

Abstract
BACKGROUND AND PURPOSE
Airway sensory nerves play a key role in respiratory cough, dyspnoea, airway hyper-responsiveness (AHR), all fundamental features of airway diseases [asthma and chronic obstructive pulmonary disease (COPD) ]. Vagally mediated airway reflexes such as cough, bronchoconstriction and chest tightness originate from stimulation of airway sensory nerve endings. The transient receptor potential vanilloid 1 receptor (TRPV1) is present on peripheral terminals of airway sensory nerves and modulation of its activity represents a potential target for the pharmacological therapy of AHR in airway disease.
EXPERIMENTAL APPROACH
As guinea pig models can provide some of the essential features of asthma, including AHR, we have established the model with some classical pharmacological agents and examined the effect of the TRPV1 antagonists, SB-705498 and PF-04065463 on AHR to histamine evoked by ovalbumin (OA) in unanaesthetized sensitized guinea pigs restrained in a double chamber plethysmograph. Specific airway conductance (sGaw) derived from the airflow was calculated as a percentage of change from baseline.
KEY RESULTS
Cetirizine and salbutamol significantly inhibited OA-evoked bronchoconstriction [sGaw area under the curve (AUC): 70 and 78%, respectively]. Atropine, SB-705498 and PF-04065463 significantly inhibited OA-evoked AHR to histamine in unanaesthetized, OA-sensitized guinea pigs (sGaw AUC: 94%, 57% and 73%, respectively). Furthermore, this effect was not related to antagonism of histamine's activity.
CONCLUSION AND IMPLICATIONS
These data suggest that TRPV1 receptors located on airway sensory nerves are important in the development of AHR and that modulation of TRPV1-receptor activity represents a potential target for the pharmacological therapy of AHR in airway disease.
Keywords: TRPV1, airway hyper-responsiveness, sensory nerves, sensitized guinea pigs
Introduction
Airway hyper-responsiveness (AHR), respiratory cough and dyspnoea are characteristic features of airway diseases such as asthma and COPD. These features are thought to derive from the key contribution of transient receptor potential vanilloid 1 (TRPV1), which is highly expressed by sensory neurons and vagal afferents that innervate the airways, to the cough reflex. It is highly likely that sensitization of these sensory endings, decreases the threshold to stimulation resulting in a non-specific AHR similar to hyperalgesia occurring in other inflamed tissues. The potential contribution of TRPV1-sensitive nerves to AHR and bronchoconstriction has led to a large degree of interest in the potential for targeting TRPV1 for the treatment of a range of respiratory diseases (Jia et al., 2005; Geppetti et al., 2006). The TRPV1 receptor is present on peripheral terminals of airway sensory nerves (Gunthorpe et al., 2002), is a member of a subgroup/superfamily of transient receptor potential (TRP) ion channels and is a non-selective, Ca2+-preferring, cation channel. The TRP vanilloid 1 (TRPV1) channel is activated by a diverse range of chemical ligands such as capsaicin (the ‘hot’ component of chilli peppers) and other vanilloids (resiniferatoxin and the cannabinoid, anandamide), as well as acid (protons, H+), physical stimuli such as heat, certain arachidonic acid derivatives and direct phosphorylation via PKC (Caterina et al., 1997; Tominaga et al., 1998; Numazaki et al., 2002; Van Der Stelt and Di Marzo, 2004). In addition, TRPV1 is also activated (directly and indirectly) by a variety of mediators thought to contribute to neuroinflammation (Tominaga and Tominaga, 2005; Szallasi et al., 2007). Thus, sensitization of the TRPV1 receptor in inflammatory diseases appears to play a role and contribute to the transduction of noxious signalling for normally innocuous stimuli, that is AHR (Adcock, 2009).
TRPV1 is known to be sensitized and/or up-regulated in respiratory diseases such as asthma and gastro-oesophageal reflux disease (GERD) (Matthews et al., 2004; Geppetti et al., 2006). Encouragingly, preclinical studies have now demonstrated the antitussive efficacy of a range of TRPV1 antagonists such as Iodo-RTX, BCTC and JNJ17203212 in a number of rodent models including capsaicin and acid-induced cough in guinea pigs (Trevisani et al., 2004; McLeod et al., 2006; Bhattacharya et al., 2007). Furthermore, TRPV1 antagonists have also been shown to be active in a model of antigen-provoked cough in guinea pigs and have been shown to attenuate the hypersensitivity to capsaicin-induced cough that develops following airway inflammation induced by administration of the noxious gas sulphur dioxide (McLeod et al., 2006; 2007). TRPV1, therefore, is linked to playing a significant role in the genesis of cough, which is arguably the most common symptom associated with pulmonary diseases, such as asthma, COPD and the common cold. However, less is known about the role of TRPV1 receptors in the AHR that develops in the airways following inflammation.
The aim of the present work was to examine the effects of the TRPV1 antagonists (Figure 1), SB-705498 [a potent selective TRPV1 antagonist with good oral bioavailability and effective at reducing hyperalgesia and allodynia in animal models (Rami et al., 2004; 2006; Gunthorpe et al., 2007) ] and PF-04065463 (a new, effective TRPV1 antagonist developed by Pfizer in collaboration with Evotec) on ovalbumin (OA)-evoked AHR to histamine in unanaesthetized, OA-sensitized guinea pigs using double chamber plethysmography and also to test the hypothesis that TRPV1 could be involved in AHR that occurs through sensitization of airway nerve endings.
Figure 1.
