition of NHE has been previously reported (Miura et al., 2001). Aiello and Cingolani (2011) and Mosca and Cingolani (2002), reported that red wine extracts conferred cardioprotection by opening mitoKATP channels. In contrast, in our preliminary experiments, we did not observe any effect of 5-hydroxydecanoate, a mitoKATP channel inhibitor, on resveratrol-induced cardioprotection. Considering that PKC is an activator of the mitoKATP channels (Sato et al., 1998), the cardioprotective effects of resveratrol may be independent of the mitoKATP channels.
Increase in ROS is a key determinant of ischaemia/reperfusion injury. On reperfusion, re-oxygenation of the cells triggers a burst of ROS; an increase in ROS favours mPTP opening (Crompton et al., 1987). In this study, we showed that resveratrol attenuated a burst of ROS in the cardiomyocytes. Hence, cardioprotection by resveratrol may be mediated by suppressing ROS increase and subsequently preventing mPTP opening. Consistent with the present result, a recent study showed that NHE inhibitors decreased myocardial superoxide production (Garciarena et al., 2008). However, the mechanisms by which resveratrol suppresses ROS increase remain elusive. Considering that several ROS-generating enzymes are Ca2+ dependent (Gordeeva et al., 2003; Brookes, 2004), the prevention of ROS increase may be a secondary effect of the inhibition of [Ca2+]i overload. The preservation of mitochondrial function is also another possible mechanism of ROS reduction. Mitochondria are a major source of ROS production, particularly in a functionally impaired condition, and mitochondrial Ca2+ overload can enhance the generation of ROS from mitochondria (Gordeeva et al., 2003; Brookes, 2004). Furthermore, resveratrol may have ROS-scavenging property as suggested in some previous reports (Ray et al., 1999; Danz et al., 2009). In our preliminary experiments, resveratrol exerted cardioprotection against exogenously added H2O2 even after the washing out of resveratrol after 1 h pre-incubation. This result excludes the direct effect of resveratrol on H2O2. However, it does not exclude a possible scavenging effect of resveratrol after NHE activation.
The plasma concentration of resveratrol in humans after an oral administration of 25 mg resveratrol is reportedly equivalent to 2 µmol·L−1 (Walle et al., 2004). Also, plasma levels after oral administration of 20 mg·kg−1 resveratrol to rats were less than 1 µmol·L−1 (Asensi et al., 2002). These concentrations were lower than those we used in the present study. Higher levels of resveratrol supplementation may be necessary to induce cardioprotection.
In conclusion, we have shown that resveratrol conferred cardioprotection against ischaemia/reperfusion injury and oxidative stress by preserving mitochondrial function and reducing ROS burst. This protection was mediated by the PKC-dependent inhibition of NHE and subsequent attenuation of [Ca2+]i overload.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (C) (22590209) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Glossary
- ΔΨm
mitochondrial inner membrane potential
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- LV
left ventricular
- LVDP
left ventricular developed pressure
- mitoKATP
mitochondrial ATP-sensitive K+ channels
- mPTP
mitochondrial permeability transition pore
- NCX
Na+-Ca2+ exchanger
- NHE
Na+-H+ exchanger
- PI
propidium iodide
- PMA
phorbol 12-myristate 13-acetate
- ROS
reactive oxygen species
- TMRE
tetramethylrhodamine ethyl ester
Conflicts of interest
None.
