

Abstract
BACKGROUND AND PURPOSE
An ATP-binding cassette (ABC) transporter, breast cancer resistance protein (BCRP)/ABCG2, limits oral bioavailability of sulphasalazine. Here we examined the effect of curcumin, the principal curcuminoid of turmeric, on oral bioavailability of microdoses and therapeutic doses of sulphasalazine in humans.
EXPERIMENTAL APPROACH
Effects of curcumin were measured on the ATP-dependent sulphasalazine uptake by hBCRP-expressing membrane vesicles and on oral bioavailability of sulphasalazine in wild-type and Bcrp(–/–) mice. Eight healthy Japanese subjects received an oral dose of sulphasalazine suspension (100 µg) or tablets (2 g) alone or after curcumin tablets (2 g). Uptake of sulphasalazine was studied in HEK293 cells transfected with the influx transporter (OATP)2B1.
KEY RESULTS
Curcumin was a potent hBCRP inhibitor in vitro (Ki 0.70 ± 0.41 µM). Curcumin increased the area under the curve (AUC)0–8 of plasma sulphasalazine eightfold in wild-type mice at 300 and 400 mg·kg−1, but not in Bcrp(–/–) mice. Curcumin increased AUC0–24 of plasma sulphasalazine 2.0-fold at microdoses and 3.2-fold at therapeutic doses in humans. Non-linearity of the dose–exposure relationship was observed between microdoses and therapeutic doses of sulphasalazine. Sulphasalazine was a substrate for OATP2B1 (Km 1.7 ± 0.3 µM). Its linear index (dose/Km) at the therapeutic dose was high and may saturate OATP2B1.
CONCLUSIONS AND IMPLICATIONS
Curcumin can be used to investigate effects of BCRP on oral bioavailability of drugs in humans. Besides the limited dissolution, OATP2B1 saturation is a possible mechanism underlying non-linearity in the dose–exposure relationship of sulphasalazine.
Keywords: sulphasalazine, curcumin, pharmacokinetic interaction, BCRP, drug absorption, in vivo inhibitor, non-linear pharmacokinetics, microdosing
Introduction
Drug-metabolizing enzymes and transporters form an essential detoxification system in the small intestine, and limit absorption of xenobiotic compounds including drugs into the circulation (Wacher et al., 2001; Ding and Kaminsky, 2003; Murakami and Takano, 2008). For drug candidates, low oral availability is sometimes attributable to the metabolism and/or active efflux in the small intestine. Even for drugs in the market, these processes serve as potential sites causing non-linearity, and intersubject variation, due to drug–drug interactions, or genetic factors. Elucidation of the effect of such enzymes and transporters contributes to selection of candidates in drug development and to safety in drug therapy.
The breast cancer resistance protein (BCRP/ABCG2) is a multi-specific ATP-binding cassette (ABC) transporter expressed at the apical membrane in the intestine, liver, kidney and placenta (Kusuhara and Sugiyama, 2007; Cusatis and Sparreboom, 2008; Vlaming et al., 2009; nomenclature follows Alexander et al., 2011). Animal studies using Bcrp(–/–) mice have shown its ability to limit oral bioavailability, to promote elimination into the bile and urine and to limit penetration into the brain and testis (Enokizono et al., 2007; Vlaming et al., 2009). Pharmacogenetic studies, which focused on the non-synonymous single nucleotide polymorphism (SNP) of BCRP (421C>A, Q141K) associated with lower protein expression of BCRP (Kondo et al., 2004; Kobayashi et al., 2005), have disclosed the importance of BCRP in limiting oral bioavailability of drugs in human small intestine. Subjects with this mutant allele show higher systemic exposure to topotecan (Sparreboom et al., 2005), sulphasalazine (Yamasaki et al., 2008) and rosuvastatin and atorvastatin (Keskitalo et al., 2009) following oral administration. Because of the broad substrate specificity of BCRP, its effect on oral bioavailability of other substrate drugs and drug candidates is a matter of great concern.
There has been growing interest in inhibitors of drug transporters in vivo, since the International Transporter Consortium published guidelines for the study of transporters in drug development, where the decision tree recommends a clinical drug–drug interaction study to elucidate the impact of transporters on the disposition and therapeutic efficacy of investigational drugs (Giacomini et al., 2010). With regard to in vivo BCRP inhibitors, a multidrug resistance modifier GF120918, was the first compound to increase the oral absorption of topotecan by twofold in cancer patients (Kuppens et al., 2007), although it has not yet been marketed. More recently, gefitinib and curcumin was shown to greatly enhance the plasma concentration of sulphasalazine following oral administration, by inhibiting Bcrp in mice (Zaher et al., 2006; Shukla et al., 2009), but their effectiveness in humans remains unknown. In spite of urgent needs, in vivo inhibitors of BCRP applicable to clinical studies have not been established.
Curcumin, the principal curcuminoid of turmeric, is a naturally occurring polyphenol. No curcumin-related toxicity was reported during phase I clinical trial where patients received oral curcumin (8 g·day−1) for 3 months (Cheng et al., 2001). This low toxicity together with its high potency in inhibiting photolabelling of BCRP with [125I]iodoarylazidoprazosin (Shukla et al., 2009), made curcumin a promising candidate for an in vivo BCRP inhibitor in humans. Therefore, this study aimed to assess the effect of curcumin using sulphasalazine as test substrate for BCRP, oral availability of which is limited by BCRP (Yamasaki et al., 2008), in healthy subjects. Clinical studies using microdoses aid rational selection of candidate drugs based on their pharmacokinetic behaviour in the early phase of drug development. However, because microdose studies are conducted at an extremely low dose (100 µg or a dose less than 1% of the dose yielding their pharmacological effects), the possible non-linearity of the pharmacokinetics at the therapeutic dose has been debated in extrapolating the microdose data to that of the therapeutic dose. To address this issue, we examined the effect of curcumin on the disposition of sulphasalazine both in a microdose and in a therapeutic dose in this study.
Methods
Animals
All animal care and experimental procedures complied with the regulations of the Committee on Ethics in the Care and use of Laboratory Animals of Hoshi University. Female Bcrp(–/–) mice, purchased from Taconic Farms (Germantown, NY, USA), were bred by Shimizu Laboratory Supplies Co., Ltd. (Kyoto, Japan). Age-matched wild-type mice (FVB strain) were purchased from CLEA Japan (Tokyo, Japan). All mice (10–40 weeks) were housed in rooms maintained at 23°C and 55 ± 5% relative humidity, and allowed free access to food and water during the acclimatization period.