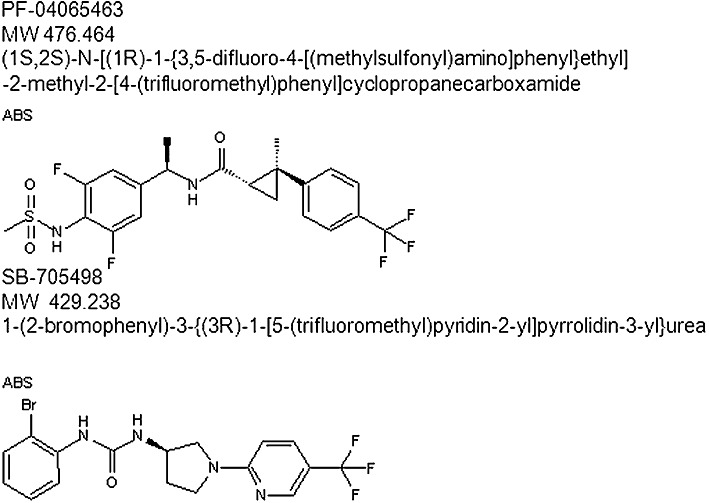
TRPV1 antagonist chemical structure.
In the first instance, the model was established and characterized with some classical pharmacological agents including a histamine H1 antagonist (cetirizine), a β2 adrenoceptor agonist (salbutamol), and a non-selective muscarinic receptor antagonist (atropine).
Methods
Animals
Male Dunkin–Hartley guinea pigs (300–350 g) from B&K Universal Ltd. (East Yorkshire, UK) were sensitized with OA [100 µg·mL−1 aluminium hydroxide (13 mg·mL−1) 1 mL per animal by i.p. route on day 0 and day 7].
Animals received food and water ad libitum. Room temperature (22 ± 2°C) and lighting (maintained on a 12-h cycle) were regulated. Experiments were subject to Home Office and local ethical approval and were carried out under the Animal (Scientific Procedures) Act, 1986, UK.
Measurement of lung function
Unanaesthetized, sensitized guinea pigs were restrained in Buxco double chambers using latex and neoprene collars on day 21. A bias flow delivered fresh air supply to the nasal chamber, preventing CO2 build-up and O2 depletion (Buxco Aerosol Delivery System AUT5110 at 2 LPM; Buxco Bias Flow Regulator PLY1020 at 2.5 LPM). As air moved in and out of the animal's chest, nasal and thoracic flow was measured by a pneumotachograph. The signal was transformed by the transducer in an electronic signal amplified (Buxco MAX 1420) and recorded by Biosystem XA software. As inspiration began in the thoracic cavity, the thoracic signal lead the nasal signal by a phase shift – the delta time (dT) value was the time delay from the thoracic to the nasal flow taken at zero crossing points. Aerosol challenges in saline were performed through a nebulizer head (Aerogen Nebulizer Head, 4–6 µMMAD), connected to the Aerosol Delivery System by the distribution reservoir (Buxco AUT1810) (Buxco Europe, Winchester, UK).
To establish if OA challenge affected the responsiveness of the airways to histamine, a subthreshold aerosol concentration of histamine was established and administered before and after OA administration. The histamine subthreshold aerosol concentration was determined by conducting a preliminary concentration-response curve to histamine (0.01, 0.03, 0.1 and 0.3 mg·mL−1 in saline). A concentration of 0.03 mg·mL−1 was chosen, which did not, in our experimental conditions, evoke a bronchoconstriction before OA challenge. In separate experiments, designed to investigate the effects of agents on histamine aerosol-evoked bronchoconstriction per se in OA-sensitized, but not -challenged guinea pigs, a dose of 0.1 mg·mL−1 in saline was used under the same experimental conditions. Finally, the concentration of OA aerosol used to induce bronchoconstriction was 1 mg·mL−1 in saline. Parameters used for these studies were 10 min of aerosol with 7–9% duty for a flow rate of 0.30–0.39 mL·min−1. The duty permits the control of the flow of the aerosol by limiting the amount of time the nebulizer produces aerosol per cycle of 6 s (i.e. 100% keeps the nebulizer in full time).
Experimental design
The experimental design was executed in two parts. This first part was designed for the characterization of the model with reference compounds such as histamine H1 antagonist (cetirizine), β2-adrenoceptor agonist (salbutamol) and non-selective muscarinic antagonist (atropine). Secondly, TRPV1 antagonists (SB-705498 and PF-04065463) were investigated. On day 21, all studies started with a saline aerosol (0.9% w v−1 NaCl) for habituation of the guinea pigs and to select the animals not responding to saline challenge. Each set of experiments had one compound only and animals were killed at the end of each study.
The effect of cetirizine was examined on day 21 in sensitized guinea pigs (Figure 2A). After saline aerosol, animals were treated with either cetirizine (10 mg·kg−1) (n = 6) or saline (1 mL·kg−1) (n = 6) i.p. 1 h before OA (1 mg·mL−1) -evoked bronchoconstriction. Salbutamol was also evaluated on day 21, after administration of a subthreshold dose of histamine (0.03 mg·mL−1) to sensitized guinea pigs (Figure 2B).
Figure 2.