References
- Aiello EA, Cingolani HE. A possible subcellular mechanism underlying the ‘French paradox’: the opening of mitochondrial KATP channels. Appl Physiol Nutr Metab. 2011;36:768–772. doi: 10.1139/h11-089. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5 th Edition) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensi M, Medina I, Ortega A, Carretero J, Bano MC, Obrador E, et al. Inhibition of cancer growth by resveratrol is related to its low bioavailability. Free Radic Biol Med. 2002;33:387–398. doi: 10.1016/s0891-5849(02)00911-5. [DOI] [PubMed] [Google Scholar]
- Avkiran M, Marber MS. Na+/H+ exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Coll Cardiol. 2002;39:747–753. doi: 10.1016/s0735-1097(02)01693-5. [DOI] [PubMed] [Google Scholar]
- Boyce SW, Bartels C, Bolli R, Chaitman B, Chen JC, Chi E, et al. Impact of sodium-hydrogen exchange inhibition by cariporide on death or myocardial infarction in high-risk CABG surgery patients: results of the CABG surgery cohort of the GUARDIAN study. J Thorac Cardiovasc Surg. 2003;126:420–427. doi: 10.1016/s0022-5223(03)00209-5. [DOI] [PubMed] [Google Scholar]
- Brookes PS. Mitochondrial nitric oxide synthase. Mitochondrion. 2004;3:187–204. doi: 10.1016/j.mito.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Yu W, Fu YC, Wang X, Li JL, Wang W. Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway. Biochem Biophys Res Commun. 2009;378:389–393. doi: 10.1016/j.bbrc.2008.11.110. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Ennis IL, Aiello EA, Perez NG. Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflugers Arch. 2011;462:29–38. doi: 10.1007/s00424-011-0930-9. [DOI] [PubMed] [Google Scholar]
- Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245:915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med. 2009;46:1589–1597. doi: 10.1016/j.freeradbiomed.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Das S, Alagappan VK, Bagchi D, Sharma HS, Maulik N, Das DK. Coordinated induction of iNOS-VEGF-KDR-eNOS after resveratrol consumption: a potential mechanism for resveratrol preconditioning of the heart. Vascul Pharmacol. 2005;42:281–289. doi: 10.1016/j.vph.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J, et al. Protein kinase C-β contributes to NADPH oxidase activation in neutrophils. Biochem J. 2000;347:285–289. [PMC free article] [PubMed] [Google Scholar]
- Garciarena CD, Caldiz CI, Correa MV, Schinella GR, Mosca SM, Chiappe de Cingolani GE, et al. Na+/H+ exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J Appl Physiol. 2008;105:1706–1713. doi: 10.1152/japplphysiol.90616.2008. [DOI] [PubMed] [Google Scholar]
- Garciarena CD, Fantinelli JC, Caldiz CI, Chiappe de Cingolani G, Ennis IL, Perez NG, et al. Myocardial reperfusion injury: reactive oxygen species vs. NHE-1 reactivation. Cell Physiol Biochem. 2011;27:13–22. doi: 10.1159/000325201. [DOI] [PubMed] [Google Scholar]
- Gordeeva AV, Zvyagilskaya RA, Labas YA. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry (Mosc) 2003;68:1077–1080. doi: 10.1023/a:1026398310003. [DOI] [PubMed] [Google Scholar]
- Hattori R, Otani H, Maulik N, Das DK. Pharmacological preconditioning with resveratrol: role of nitric oxide. Am J Physiol Heart Circ Physiol. 2002;282:H1988–H1995. doi: 10.1152/ajpheart.01012.2001. [DOI] [PubMed] [Google Scholar]
- Jhumka Z, Pervaiz S, Clement MV. Resveratrol regulates the expression of NHE-1 by repressing its promoter activity: critical involvement of intracellular H2O2 and caspases 3 and 6 in the absence of cell death. Int J Biochem Cell Biol. 2009;41:945–956. doi: 10.1016/j.biocel.2008.09.028. [DOI] [PubMed] [Google Scholar]