In vitro inhibition of ATP-dependent uptake of sulphasalazine by BCRP using curcumin
The studies of sulphasalazine (0.9 µM) transport using membrane vesicles expressing hBCRP were performed using a rapid filtration technique (Kondo et al., 2004). All experiments were completed within 25 min after addition of curcumin to the transport medium, during which more than 80% of the curcumin remained intact. Sulphasalazine retained on the membrane filter was recovered in methanol by sonication for 15 min then vortexing for 10 min. After centrifugation and evaporation, the residue was reconstituted in methanol/10 mM ammonium acetate (40:60, v/v), and subjected to analysis using high-performance LC/MS (LCMS-2010EV, Shimadzu Co., Kyoto, Japan). Chromatographic separation was performed at 40°C on a Capcell Pak C18 column (MGII, 3 µm, 2.0 mm × 50 mm; Shiseido, Tokyo, Japan) under isocratic conditions at a flow rate of 0.3 mL·min−1. The mobile phase consisted of methanol and 10 mM ammonium acetate (40/60, v/v). Sulphasalazine was detected by selected ionization monitoring in negative mode (m/z 397.05).
To calculate the IC50 for the ATP-dependent uptake of sulphasalazine, the difference between the uptake in the presence of AMP and that in the presence of ATP, was fitted to the following equation:
| (1) |
where CLuptake (+Inhibitor) and CLuptake (control) are the uptake clearance in the presence or absence of curcumin, and I is the curcumin concentration. Fitting was performed by the non-linear least-squares method using the MULTI program (Yamaoka et al., 1986) and the Damping Gauss–Newton algorithm.
In vivo pharmacokinetic study of sulphasalazine with curcumin in wild-type and Bcrp(–/–) mice
Curcumin and sulphasalazine were dissolved in PBS containing 0.5% methylcellulose. After an overnight fast, mice were given curcumin orally at a dose of 150, 300 or 400 mg·kg−1 (4 µL·g−1 body weight) or vehicle using a stomach sonde needle. At 1 h after oral administration of curcumin solution or buffer, mice received sulphasalazine orally at a dose of 10 mg·kg−1 body weight. Blood was collected from the tail vein at 0.25, 0.5, 1, 4, 6 and 8 h. After protein precipitation by acetonitrile and centrifugation, the supernatants of the plasma samples were evaporated at 40°C under nitrogen, and the residue was reconstituted with the mobile phase and subjected to LC/MS/MS analysis on the API4000 system (Applied Biosystems, Foster City, CA, USA) equipped with the Prominence LC system (Shimadzu Co.). Chromatographic separation was performed at 40°C on a Capcell Pak C18 column (MGII) under isocratic conditions at a flow rate of 0.2 mL·min−1. The mobile phase consisted of acetonitrile and 10 mM ammonium acetate (pH 8) (30/70, v/v). The ion spray interface was operated in the negative ion mode. The mass transition was from m/z 397 to 197 for sulphasalazine.
Human subjects
This study was approved by the Ethics Committee of Osaka Pharmacology Clinical Research Hospital and Graduate School of Pharmaceutical Sciences, and was conducted in accordance with the Declaration of Helsinki and current Japanese ethical guidelines for clinical research. Eight healthy Japanese male volunteers carrying the CC genotype of the BCRP gene at the position of SNP 421C>A (rs2231142) were enrolled in this study after giving written informed consent. Each subject was physically normal by clinical examination and routine clinical testing, and had no history of significant medical illness or hypersensitivity to any drugs.
Study design
This was a single-arm and four-phase study. The first half of the study was the microdose pharmacokinetic study, and the second half was the pharmacokinetic study at a therapeutic dose. Assuming the volume of the intestinal lumen (2.8–11 L, Tachibana et al., 2009), the lowest effective dose of curcumin was estimated to be 2.8 mg to inhibit BCRP in the small intestine. Because a large dose of curcumin (>150 mg·kg−1) is necessary to inhibit Bcrp in mouse (Figure 2; Shukla et al., 2009), the dose of curcumin used in human studies was set at 2 g, the highest dose approved by the Ethics Committees. This dose of curcumin was lower than the highest dose (8 g per day for 3 months) under which subjects did not display any adverse effects (Cheng et al., 2001).
Figure 2.
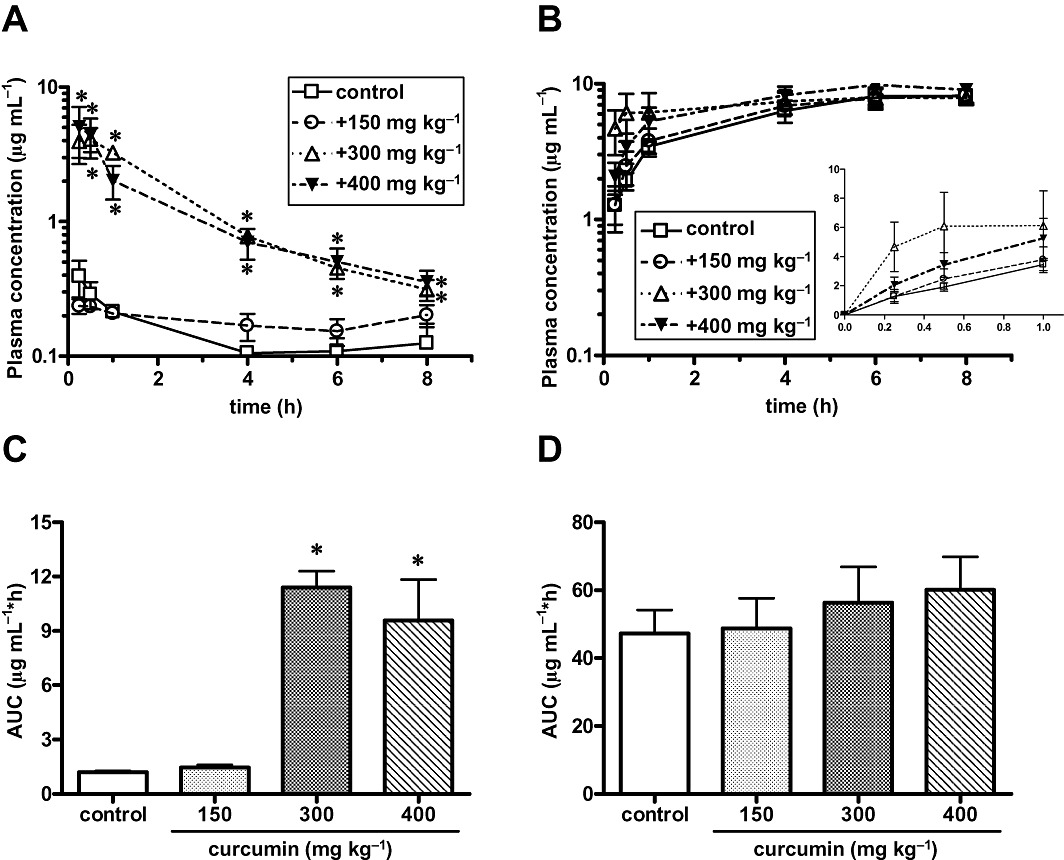
Effect of curcumin on systemic exposure to sulphasalazine following oral administration in wild-type and Bcrp(–/–) mice. Female wild-type (panel A) and Bcrp(–/–) mice (panel B) were given oral sulphasalazine (10 mg·kg−1) alone, or with a pretreatment of curcumin at 150, 300 and 400 mg kg−1, 1 h before the sulphasalazine dose. Plasma concentrations of sulphasalazine were determined at the times shown. Data shown are mean (±SEM) values of the plasma concentrations (n= 5 or 6). In (C) and (D) the AUC0–8 corresponding to the data in (A) and (B) respectively, was calculated by the trapezoidal method. *P < 0.05, significantly different from control; one-way anova with Dunnett's post hoc multiple comparison test.