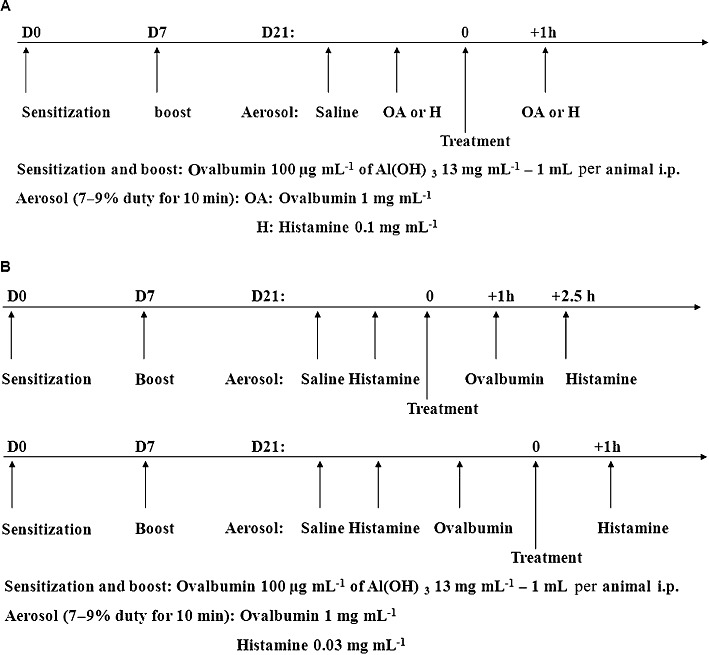
(A) Protocol employed for studying the effect of cetirizine on OA-induced bronchoconstriction. Also the same timings were used to study vehicle, SB-705498, PF-04065463 or atropine on histamine-induced bronchoconstriction. (B) Protocol employed for studying the responsiveness to a subthreshold aerosol of histamine before and after OA. Treatment 1 h before OA, 2.5 h before histamine with vehicle, SB-705498, PF-04065463, atropine or salbutamol in upper panel. Treatment after OA (1 h before histamine) with vehicle, atropine or SB-705498 in lower panel.
Animals were placed in an anaesthetic chamber containing 3.5% halothane delivered in 4 L·min−1 O2 to promote general anaesthesia and enable intra-tracheal treatment (i.t.) with a Hamilton syringe. The control group (n = 10) was treated with saline (100 µL per animal) and the treated group (n = 8) was dosed with salbutamol (3 mg·mL−1) i.t. 1 h before OA (1 mg·mL−1)-induced bronchoconstriction and 2 h 30 min before AHR to histamine was evaluated. The i.t. route was chosen to exclude nasal inhalation and to verify that the bronchoconstriction measured in this model was derived from the airways and not occurring as a consequence of changes in nasal resistance. Finally, the effects of either atropine (10 mg·kg−1) or saline (1 mL·kg−1) i.p. were examined in 2 separate studies. In the 1st experiment (Figure 2B), after administration of a subthreshold dose of histamine (0.03 mg·mL−1), control (n = 8) and treated (n = 8) guinea pigs were dosed with atropine at 1 h before OA (1 mg·mL−1)-evoked bronchoconstriction and 2 h 30 min before AHR to histamine. In the 2nd study (Figure 2A), after saline and histamine (0.1 mg·mL−1)-induced bronchoconstriction challenges, control (n = 8) and treated (n = 8) animals were dosed 1 h before histamine (0.1 mg·mL−1)-induced bronchoconstriction.
The effects of either SB-705498 10 mg·kg−1 p.o. or methylcellulose (0.5% w v−1 methylcellulose/0.1% w v−1 Tween 80) 1 mL·kg−1 p.o. were evaluated in 3 different studies. In the 1st study (Figure 2B), after administration of a subthreshold dose of histamine (0.03 mg·mL−1), control (n = 11) and treated (n = 8) groups were dosed 1 h before OA (1 mg·mL−1)-induced bronchoconstriction. AHR to histamine was then evaluated 2 h 30 min after oral dosing. In the 2nd study (Figure 2B), all animals were challenged with saline, a subthreshold dose of histamine (0.03 mg·mL−1) and OA (1 mg·mL−1). At that time, they were separated into 2 groups: control group (n = 9) and treated group (n = 11). Guinea pigs were dosed 45 min after OA (1 mg·mL−1)-induced bronchoconstriction and 1 h before AHR to histamine was evaluated. Finally in the 3rd study (Figure 2A), after saline and histamine (0.1 mg·mL−1)-induced bronchoconstriction challenges, control (n = 7) and treated (n = 8) animals were dosed 1 h before histamine (0.1 mg·mL−1)-evoked bronchoconstriction. Also, the effects of either PF-04065463 10 mg·kg−1 p.o. or methylcellulose 1 mL·kg−1 p.o. were evaluated in 2 different studies. In the 1st study (Figure 2B), after a subthreshold aerosol dose of histamine (0.03 mg·mL−1), control (n = 16) and treated (n = 16) guinea pigs were dosed 1 h before OA (1 mg·mL−1)-induced bronchoconstriction; followed by AHR to histamine (0.03 mg·mL−1) 2 h 30 min after dosing. In the 2nd study (Figure 2A), after saline and histamine (0.1 mg·mL−1)-induced bronchoconstriction challenges, control (n = 11) and treated (n = 13) animals were dosed 1 h before histamine (0.1 mg·mL−1)-evoked bronchoconstriction.
Data analysis and statistics
Different respiratory parameters were derived from the airflow, such as respiratory rate, specific air resistance (sRaw), specific airway conductance (sGaw). In this study, we have presented the data as sGaw, which is used in the clinic. Each session started with 3 min of baseline recording of the airflow. All measurements were calculated as percentage change from the baseline. A negative value of sGaw (percentage of change from baseline versus time) indicated bronchoconstriction. Data acquisition was performed for 10 min of aerosol plus 20 min of airflow recording in one session. sGaw area under the curve (AUC) ± SEM was analysed after the data had been recorded for 30 min. Statistical comparisons were performed using one-way anova on a log scale. Values of P < 0.05 were considered significant.
Compounds and materials
OA (albumin from chicken egg white), histamine (histamine biphosphate monohydrate), aluminium hydroxide, Tween 80, halothane (2-bromo-2-1,1,1-triofluoroethane), salbutamol, atropine, and methylcellulose (Methocel K4M), cetirizine (cetirizine dihydrochloride – Fluka) were from Sigma –Aldrich Company Ltd UK. SB-705498 and PF-04065463 were synthesized in the laboratories of Pfizer Global R&D.
The drug/molecular target nomenclature conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Results
Effect of cetirizine, salbutamol and atropine on OA-evoked bronchoconstriction and AHR to histamine
In the first part of the study, the model was validated by evaluating the effects of reference compounds on OA-induced bronchoconstriction and/or OA-evoked AHR to histamine in OA-sensitized guinea pigs.