- Knight DR, Smith AH, Flynn DM, MacAndrew JT, Ellery SS, Kong JX, et al. A novel sodium-hydrogen exchanger isoform-1 inhibitor, zoniporide, reduces ischemic myocardial injury in vitro and in vivo. J Pharmacol Exp Ther. 2001;297:254–259. [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Maly K, Strese K, Kampfer S, Ueberall F, Baier G, Ghaffari-Tabrizi N, et al. Critical role of protein kinase C α and calcium in growth factor induced activation of the Na+/H+ exchanger NHE1. FEBS Lett. 2002;521:205–210. doi: 10.1016/s0014-5793(02)02867-3. [DOI] [PubMed] [Google Scholar]
- Miura T, Liu Y, Goto M, Tsuchida A, Miki T, Nakano A, et al. Mitochondrial ATP-sensitive K+ channels play a role in cardioprotection by Na+-H+ exchange inhibition against ischemia/reperfusion injury. J Am Coll Cardiol. 2001;37:957–963. doi: 10.1016/s0735-1097(00)01183-9. [DOI] [PubMed] [Google Scholar]
- Mosca SM, Cingolani HE. Cardioprotection from ischemia/reperfusion induced by red wine extract is mediated by KATP channels. J Cardiovasc Pharmacol. 2002;40:429–437. doi: 10.1097/00005344-200209000-00012. [DOI] [PubMed] [Google Scholar]
- Motegi K, Tanonaka K, Takenaga Y, Takagi N, Takeo S. Preservation of mitochondrial function may contribute to cardioprotective effects of Na+/Ca2+ exchanger inhibitors in ischaemic/reperfused rat hearts. Br J Pharmacol. 2007;151:963–978. doi: 10.1038/sj.bjp.0707321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PS, Maulik G, Cordis GA, Bertelli AA, Bertelli A, Das DK. The red wine antioxidant resveratrol protects isolated rat hearts from ischemia reperfusion injury. Free Radic Biol Med. 1999;27:160–169. doi: 10.1016/s0891-5849(99)00063-5. [DOI] [PubMed] [Google Scholar]
- Rupprecht HJ, vom Dahl J, Terres W, Seyfarth KM, Richardt G, Schultheibeta HP, et al. Cardioprotective effects of the Na+/H+ exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation. 2000;101:2902–2908. doi: 10.1161/01.cir.101.25.2902. [DOI] [PubMed] [Google Scholar]
- Sabri A, Byron KL, Samarel AM, Bell J, Lucchesi PA. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ Res. 1998;82:1053–1062. doi: 10.1161/01.res.82.10.1053. [DOI] [PubMed] [Google Scholar]
- Sato T, O'Rourke B, Marbán E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circ Res. 1998;83:110–114. doi: 10.1161/01.res.83.1.110. [DOI] [PubMed] [Google Scholar]
- Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, et al. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–268. [PubMed] [Google Scholar]
- Slater SJ, Seiz JL, Cook AC, Stagliano BA, Buzas CJ. Inhibition of protein kinase C by resveratrol. Biochim Biophys Acta. 2003;1637:59–69. doi: 10.1016/s0925-4439(02)00214-4. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, Hearse DJ, Avkiran M. Regulation of sarcolemmal Na+/H+ exchange by hydrogen peroxide in adult rat ventricular myocytes. Cardiovasc Res. 2002;53:470–480. doi: 10.1016/s0008-6363(01)00464-3. [DOI] [PubMed] [Google Scholar]
- Teshima Y, Akao M, Jones SP, Marbán E. Cariporide (HOE642), a selective Na+-H+ exchange inhibitor, inhibits the mitochondrial death pathway. Circulation. 2003;108:2275–2281. doi: 10.1161/01.CIR.0000093277.20968.C7. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- Thuc LC, Teshima Y, Takahashi N, Nagano-Torigoe Y, Ezaki K, Yufu K, et al. Mitochondrial KATP channels-derived reactive oxygen species activate pro-survival pathway in pravastatin-induced cardioprotection. Apoptosis. 2010;15:669–678. doi: 10.1007/s10495-010-0473-0. [DOI] [PubMed] [Google Scholar]
- Walle T, Hsieh F, DeLegge MH, Oatis JE, Jr, Walle UK. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Hirata T, Akita Y, Mizukami Y, Yamaguchi K, Sorimachi Y, et al. Translocation of protein kinase C-α, δ and ε isoforms in ischemic rat heart. Biochim Biophys Acta. 1996;1317:36–44. doi: 10.1016/0925-4439(96)00035-x. [DOI] [PubMed] [Google Scholar]
- Yoshitomi O, Akiyama D, Hara T, Cho S, Tomiyasu S, Sumikawa K. Cardioprotective effects of KB-R7943, a novel inhibitor of Na+/Ca2+ exchanger, on stunned myocardium in anesthetized dogs. J Anesth. 2005;19:124–130. doi: 10.1007/s00540-004-0290-0. [DOI] [PubMed] [Google Scholar]
- Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]