The microdose of sulphasalazine was prepared as follows: Salazopyrin® tablets were pulverized and dissolved in water containing 0.25 mM NaHCO3 and 10.4% ethanol followed by a 30-fold dilution using water. Sulphasalazine (100 µg) was administered orally after an overnight fast in period 1 and period 2 with a washout period of 7 days, and a therapeutic dose of sulphasalazine (2 g, 4 tablets of Salazopyrin®) was given orally in period 3 and period 4 with a washout period of 7 days. In period 2 and 4, the volunteers received 2 g curcumin (18 tablets) 30 min before sulphasalazine administration. Blood samples were taken by direct venepuncture (sodium heparin anticoagulant) before dosing and at 0.5, 1, 2, 3, 4, 6, 9, 12 and 24 h after dosing. Blood samples were centrifuged to produce plasma, and stored at −80°C until quantification. The study was registered in the UMIN Clinical Trials Registry at http://www.umin.ac.jp/ctr/index.htm (UMIN000002715).
Quantification of sulphasalazine and curcumin in human plasma samples
After addition of the internal standard (IS, probenecid), sulphasalazine and IS in human plasma were extracted using t-butyl methyl ether followed by evaporation of the organic layer. The residue was dissolved in distilled water containing acetonitrile and formic acid (600:400:2, v/v/v), and subjected to quantitation on an API4000 mass spectrometer equipped with an Alliance 2690 separation module (Waters Corporation, Milford, MA, USA). Chromatographic separation was performed at 40°C on a Gemini C18 column (3 µm, 2.0 mm × 50 mm, Phenomenex, Torrance, CA, USA) under gradient conditions at a flow rate of 0.3 mL·min−1. The mobile phases consisted of 10 mM ammonium acetate/25% ammonia solution (2000:1, v/v) and acetonitrile. The turbo ion spray interface was operated in the negative ion mode. The mass transition was from m/z 397 to 197 for sulphasalazine and from m/z 284 to 240 for the IS. The calibration curves were linear in the range of 10–10 000 pg·mL−1 (low range) using 500 µL of plasma and in the range of 20–20 000 ng·mL−1 (high range) using 20 µL of plasma, respectively.
Curcumin and IS (4-hydroxybenzophenone) in human plasma were extracted using ethyl acetate following protein precipitation followed by evaporation of the organic layer. The residue was dissolved in distilled water containing acetonitrile and formic acid (100:170:0.1, v/v/v). Curcumin was analysed using the LCMS-2010EV. Chromatographic separation was performed at 40°C on an Atlantis® dC18 column (3 µm, 2.1 mm × 150 mm, Waters) under isocratic conditions at a flow rate of 0.2 mL·min−1. The mobile phase consisted of acetonitrile and 0.1% formic acid (63:37, v/v). Detection was performed by selected ionization monitoring in positive mode (m/z 369.15 and m/z 199.00 for curcumin and IS, respectively). The analytical method was validated in terms of selectivity and linearity (1–50 ng·mL−1 in plasma, correlation coefficient (r) ≥ 0.99).
Determination of pharmacokinetic parameters of sulphasalazine in healthy subjects
Cmax was obtained directly from the data. The area under the curve (AUC)0–24 was calculated by the linear trapezoidal rule. We calculated the oral clearance (CLtot/F) using the following equation:
The Ke was estimated using least-squares regression analysis from the terminal post-distribution phase of the concentration–time curve.
Determination of solubility of sulphasalazine in inorganic buffer at different pH
The solubility of sulphasalazine was examined at different pH (pH 4.0 and 5.0 in 0.1 M acetate buffer, pH 5.0–8.0 in 0.1 phosphate buffer). Buffer containing sulphasalazine (100 mg) was continuously stirred for 3 days at 25°C. After centrifugation and filtration through a 0.2 µm membrane filter, the specimens were subjected to LC/MS/MS analysis as described later.
Construction of a stable transfectant of human organic anion-transporting polypeptide (OATP)2B1
The full open reading frame of human OATP2B1 cDNA (NM_007256) was amplified by PCR using a fetal brain cDNA library as the template and ligated into a pcDNA3.1(+) vector (Invitrogen, Carlsbad, CA, USA). The PCR primer pairs were 5′-atgggacccaggatagg-3′ and 5′-ctcacactcgggaatcctct-3′. The OATP2B1 open reading frame inserted in the sense orientation was verified by direct sequencing and found to fully match the published reference sequence. HEK293 cells were then transfected with OATP2B1 cDNA packaged into the pcDNA3.1(+) vector, along with Lipofectamine LTX according to the manufacturer's protocols. The transfected cells were selected with Geneticin and cloned using the penicillin cup method. The transport activity of the cloned cells was confirmed.
Determination of the uptake of sulphasalazine by OATP2B1-HEK
Cells were seeded 72 h before the transport assay in poly-l-lysine- and poly-l-ornithine-coated 12-well plates at a density of 1.5 × 105 cells per well. For the transport study, the cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before the transport assay to induce expression of the transporters. The in vitro transport study using HEK293 cells expressing OATP2B1 was performed as described previously (Shimizu et al., 2005). After termination of the uptake, cells were recovered in a test tube and sulphasalazine was extracted by acetonitrile. After three washes with ice-cold buffer, cells were disrupted in sterile distilled water containing acetonitrile (67%, v/v) using a tip sonicator, and the lysates were centrifuged at 15 000×g for 5 min at 4°C. The supernatants were subjected to LC/MS/MS analysis.
Quantitation of sulphasalazine associated with cell specimens was performed using an AB SCIEX QTRAP 5500 mass spectrometer (AB SCIEX, Foster City, CA, USA) equipped with a Prominence LC system (Shimadzu). Chromatographic separation was performed at 40°C on an Atlantis T3 column (5 µm, 2.1 mm × 50 mm, Waters, Tokyo, Japan) under gradient conditions at a flow rate of 0.4 mL·min−1. Mobile phases were acetonitrile and 0.1% formic acid. Detection was performed by selected ionization monitoring in a positive mode (m/z 398.892 for the parent ion, and m/z 196.6 for the daughter ion).