We investigated the effect of the histamine H1 antagonist, cetirizine. In vehicle-treated control animals, OA caused bronchoconstriction with a slow decrease in sGaw reaching a maximum bronchoconstriction (−71%) at 18 min which was maintained over 30 min (−47%) (vehicle sGaw AUC: −1533.45 ± 185.2). However, OA challenge produced only a small fall in sGaw with a plateau from 5 to 30 min (−14%; −25%) in animals treated with cetirizine (Figure 3A) (cetirizine sGaw AUC: −462.86 ± 131.6). Cetirizine significantly inhibited OA-induced bronchoconstriction (70% inhibition; sGaw AUC one-way anova on log scale P < 0.001).
Figure 3.
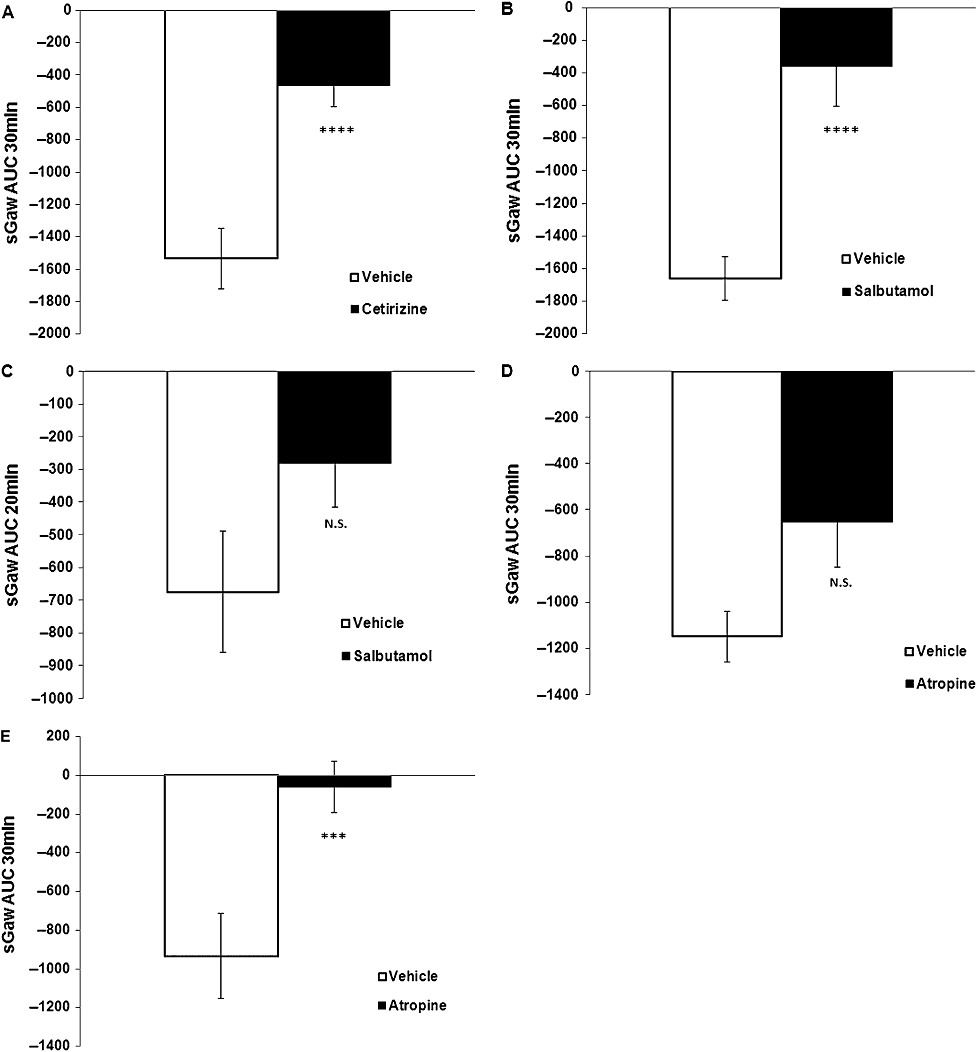
(A) Effect of cetirizine 10 mg·kg−1 i.p. (n = 6) on OA-evoked bronchoconstriction (sGaw AUC for 30 min = −463 ± 132) compared with saline 1 mL·kg−1 i.p. (n = 6) (sGaw AUC for 30 min = −1533 ± 185). Animals dosed 1 h before OA. Negative sGaw AUC ± SEM values show a bronchoconstriction. ****P < 0.001: significant inhibition of 70%. (B) Effect of salbutamol 3 mg·mL−1 i.t. (n = 8) on OA-evoked bronchoconstriction (sGaw AUC for 30 min = −359 ± 246) compared with saline 100 µL per animal i.t. (n = 10) (sGaw AUC for 30 min = −1661 ± 133). Animals dosed 1 h before OA. Negative sGaw AUC ± SEM values show a bronchoconstriction. ****P < 0.001: significant inhibition of 78%. (C) Lack of effect of salbutamol 3 mg·mL−1 i.t. (n = 8) on OA-evoked AHR to a subthreshold dose of histamine (sGaw AUC for 20 min = −282 ± 135) compared with saline, 100 µL per animal i.t. (n = 10) (sGaw AUC for 20 min = −674 ± 186). Animals were dosed 2.5 h before histamine. Negative sGaw AUC ± SEM values show a bronchoconstriction. P > 0.05: non-significant inhibition of 58%. (D) Lack of effect of atropine 10 mg·mL−1 i.p. (n = 8) on OA-induced bronchoconstriction (sGaw AUC for 30 min = −652 ± 199) compared with saline 1 mL·kg−1 i.p. (n = 8) (sGaw AUC for 30 min = −1150 ± 110). Animals were dosed 1 h before OA. Negative sGaw AUC ± SEM values show a bronchoconstriction. P > 0.05: non-significant inhibition of 43%. (E) Effect of atropine 10 mg·mL−1 i.p. (n = 8) on OA-evoked AHR to histamine (sGaw AUC for 30 min = −60 ± 133) compared with saline 1 mL·kg−1 i.p. (n = 8) (sGaw AUC for 30 min = −935 ± 219). Animals were dosed 1 h before OA and 2.5 h before AHR to a subthreshold dose of histamine. Negative sGaw AUC ± SEM values show a bronchoconstriction. ***P < 0.005: significant inhibition of 94%.
When the β2-adrenoceptor agonist, salbutamol was firstly evaluated following intra-tracheal administration with OA challenge, sGaw fell to −69% at 17 min and maintained to −55% up to 30 min in the vehicle group (vehicle sGaw AUC: −1660.7 ± 133.1). In treated animals, sGaw was not reduced by OA, staying quite constant between 0 and −31% for 30 min (salbutamol sGaw AUC: −358.5 ± 245.7). Salbutamol (Figure 3B) produced a statistically significant inhibition of OA-evoked bronchoconstriction (78% inhibition; one-way sGaw AUC on log scale P < 0.001). Following OA challenge there was a marked AHR to histamine in vehicle-treated animals, sGaw fell to −50% at 5 min and returned to baseline and over (+13%) at 25 min in control animals. In the salbutamol-treated group sGaw fell to −22% at 8 min and decreased slowly to −36% over 30 min. sGaw AUC for 20 min was −674.1 ± 185.9 for vehicle and −281.8 ± 134.8 for salbutamol. Although salbutamol reduced the bronchoconstriction (AHR) by 58% (Figure 3C) it was not significantly different from vehicle (sGaw AUC one-way anova on log scale P > 0.05).
The effects of the non-selective muscarinic antagonist, atropine (i.p.) were examined on OA-evoked bronchoconstriction, OA-evoked AHR to histamine and on histamine-induced bronchoconstriction in separate animals. Following OA challenge the sGaw decreased slowly to a maximum of −53% at 15 min and was maintained up to −31% over 30 min in vehicle control guinea pigs (vehicle sGaw AUC: −1149.5 ± 110.2). Furthermore in treated animals, sGaw reached a plateau of −22%; −30% from 7 min up to 30 min (atropine AUC: −651.7 ± 198.6). Although atropine reduced the OA-evoked bronchoconstriction (Figure 3D) the effect was not significant (43% inhibition; sGaw AUC one-way anova on log scale P > 0.05). However, when we measured AHR to histamine, sGaw dropped quickly to −54% at 4 min with a maximum of −64% at 8 min and came back to a plateau of −10% from 21 min to 30 min in control group (vehicle sGaw AUC: −934.9 ± 218.8). In guinea pigs treated with atropine, sGaw was only slightly reduced following histamine (−13%; +9%) for 30 min (atropine sGaw AUC: −59.8 ± 133.1). Atropine produced a marked significant inhibition of the AHR to histamine (94% inhibition; sGaw AUC one-way anova on log scale p < 0.005) (Figure 3E).
Atropine was also evaluated on histamine-induced bronchoconstriction. sGaw dropped to −74% at 6 min for 6 min and returned slowly to baseline at 30 min in control guinea pigs (vehicle sGaw AUC: −1077.4 ± 59.6). sGaw fell to a maximum of −57% at 12 min then returned to baseline at 30 min in the treated group (atropine sGaw AUC: −747.7 ± 136.9). Atropine did not have any significant effect on histamine–induced bronchoconstriction (31% inhibition; sGaw AUC one-way anova on log scale P > 0.05).
Effect of SB-705498 and PF-0406563 on OA-evoked bronchoconstriction and AHR to histamine
In the 2nd part of these studies we evaluated the effects of the TRPV1 antagonists administered p.o. In the 1st series of experiments the animals were treated with SB-705498 10.0 mg·kg−1. OA caused bronchoconstriction with a slow decrease in sGaw that reached a plateau from 7 min (−70%; −75%) and was maintained for 30 min in the vehicle-treated group (vehicle sGaw AUC −1916.2 ± 148.2) and in the SB-705498-treated group (SB-705498 sGaw AUC −1955.06 ± 62.08). This effect of SB-705498 was not significantly different from vehicle (sGaw AUC one-way anova on log scale P > 0.05). The AHR to histamine showed a quicker drop for the vehicle group with a plateau at 4–15 min (−50%; −60%) which returned slowly back to the baseline (−17%) at 30 min. However in the animals treated with SB-705498 the AHR to histamine had a shorter and smaller plateau at 7–14 min (−32%; −41%) and was back to baseline at 21 min (−2%) (Figure 4A). SB-705498 significantly inhibited, by 57%, the sGaw of the AHR to histamine (sGaw AUC one-way anova on log scale P < 0.05) (Figure 4B).
Figure 4.