Data analysis
Data are shown as means ± SEM except for results in Figure 4 which are shown as box and whisker plots. The horizontal line within each box represents the median and the box edges represent the lower and upper quartiles, respectively. The whiskers extend from the lower and upper quartiles to the furthest data points still within a distance of 1.5 interquartile ranges from the lower and upper quartiles. Statistical significance was examined by one-way analysis of variance (ANOVA) followed by Dunnett's post hoc multiple comparison tests, by paired t-test or by one-way ANOVA followed by Tukey's multiple comparison tests. Differences were considered to be significant at P < 0.05
Figure 4.
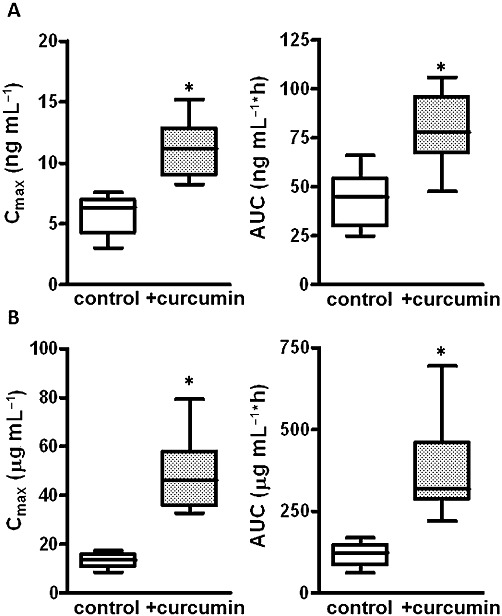
Effect of curcumin on Cmax and AUC of sulphasalazine given orally at a microdose and at a therapeutic dose. Cmax and AUC at a microdose (A) and a therapeutic dose (B) were calculated based on data shown in Figure 3. Values are expressed as box (lower and upper quartiles) and whisker (the furthest data points still within a distance of 1.5 interquartile ranges from the lower and upper quartiles) plots with medians shown as the horizontal line. *P < 0.05, significantly different from control; paired t-test.
Materials
Salazopyrin® (Pfizer, Tokyo, Japan) was used in the clinical study. Sulphasalazine and oestrone-3-sulphate were purchased from Sigma-Aldrich (St Louis, MO, USA), and atorvastatin was from Toronto Research Chemicals (Ontario, Canada). [3H]Oestrone-3-sulphate was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA, USA). All other chemicals of the highest grade commercially available were obtained from Wako Pure Chemical Industries (Kyoto, Japan). Curcumin tablets were manufactured by API (Gifu, Japan) and contained about 111 mg of curcumin per tablet (350 mg total weight of tablet) and maltitol 23.8% (w/w), sucrose fatty acid esters 2.5% (w/w), highly dispersed silicon dioxide 2% (w/w) and starch 31.2% (w/w). Disintegration of the tablet was confirmed by a conventional method (<9 min).
Results
Effect of curcumin on the ATP-dependent uptake of sulphasalazine by hBCRP
Addition of ATP significantly stimulated the uptake of sulphasalazine by membrane vesicles expressing hBCRP for 10 min compared with AMP. The uptake of sulphasalazine in the presence of AMP and ATP (µL/10 min·mg−1 protein) was 352 ± 6 and 868 ± 104, respectively (P < 0.05). Curcumin inhibited the ATP-dependent uptake in a concentration-dependent manner with an IC50 value of 1.60 ± 0.93 µM (Figure 1). Using the previously reported Km of sulphasalazine (0.70 µM) (Jani et al., 2009), the Ki of curcumin was calculated to be 0.70 ± 0.41 µM.
Figure 1.
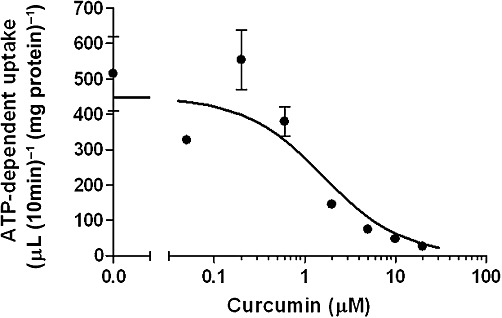
Effect of curcumin on the ATP-dependent uptake of sulphasalazine by hBCRP. Membrane vesicles expressing hBCRP were incubated with sulphasalazine (0.9 µM) in the presence of ATP or AMP for 10 min at 37°C in the absence or presence of curcumin at the designated concentrations. Data shown are mean(±SEM) values of the ATP-dependent uptake of sulphasalazine, which was obtained by subtracting the uptake in the presence of AMP from that in the presence of ATP (n= 3). A calculated curve is shown.
Effect of curcumin on the plasma concentrations of sulphasalazine in wild-type and Bcrp(–/–) mice
Curcumin was given orally to wild-type and Bcrp(–/–) mice 1 h before oral administration of sulphasalazine. Curcumin at a dose of 150 mg·kg−1 did not affect the Cmax of sulphasalazine, but it prolonged the systemic exposure, although the difference was not statistically significant. Oral dosing of curcumin at a dose of 300 and 400 mg·kg−1 markedly elevated the plasma concentrations of sulphasalazine in wild-type mice (Figure 2). The mean AUC0–8 of plasma sulphasalazine concentrations was 8.5- and 8.0-fold higher in wild-type mice given at a dose of 300 and 400 mg·kg−1, respectively, compared with vehicle control. The plasma concentration of sulphasalazine was markedly higher in Bcrp(–/–) mice than in wild-type mice under control condition (Figure 2). Bcrp(–/–) mice treated with oral curcumin (400 mg·kg−1) showed a slight increase in the plasma concentration of sulphasalazine at 0.25 and 0.5 h, but because of large variations, the difference was not statistically significant.
Effect of curcumin on the systemic exposure to sulphasalazine at a microdose and a therapeutic dose
No clinically undesirable signs and symptoms possibly attributable to the administration of sulphasalazine and curcumin were identified during the study. All subjects successfully completed the study according to the protocol. During the study period, the concentrations of curcumin in the plasma samples were below 0.5 ng·mL−1 (lower limit of quantification) or were undetectable except for two plasma samples taken at 2 h (1.7 and 1.1 ng·mL−1).
Curcumin treatment significantly elevated the plasma concentrations of sulphasalazine given either as a microdose or as a therapeutic dose orally (Figure 3). Both Cmax and AUC0–24 of sulphasalazine was significantly greater in the curcumin-treated group than in the control group in both the microdose and therapeutic dose studies (Figure 4), whereas there was no difference in the elimination constants (Ke) (Table 1). The mean AUC0–24 of plasma sulphasalazine concentrations were 3.2- and 1.8-fold larger in curcumin-treated groups at the microdose and therapeutic dose of sulphasalazine, respectively. The effect of BCRP inhibition was more prominent in the therapeutic dose study than in the microdose study (Figure 4).
Figure 3.
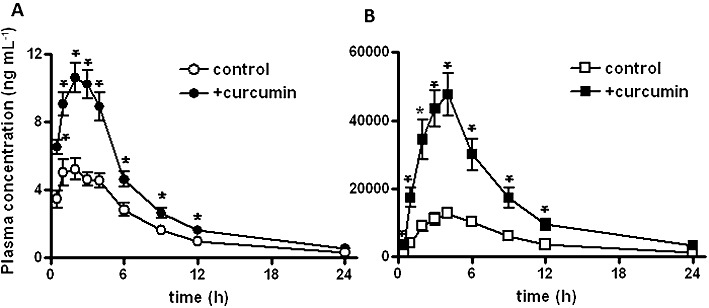
Effect of curcumin on the systemic exposure to sulphasalazine following oral administration in healthy subjects. Curcumin (2 g) was given orally to healthy subjects 30 min before oral sulphasalazine (100 µg panel A and 2 g panel B). Plasma concentrations of sulphasalazine were determined at the times shown. Data shown are mean(±SEM) values of the plasma concentrations (n= 8). *P < 0.05, significantly different from control; paired t-test.
Table 1.
Pharmacokinetic parameters of sulphasalazine (SASP) given orally at a microdose (MD) and at a therapeutic dose (ThD) alone or after pretreatment with oral curcumin
| Period | n | Cmax/Dose (×10−6 mL−1) | AUC0–24/Dose (×10−5 mL−1·hr) | Ke (hr–1) | CLtot/F (hr–1) | |
|---|---|---|---|---|---|---|
| Period1 | SASP (MD) | 8 | 57.2 ± 6.0 | 43.5 ± 5.3 | 0.106 ± 0.003 | 2.58 ± 0.37 |
| Period2 | SASP (MD) + curcumin | 8 | 112 ± 9* | 79.4 ± 6.8* | 0.101 ± 0.003 | 1.34 ± 0.13* |
| Period3 | SASP (ThD) | 8 | 6.58 ± 0.58 | 5.89 ± 0.71 | 0.112 ± 0.003 | 19.2 ± 2.8† |
| Period4 | SASP (ThD) + curcumin | 8 | 24.5 ± 2.9* | 19.0 ± 2.7* | 0.108 ± 0.003 | 5.91 ± 0.70*,† |
Each value represents the mean ± SE.
Significantly different from values in the control group as determined by paired t-test (P < 0.05).
Significantly different from values in the microdose study as determined by paired t-test (P < 0.05).
When the plasma concentration-time profile of sulphasalazine was compared in both control groups, the dose-normalized plasma concentration was significantly higher at the microdose than at the therapeutic dose (Figure 5). AUC0–24 was 7.4-fold higher in the control group of the microdose study than in the control group of the therapeutic dose study (P < 0.05), while the difference was 4.1-fold in the curcumin-treated groups.
Figure 5.
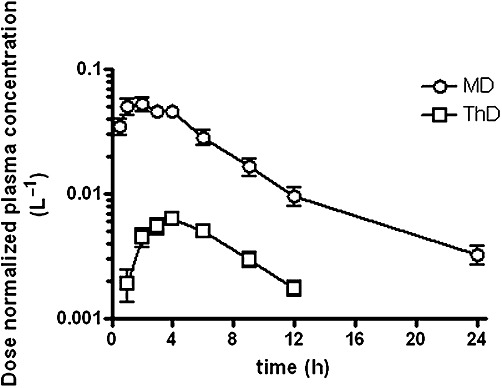
Comparison of dose-normalized plasma concentration-time profile of sulphasalazine in the control groups. Plasma concentrations of sulphasalazine in subjects given an oral microdose (MD) and a therapeutic dose (ThD) of sulphasalazine alone, were divided by the doses. Data are taken from Figure 3.
Solubility of sulphasalazine
The solubility of sulphasalazine in the intestinal lumen was estimated by a conventional solubility test. The maximum concentrations of sulphasalazine achievable were 0.0243, 0.209, 1.66, 19.3 and 17.1 mM at pH 4.0, 5.0, 6.0, 7.0 and 8.0, respectively. A simulation with GastroPlus (Simulations Plus, Inc., Lancaster, CA, USA) using the solubility obtained predicted that the entire amount of sulphasalazine is dissolved during the transition through the small intestine even at the therapeutic dose (data not shown).
OATP2B1-mediated uptake of sulphasalazine in HEK293 cells
The uptake of oestrone-3-sulphate by OATP2B1-HEK was significantly greater than that by mock transfected cells. It was markedly inhibited by an OATP2B1 substrate, atorvastatin (Figure 6). The Ki of atorvastatin has been reported to be 0.7 µM (Grube et al., 2006). It is reasonable that the uptake of oestrone-3-sulphate by OATP2B1 was markedly inhibited in the presence of 20 µM atorvastatin. The uptake of sulphasalazine by OATP2B1-HEK cells was significantly greater than that by mock transfected cells (Figure 6) and markedly inhibited by atorvastatin. The uptake of sulphasalazine by OATP2B1 was saturable with Km and Vmax of 1.73 ± 0.30 µM and 9.72 ± 0.98 pmol·min-1·(mg protein)−1.
Figure 6.
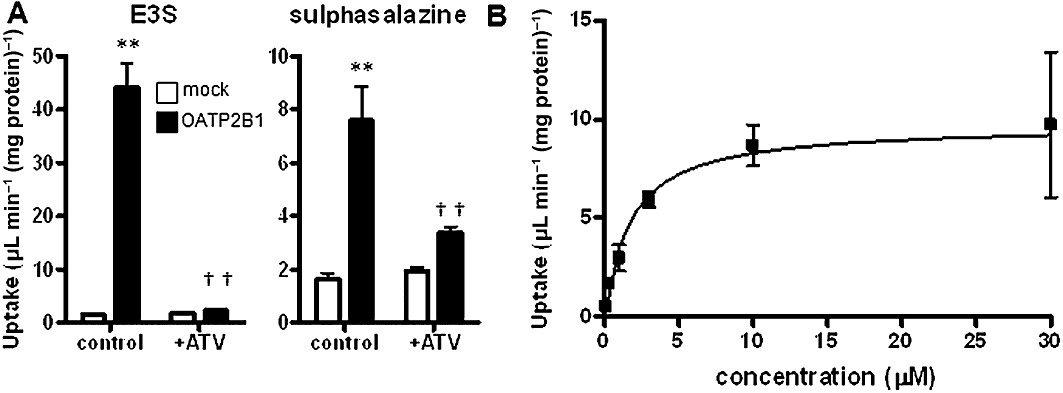
OATP2B1-mediated uptake of sulphasalazine in HEK293 cells. (A) The uptake of [3H]oestrone-3-sulphate (E3S; 0.1 nM) and sulphasalazine (0.1 µM) was determined in OATP2B1-expressing HEK293 cells and mock transfected cells (control) for 2 min at 37°C in the absence and presence of atorvastatin (ATV; 20 µM). Data shown are mean (±SEM) values (n= 3). **P < 0.01 (mock control vs. OATP2B1-HEK significantly different from control), ††P < 0.01 (significant effect of atorvastatin); one-way anova followed by Tukey's multiple comparison tests. (B) The uptake of sulphasalazine was determined in OATP2B1-HEK and mock transfected cells for 2 min at 37°C at various substrate concentrations (0.1, 0.3, 1.0, 3.0, 10, 30 and 100 µM). Specific uptake by OATP2B1 was obtained by subtracting the uptake by mock transfected cells from the uptake by OATP2B1-HEK. A calculated curve is shown.