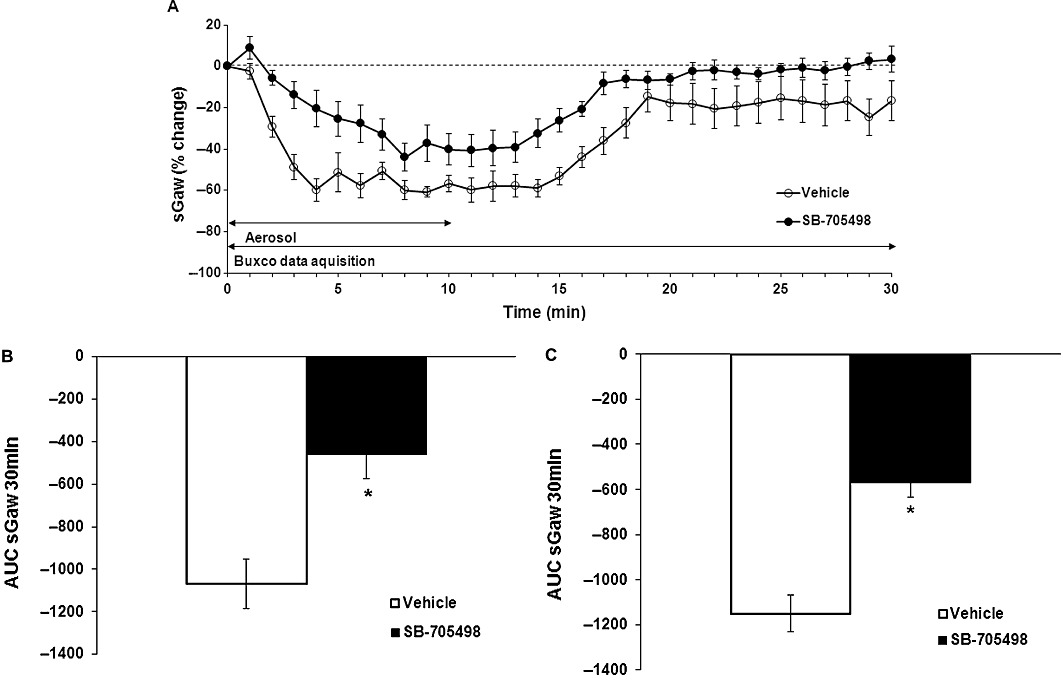
(A) Effect of SB-705498 10 mg·kg−1 p.o. (n = 11) on OA-evoked AHR to histamine (0.03 mg·mL−1) compared with methylcellulose 1 mL·kg−1 p.o. (n = 8) expressed as % change from the baseline sGaw values (10-min aerosol exposure then 20 min of recording) ± SEM. Negative values show a bronchoconstriction. (B) Effect of SB-705498 10 mg·kg−1 p.o. (n = 11) on OA-evoked AHR to histamine (0.03 mg·mL−1) (sGaw AUC for 30 min = −462 ± 114) compared with methylcellulose 1 mL·kg−1 p.o. (n = 8) (sGaw AUC for 30 min −1070 ± 116). Animals were dosed 1 h before OA and 2.5 h before a subthreshold dose of histamine. Negative sGaw AUC ± SEM values show a bronchoconstriction. *P < 0.05: significant inhibition of 57%. (C) Effect of SB-705498 10 mg·kg−1 p.o. (n = 9) on OA-evoked AHR to histamine (0.03 mg·mL−1) (sGaw AUC for 30 min = −569 ± 64) compared with methylcellulose 1 mL·kg−1 p.o. (n = 11) (sGaw AUC for 30 min −1151 ± 82). Animals were dosed after OA and 1 h before a subthreshold aerosol dose of histamine. Negative sGaw AUC ± SEM values show a bronchoconstriction. *P < 0.05: significant inhibition of 50%.
In the 2nd separate set of experiments, OA caused bronchoconstriction with a plateau from 6 min which was maintained over 30 min (−54%; −66%) for all animals. After OA challenge, guinea pigs were separated into 2 groups: vehicle group and SB-705498-treated group. The bronchoconstriction induced by a previous subthreshold dose of histamine showed a quick drop at 6 min (−54%; −65%) with a plateau up to 14 min then back to −25% at 30 min for the control group (vehicle sGaw AUC: −1151.22 ± 81.25). In the SB-705498-treated animals, sGaw showed a shorter and smaller plateau 10–13 min (−27%; −23%) and this was maintained at 16% over 30 min (SB-705498 sGaw AUC: −568.65 ± 64.39). Once again, SB-705498 caused a significant inhibition of AHR to histamine (50% inhibition, sGaw AUC one-way anova on log scale P < 0.05) (Figure 4C).
In separate sensitized animals not challenged with OA, a larger dose of histamine caused bronchoconstriction per se. sGaw fell to −60% at 6 min for a plateau (9–14 min) at −76% for both groups. In the control group, sGaw came back slowly to −9% at 30 min (vehicle sGaw AUC: −1105.8 ± 143.3). While in the SB-705498-treated group, sGaw went back to −32% at 30 min (−1441.9 ± 129.3). Thus, histamine-induced bronchoconstriction was not affected by SB-705498 (AUC one-way anova on log scale P > 0.05; Figure 5).
Figure 5.
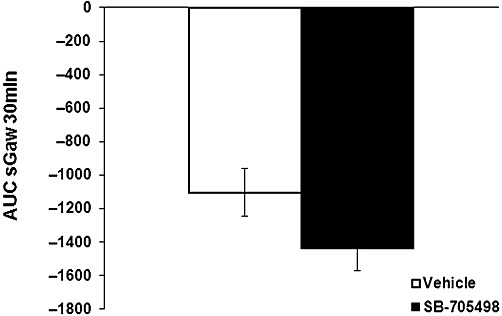
Lack of effect of SB-705498 10 mg·kg−1 p.o. (n = 8) on histamine-induced bronchoconstriction (sGaw AUC for 30 min = −1442 ± 129) compared with methylcellulose 1 mL·kg−1 p.o. (n = 7) (sGaw AUC for 30 min −1106 ± 143). Animals were dosed 1 h before histamine (0.1 mg·mL−1) aerosol. Negative sGaw AUC ± SEM values show a bronchoconstriction. SB-705498 had no antagonist activity against this effect of histamine.