Discussion and conclusions
BCRP is an ABC transporter, limiting oral bioavailability of drugs by active extrusion to the lumen of intestine. The purpose of this study was to examine the effect of curcumin on pharmacokinetics of oral sulphasalazine, a test substrate of BCRP in healthy subjects as well as experimental animals. A microdose and a therapeutic dose study in healthy subjects were designed to examine the linearity of pharmacokinetics of oral sulphasalazine, and dose dependence on the magnitude of drug–drug interaction.
The inhibitory potency of curcumin was examined in vitro and in vivo in mice. The IC50 of curcumin determined for the ATP-dependent uptake of sulphasalazine by hBCRP is comparable with the previously reported value, which was determined against photolabelling of BCRP with [125I]iodoarylazidoprazosin (Shukla et al., 2009). Consistent with previous mouse studies (Zaher et al., 2006; Shukla et al., 2009), curcumin increased the systemic exposure of sulphasalazine in a dose-dependent manner in wild-type mice. Because the plasma concentration of sulphasalazine in Bcrp(–/–) mice was lower than that in wild-type mice given 300 and 400 mg of curcumin and curcumin slightly increased the plasma concentration of sulphasalazine by 1 h after administration to Bcrp(–/–) mice, it is possible that other transporter(s) delays the absorption and/or limits the oral bioavailability of sulphasalazine. It is also suggested that sulphasalazine undergoes active efflux by multidrug resistance-associated protein 2 (MRP2) as well as Bcrp in rat intestine and Caco-2 cells (Dahan and Amidon, 2009). However, because of the marginal effect of curcumin on the AUC0–8 of sulphasalazine in Bcrp(–/–) mice compared with wild-type mice, most of the effect of curcumin observed in wild-type mice is attributable to Bcrp inhibition.
Curcumin significantly elevated the plasma concentrations of sulphasalazine following oral administration at both microdose and therapeutic dose in healthy subjects (Figure 3). Notably, curcumin showed 10-fold higher potency for in vivo inhibition of BCRP in humans than in mice. Cmax of sulphasalazine in the subjects given a therapeutic dose of sulphasalazine with pretreatment of oral curcumin was similar to the value detected in subjects homozygous for the BCRP SNP (421AA) (49 vs. 41 µg·mL−1) (Yamasaki et al., 2008). Thus, the magnitude of reduction of BCRP activity by curcumin at the current dose is probably similar to that caused by the SNP. The plasma concentration of curcumin was below the limit of quantification, which is in a good agreement with previous report (Vareed et al., 2008). Therefore, curcumin is not suitable to inhibit BCRP involved in active efflux at the blood–brain barrier or systemic elimination by the liver and kidney at the current dose, although inhibition of BCRP in the liver during its first pass remains unclear. Overall, curcumin was an effective in vivo BCRP inhibitor and could be used to investigate the effect of BCRP on the absorption of drugs or drug candidates in clinical studies. This type of study would help to understand the mechanism underlying poor oral absorption and inter-individual variation in the oral availability of BCRP substrate drugs.
However the specificity of curcumin in terms of other transporters and enzymes in the small intestine must be considered. For instance, because the IC50 of curcumin for MRP2 is 5 µM (Wortelboer et al., 2005), curcumin may inhibit MRP2 in the small intestine, as well as BRCP. Currently, the importance of MRP2 in the small intestine has not been established. Abundance of MRP2 mRNA in the intestine is still to be established. In the jejunum it is similar to that of BCRP (Taipalensuu et al., 2001), but somewhat lower in the intestine, compared with BCRP and P-gp (Englund et al., 2006). Further pharmacokinetic interaction studies using in vivo MRP2 test substrates will be needed to elucidate the selectivity of curcumin.
The microdose study provided two pharmacokinetic characteristics of sulphasalazine: lower impact of BCRP inhibition at microdose than at therapeutic dose (Table 1), and non-linearity in AUC of plasma sulphasalazine concentrations (Figure 5). The apparent difference in the effect of BCRP inhibition between microdose and therapeutic dose studies is attributable to the non-linearity in its oral bioavailability. Based on the recovery of sulphasalazine in the urine and bile at the therapeutic dose (4.5% and 2.5%, respectively, Azadkhan et al., 1982), the fraction of sulphasalazine absorbed is estimated to be 50% at the microdose. This limits the magnitude of interaction with curcumin to 2 at most. There are three possible mechanisms underlying the non-linear pharmacokinetics of sulphasalazine: limited dissolution of sulphasalazine, regional difference in transporter expression and saturation of the influx transporter. Although the simulation using GastroPlus® suggests that sulphasalazine can be solubilized in the lumen at a therapeutic dose during transit through the small intestine, dissolution is still considered a limiting factor in sulphasalazine absorption. Formulations of sulphasalazine were different in the microdose and therapeutic dose, solution and tablet, respectively. It is possible that such difference alters the site of sulphasalazine absorption in the small intestine, leading to the difference in its oral availability because of regional difference in the expression of metabolic enzymes and transporters, microenvironment and transit time. As far as BCRP mRNA expression is concerned, its regional difference, if any, is marginal. Gutmann et al., (2005) reported that BCRP mRNA expression is similar between the duodenum and ileum, while Englund et al. (2006) reported that it is twofold higher in the ileum than in the jejunum. We need further clarification of the regional difference in the sulphasalazine absorption along the intestine.
Saturation of the influx transporter for sulphasalazine is another possibility. There have been several reports showing the involvement of multi-specific drug transporters, such as OATP1A2 and OATP2B1, in the intestinal absorption of drugs based on a pharmacokinetic interaction with grapefruit juice and its ingredient, naringenin, which lowers oral availability of fexofenadine, and β-blockers, such as talinolol and celiprolol (Glaeser et al., 2007; Bailey, 2010; Tapaninen et al., 2011). Furthermore, a genotype of OATP2B1 is associated with variation of oral availability of S-fexofenadine (Akamine et al., 2010), fexofenadine (Imanaga et al., 2011) and celiprolol (Ieiri et al., in press), although the effect of the SNP reported by Akamine et al. (2010) is opposite to the reports by others (Imanaga et al., 2011; Ieiri et al., in press). We found sulphasalazine to be a substrate of OATP2B1 in vitro (Figure 5). OATP2B1 mRNA is expressed in the jejunum and ileum with similar abundance (Englund et al., 2006). It is possible that intestinal absorption of sulphasalazine involves OATP2B1-mediated influx. Furthermore, assuming the apparent volume of intestinal lumen to be 2.8–11 L (Tachibana et al., 2009) or 0.7–2.8 L (Tachibana et al., 2012) based on the drug–drug interaction and non-linearity in oral bioavailability of CYP3A4 and P-glycoprotein substrates, the effective concentration of sulphasalazine in the intestinal lumen at the therapeutic dose is estimated to be 0.46–1.8 or 1.8–7.1 mM, respectively, which are far greater than its Km for OATP2B1. The corresponding values at microdose were 0.023–0.090 and 0.090–0.35 µM, respectively, lower than its Km value. This led us to consider the possibility of OATP2B1 saturation at the therapeutic dose of sulphasalazine as part of the mechanism underlying the non-linearity. If this is true, sulphasalazine should inhibit the absorption of drugs that are OATP2B1 substrates; however, there is no report on this interaction. Further studies are necessary to support this hypothesis.
Because sulphasalazine is a high affinity substrate of BCRP (Jani et al., 2009), saturation of BCRP in the intestine should be discussed at the therapeutic dose. The maximum concentration of sulphasalazine we could assume in the lumen exceeds its Km for BCRP. The SNP of BCRP is associated with an intersubject difference in the systemic exposure of sulphasalazine in subjects given a therapeutic dose of sulphasalazine orally and this finding suggests that BCRP is not totally saturated. Unlike the influx transporter, magnitude of BCRP saturation depends on the intracellular unbound concentration. Because of its limited dissolution in the lumen or saturation of the influx transporter, or both, the intracellular unbound concentration may not be high enough to saturate the efflux transporter. Dahan and Amidon (2009) reported non-linearity in the permeability coefficient of sulphasalazine using in situ single-pass rat jejunal perfusion. The permeability became higher along with the substrate concentration, suggesting the saturation of Bcrp-mediated efflux in rats. There may be a species difference between rats and humans in the BCRP efflux activity or in the influx activity by OATPs in the intestine, or both.
Sulphasalazine has long been used in the treatment of inflammatory bowel diseases such as ulcerative colitis and Crohn's disease and rheumatoid disease (Peppercorn, 1984; Rains et al., 1995). After oral administration, sulphasalazine is broken down into sulphapyridine and 5-aminosalicylic acid by bacterial azoreductases in the colon and cecum (Peppercorn and Goldman, 1972; Houston et al., 1982). The targeting of sulphasalazine to the colon is critical for demonstrating its pharmacological action. In addition to the active efflux into the lumen and limited dissolution, saturation of OATP2B1 may contribute to deliver sulphasalazine to its target site. This provides a rationale for relatively high doses of sulphasalazine employed for the treatment of inflammatory bowel diseases.
In conclusion, curcumin is a useful in vivo inhibitor of BCRP in studies assessing the importance of this transporter to the oral availability of drugs and drug candidates in humans. The intestinal absorption of sulphasalazine may involve OATP2B1 in its influx process. Besides the limited dissolution, OATP2B1 saturation is suggested as another possible mechanism underlying the non-linearity in the dose–exposure relationship of sulphasalazine.
Acknowledgments
The authors would like to thank Dr Hiroyuki Hanai (Hamamatsu Minami Hospital, Hamamatsu Japan) for providing information about the formulation of curcumin, Mayuko Miyagawa (The University of Tokyo, Tokyo, Japan) for her technical support; Dr Yasuhisa Adachi and Miki Fujishima (Sekisui Medical, Japan) for supplying HEK293 cells stably expressing OATP2B1, Mr Masato Terakawa (Rabiton, Osaka, Japan) for his support to conduct clinical studies, and Professor Kozo Takayama (Hoshi University, Tokyo, Japan) and Dr Makiko Kusama (The University of Tokyo) for their fruitful suggestions.
This study is a part of a research project for the ‘Establishment of Evolutional Drug Development with the Use of Microdose Clinical Trials’ sponsored by the New Energy and Industrial Technology Development Organization. This work was also supported by a Grant-in-Aid for Scientific Research on Innovative Areas HD-Physiology [Grant 23136101] from the Ministry of Education, Science, and Culture of Japan (to H.K.).
Glossary
- ABC
ATP-binding cassette
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- CLtot
total body clearance
- Ki
inhibition constant Km, Michaelis constant
- MRP2
multidrug resistance-associated protein 2
- OATP
organic anion-transporting polypeptide
- SNP
single nucleotide polymorphism
Conflicts of interest
The authors declare that they have no conflict of interest.
References
- Akamine Y, Miura M, Sunagawa S, Kagaya H, Yasui-Furukori N, Uno T. Influence of drug-transporter polymorphisms on the pharmacokinetics of fexofenadine enantiomers. Xenobiotica. 2010;40:782–789. doi: 10.3109/00498254.2010.515318. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azadkhan AK, Truelove SC, Aronson JK. The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol. 1982;13:523–528. doi: 10.1111/j.1365-2125.1982.tb01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DG. Fruit juice inhibition of uptake transport: a new type of food–drug interaction. Br J Clin Pharmacol. 2010;70:645–655. doi: 10.1111/j.1365-2125.2010.03722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21:2895–2900. [PubMed] [Google Scholar]
- Cusatis G, Sparreboom A. Pharmacogenomic importance of ABCG2. Pharmacogenomics. 2008;9:1005–1009. doi: 10.2217/14622416.9.8.1005. [DOI] [PubMed] [Google Scholar]
- Dahan A, Amidon GL. Small intestinal efflux mediated by MRP2 and BCRP shifts sulfasalazine intestinal permeability from high to low, enabling its colonic targeting. Am J Physiol Gastrointest Liver Physiol. 2009;297:G371–G377. doi: 10.1152/ajpgi.00102.2009. [DOI] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- Englund G, Rorsman F, Rönnblom A, Karlbom U, Lazorova L, Gråsjö J, et al. Regional levels of drug transporters along the human intestinal tract: co-expression of ABC and SLC transporters and comparison with Caco-2 cells. Eur J Pharm Sci. 2006;29:269–277. doi: 10.1016/j.ejps.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Enokizono J, Kusuhara H, Sugiyama Y. Effect of breast cancer resistance protein (Bcrp/Abcg2) on the disposition of phytoestrogens. Mol Pharmacol. 2007;72:967–975. doi: 10.1124/mol.107.034751. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaeser H, Bailey DG, Dresser GK, Gregor JC, Schwarz UI, McGrath JS, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81:362–370. doi: 10.1038/sj.clpt.6100056. [DOI] [PubMed] [Google Scholar]