PF-0406563 was evaluated in two sets of experiment. In the first set, in the control group, OA induced a bronchoconstriction with sGaw dropping to −62% at 8 min with a maximum of −65% at 19 min then it slowly returned to baseline (−59%) at 30 min (vehicle sGaw AUC: −1618 ± 105.9). In the PF-0406563-treated group, sGaw was reduced to −52% at 8 min to reach a maximum of −58% at 14 min then returned to −38% at 30 min (PF-0406563 sGaw AUC: −1244.5 ± 175.9). PF-4065463 did not have any effect on OA-evoked bronchoconstriction (23% inhibition, sGaw AUC one-way anova on log scale P > 0.05). The AHR to histamine showed a quicker drop for the vehicle group with a plateau of sGaw at 5 min (−47%) with a maximum at 15 min (−50%), which then returned to baseline at 30 min (−9%) (vehicle sGaw AUC: −850.5 ± 132.7). PF-4065463 significantly inhibited the AHR to histamine (73% inhibition, sGaw AUC one-way anova on log scale P < 0.005): sGaw was reduced to −28% at 9 min for 5 min plateau and returned to baseline at 20 min (PF-4065463 sGaw AUC −231.6 ± 140.5) (Figure 6).
Figure 6.
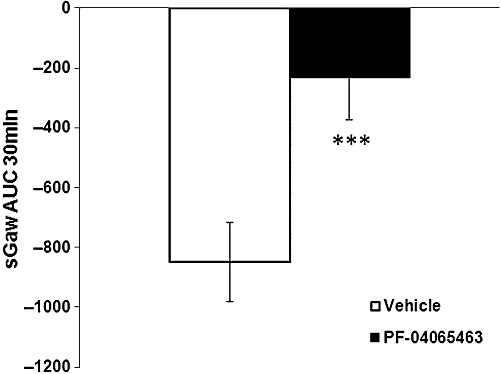
Effect of PF-04065463 10 mg·kg−1 p.o. (n = 16) on OA-evoked AHR to histamine (0.03 mg·mL−1) (sGaw AUC for 30 min = −232 ± 141) compared with methylcellulose 1 mL·kg−1 p.o. (n = 16) (sGaw AUC for 30 min −851 ± 133). Animals were dosed 1 h before OA and 2.5 h before a subthreshold dose of histamine. Negative sGaw AUC ± SEM values show a bronchoconstriction. ***P < 0.005: significant inhibition of 73%.
In separate sensitized animals not challenged with OA, a larger dose of histamine caused bronchoconstriction per se. sGaw fell to −75% at 11 min, and returned to baseline (−3%) at 30 min in the control group (vehicle sGaw AUC: (−1087.5 ± 183.6). In the PF-04065463-treated group, sGaw decreased to −58% at 13 min then came back slowly to −13% at 30 min (−1070.3 ± 185.9). Thus, histamine-induced bronchoconstriction was not affected by PF-4065463 (sGaw AUC one-way anova on log scale P > 0.05).
Discussion and conclusions
It is well known that OA-evoked bronchoconstriction in the OA-sensitized guinea pig reproduces many features of the airway responses to allergen observed in humans, including AHR (Toward and Broadley, 2004). AHR, airway inflammation and cough may be the consequence of the sensitization of airway sensory nerves, and increasing evidence suggests a significant role for TRPV1 in the activity of airway sensory nerves, particularly in relation to cough (Adcock, 2009; Nasra and Belvisi, 2009). The TRPV1 antagonists used in this study, SB-705498 (Rami et al., 2004; 2006; Gunthorpe et al., 2007) and PF-04065463 (unpublished data) have been extensively studied in a range of models of pain and hyperalgesia in various species; however, their effects in the airways have not been tested previously. The objective of the present study was to examine the role of TRPV1 receptors in an ‘allergic’ disease model of AHR initially established and characterized with some classical pharmacological agents. Exposure of OA-sensitized guinea pigs to aerosolized OA caused an immediate early-phase bronchoconstriction that lasted no longer than 30 min. This was significantly inhibited by cetirizine, indicating that the early phase of OA bronchoconstriction was mediated largely by the release of histamine acting on histamine-H1 receptors on the airway smooth muscle (Toward and Broadley, 2004). Likewise salbutamol, by a direct relaxant effect on the airways, also significantly inhibited the OA-evoked bronchoconstriction. Salbutamol administered by the i.t. route, also verified that the measurements of sGaw in this model were derived from the airways and did not occur as a consequence of changes in nasal resistance. Although atropine appeared to reduce OA-evoked bronchoconstriction, this effect was not significant. This lack of effect of atropine was not surprising because there is no evidence to suggest that atropine inhibits the early phase of OA-evoked bronchoconstriction in guinea pigs nor, in the present study, did atropine produce any anti-histamine actions at the dose tested.