- Grube M, Köck K, Oswald S, Draber K, Meissner K, Eckel L, et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin Pharmacol Ther. 2006;80:607–620. doi: 10.1016/j.clpt.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Gutmann H, Hruz P, Zimmermann C, Beglinger C, Drewe J. Distribution of breast cancer resistance protein (BCRP/ABCG2) mRNA expression along the human GI tract. Biochem Pharmacol. 2005;70:695–699. doi: 10.1016/j.bcp.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Houston JB, Day J, Walker J. Azo reduction of sulphasalazine in healthy volunteers. Br J Clin Pharmacol. 1982;14:395–398. doi: 10.1111/j.1365-2125.1982.tb01997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieiri I, Doi Y, Maeda K, Sasaki T, Kimura M, Hirota T, et al. Microdosing clinical study: pharmacokinetic, pharmacogenomic (SLCO2B1), and interaction (grapefruit juice) profiles of celiprolol following the oral microdose and therapeutic dose. J Clin Pharmacol. doi: 10.1177/0091270011408612. (in press) DOI: 10.1177/0091270011408612. [DOI] [PubMed] [Google Scholar]
- Imanaga J, Kotegawa T, Imai H, Tsutsumi K, Yoshizato T, Ohyama T, et al. The effects of the SLCO2B1 c.1457C>T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet Genomics. 2011;21:84–93. doi: 10.1097/fpc.0b013e32834300cc. [DOI] [PubMed] [Google Scholar]
- Jani M, Szabo P, Kis E, Molnar E, Glavinas H, Krajcsi P. Kinetic characterization of sulfasalazine transport by human ATP-binding cassette G2. Biol Pharm Bull. 2009;32:497–499. doi: 10.1248/bpb.32.497. [DOI] [PubMed] [Google Scholar]
- Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86:197–203. doi: 10.1038/clpt.2009.79. [DOI] [PubMed] [Google Scholar]
- Kobayashi D, Ieiri I, Hirota T, Takane H, Maegawa S, Kigawa J, et al. Functional assessment of ABCG2 (BCRP) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos. 2005;33:94–101. doi: 10.1124/dmd.104.001628. [DOI] [PubMed] [Google Scholar]
- Kondo C, Suzuki H, Itoda M, Ozawa S, Sawada J, Kobayashi D, et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res. 2004;21:1895–1903. doi: 10.1023/b:pham.0000045245.21637.d4. [DOI] [PubMed] [Google Scholar]
- Kuppens IE, Witteveen EO, Jewell RC, Radema SA, Paul EM, Mangum SG, et al. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin Cancer Res. 2007;13:3276–3285. doi: 10.1158/1078-0432.CCR-06-2414. [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Sugiyama Y. ATP-binding cassette, subfamily G (ABCG family) Pflugers Arch. 2007;453:735–744. doi: 10.1007/s00424-006-0134-x. [DOI] [PubMed] [Google Scholar]
- Murakami T, Takano M. Intestinal efflux transporters and drug absorption. Expert Opin Drug Metab Toxicol. 2008;4:923–939. doi: 10.1517/17425255.4.7.923. [DOI] [PubMed] [Google Scholar]
- Peppercorn MA. Sulfasalazine. Pharmacology, clinical use, toxicity, and related new drug development. Ann Intern Med. 1984;101:377–386. doi: 10.7326/0003-4819-101-3-377. [DOI] [PubMed] [Google Scholar]
- Peppercorn MA, Goldman P. The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther. 1972;181:555–562. [PubMed] [Google Scholar]
- Rains CP, Noble S, Faulds D. Sulfasalazine. A review of its pharmacological properties and therapeutic efficacy in the treatment of rheumatoid arthritis. Drugs. 1995;50:137–156. doi: 10.2165/00003495-199550010-00009. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, et al. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab Dispos. 2005;33:1477–1481. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- Shukla S, Zaher H, Hartz A, Bauer B, Ware JA, Ambudkar SV. Curcumin inhibits the activity of ABCG2/BCRP1, a multidrug resistance-linked ABC drug transporter in mice. Pharm Res. 2009;26:480–487. doi: 10.1007/s11095-008-9735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, et al. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol Ther. 2005;4:650–658. doi: 10.4161/cbt.4.6.1731. [DOI] [PubMed] [Google Scholar]
- Tachibana T, Kato M, Sugiyama Y. Prediction of nonlinear intestinal absorption of CYP3A4 and P-glycoprotein substrates from their in vitro Km values. Pharm Res. 2012;29:651–668. doi: 10.1007/s11095-011-0579-2. [DOI] [PubMed] [Google Scholar]
- Tachibana T, Kato M, Watanabe T, Mitsui T, Sugiyama Y. Method for predicting the risk of drug–drug interactions involving inhibition of intestinal CYP3A4 and P-glycoprotein. Xenobiotica. 2009;39:430–443. doi: 10.1080/00498250902846252. [DOI] [PubMed] [Google Scholar]
- Taipalensuu J, Törnblom H, Lindberg G, Einarsson C, Sjöqvist F, Melhus H, et al. Correlation of gene expression of ten drug efflux proteins of the ATP-binding cassette transporter family in normal human jejunum and in human intestinal epithelial Caco-2 cell monolayers. J Pharmacol Exp Ther. 2001;299:164–170. [PubMed] [Google Scholar]
- Tapaninen T, Neuvonen PJ, Niemi M. Orange and apple juices greatly reduce the plasma concentrations of the OATP2B1 substrate aliskiren. Br J Clin Pharmacol. 2011;71:718–726. doi: 10.1111/j.1365-2125.2010.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vareed SK, Kakarala M, Ruffin MT, Crowell JA, Normolle DP, Djuric Z, et al. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol Biomarkers Prev. 2008;17:1411–1417. doi: 10.1158/1055-9965.EPI-07-2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaming ML, Lagas JS, Schinkel AH. Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev. 2009;61:14–25. doi: 10.1016/j.addr.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Wacher VJ, Salphati L, Benet LZ. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv Drug Deliv Rev. 2001;46:89–102. doi: 10.1016/s0169-409x(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Wortelboer HM, Usta M, van Zanden JJ, van Bladeren PJ, Rietjens IM, Cnubben NH. Inhibition of multidrug resistance proteins MRP1 and MRP2 by a series of alpha,beta-unsaturated carbonyl compounds. Biochem Pharmacol. 2005;69:1879–1890. doi: 10.1016/j.bcp.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Yamaoka K, Tanaka H, Okumura K, Yasuhara M, Hori R. An analysis program MULTI(ELS) based on extended nonlinear least squares method for microcomputers. J Pharmacobiodyn. 1986;9:161–173. [PubMed] [Google Scholar]
- Yamasaki Y, Ieiri I, Kusuhara H, Sasaki T, Kimura M, Tabuchi H, et al. Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin Pharmacol Ther. 2008;84:95–103. doi: 10.1038/sj.clpt.6100459. [DOI] [PubMed] [Google Scholar]
- Zaher H, Khan AA, Palandra J, Brayman TG, Yu L, Ware JA. Breast cancer resistance protein (Bcrp/abcg2) is a major determinant of sulfasalazine absorption and elimination in the mouse. Mol Pharm. 2006;3:55–61. doi: 10.1021/mp050113v. [DOI] [PubMed] [Google Scholar]