A marked increased airway response to a low dose of histamine (AHR) was observed 1.5 h after the early-phase bronchoconstriction evoked by OA inhalation. The mechanism of the OA-induced AHR appears to be independent of residual bronchoconstriction, which would exaggerate contractility of the airways to histamine (Pare and Hogg, 1989), as sGaw had recovered to baseline values at the time of assessing AHR. Tachykinins released from sensory nerve endings cause neurogenic inflammation and cholinergic hypersensitivity and could contribute to the AHR in this study (Schuiling et al., 1999). Another more likely explanation for the AHR is that following OA challenge, a myriad of inflammatory mediators released from mast cells and other inflammatory cells may sensitize and/or expose airway sensory nerve endings that would potentiate local axon reflex responses and the vagal-cholinergic reflex to subsequent activation. Indeed, it is becoming increasingly evident that pulmonary inflammation alters the excitability of afferent airway nerves that are important in the initiation of cough (Carr and Undem, 2001; Lee and Widdicombe, 2001). Furthermore, in our study, AHR to histamine was almost completely abolished by atropine when it was administered before OA (94%) suggesting the involvement of a vagal/cholinergic reflex. The anti-AHR effect of atropine was independent of residual bronchodilator activity, because after treatment with atropine, there was no functional antagonism of the bronchoconstriction by a higher dose of inhaled histamine in non-OA–challenged animals. Interestingly, when atropine was administered after OA challenge, AHR to histamine was significantly inhibited but only by 48%. This might be due to inadequate exposure; although, as atropine is a centrally penetrant drug, we cannot rule out the possibility that it could have prevented a centrally initiated cascade (when administered before OA) resulting in enhanced responsiveness to histamine caused by OA. Administration of atropine following OA would not be expected to block the central component of this cascade. Such an action would depend on the presence of peripheral non-cholinergic mediators that are released in response to central vagal drive. However, further investigations are necessary to elucidate this possibility. The lack of significant inhibition of AHR to histamine by salbutamol may be explained by its short-acting nature, resulting in insufficient exposure at the time histamine was aerosolized (2.5 h). Both TRPV1 antagonists SB-705498 (57%) and PF-04065463 (73%) significantly inhibited AHR to histamine when administered before the OA challenge. SB-705498 also caused similar inhibition (50%) of the AHR to histamine even when administered after the OA challenge. The anti-AHR effect of these TRPV1 antagonists was independent of a residual bronchodilator activity, because after treatment there was no functional antagonism of the bronchoconstriction by a higher dose of inhaled histamine in non-OA–challenged animals. Furthermore, there is no evidence to suggest that TRPV1 receptors are localized on airway smooth muscle and physiological antagonism (bronchodilatation) by the TRPV1 antagonists cannot explain their effects observed in this study. Likewise, there is no evidence to suggest that TRPV1 antagonists inhibit mediator release from mast cells and, indeed, both the TRPV1 antagonists examined had no effect on the OA-evoked bronchoconstriction. The doses chosen for both compounds were based on separate pharmacokinetic studies. With SB-705498, the plasma concentrations required for an effect in our study were consistent with activity at the target receptor in the guinea pig (Rami et al., 2006). The selectivity of SB-705498 was defined by broad receptor profiling (CEREP) and other in vitro assays, where it showed little or no activity on a wide range of ion channels, receptors and enzymes (Gunthorpe et al., 2005). Although we have no in vitro potency data for PF-04065463 in the guinea pig and cannot predict what multiples were achieved, the plasma exposure levels and free concentrations (in the range of 150 nM at the time when AHR to histamine was measured) were greatly in excess of the Ki values observed in human and rat tissues (0.6 and 4.4 nM, respectively, unpublished observations). The species difference index of PF-04065463 between human and rat was calculated as 7.6. In addition, PF-04065463 inhibited proton (pH 5.8) activation of human and rat TRPV1 – IC50 values were 0.664 and 7.52 nM, respectively (unpublished observations). Thus, we can be fairly certain that the receptor would have been targeted. As with SB-705498, the selectivity of PF-04065463 was defined by broad receptor profiling (CEREP, unpublished observations). The selectivity profile of PF-04065463 against other TRP members such as TRPM8, TRPV4 or TRPA1 was not assessed. These three TRP members are all present in the lungs and there is evidence that TRPV4 may be important in AHR (Nilius et al., 2007) and also in ventilator-induced lung injury, both of which may be potential targets for TRPV4 antagonists. In addition, recent evidence suggests that TRPA1 receptors may be important in the late asthmatic response (LAR) in rat and mouse (Raemdonck et al., 2011). Activation of TRPA1 channels on airway sensory nerves by allergen could initiate a central reflex event leading to a parasympathetic cholinergic constrictor response manifesting as the LAR. Interestingly, in these studies, TRPV1 antagonists had no effect on the LAR, but the latter was blocked by a TRPA1 antagonist (HC-030031) in both species. In view of this evidence, it would be interesting to extend our studies to investigate the involvement of TRPV1 and TRPA1 channels in the LAR and the associated reflex responses in our model. Less is known about the role of TRPM8 receptors in the lung, which appear to be localized in epithelial cells. Although the effect of PF-04065463 was not examined on other TRP channels, SB-705498 shows little or no activity at a range of closely related targets such as the swelling/osmotically sensitive and heat-gated channel TRPV4, the cold- and menthol-sensitive receptor TRPM8, in both cases it exhibited greater than 100-fold selectivity for TRPV1 (Gunthorpe et al., 2007).
Although it is possible that the in vivo effects of SB-705498 and PF-04065463 may be mediated via a non-TRPV1 mechanism, this is unlikely for the following reasons. Firstly, both compounds behave as highly selective TRPV1 antagonists. Secondly, and as mentioned above, they did not significantly displace radioligand binding to a panel of receptors and transporters (CEREP). Finally, the plasma concentrations required for effects in our study were consistent with activity at the target receptor. These data also indicate that at the doses of SB-705498 and PF-04065463 used, the compounds are capable of reaching the airway sensory nerves if, indeed, these nerves are responsible for the AHR, and exerting a TRPV1 antagonist effect in vivo. Thus, we believe that the inhibition of AHR to histamine by SB-705498 and PF-04065463 observed in the present study is a consequence of TRPV1 antagonism.
TRPV1 is expressed predominantly in non-myelinated (C-fibre) afferents, which represent approximately 75% of the afferents fibres in the pulmonary branch of the vagus nerve (Ho et al., 2001; Watanabe et al., 2006) and also on Aδ-fibres responding to chemical, mechanical and thermal stimuli (Adcock, 2009). Thus, sensitization of TRPV1 receptors on the sensory endings by inflammatory mediators released following OA challenge could decrease the threshold to stimulation resulting in a non-specific AHR. It is well documented that a number of endogenous inflammatory mediators can modulate the sensitivity of TRPV1 during tissue inflammation. The exact mechanisms underlying the sensitization of TRPV1 are not yet fully understood, but several signal transduction pathways are known to be involved. In addition to capsaicin, the TRPV1 channel is activated by a number of different stimuli including heat, acid, certain arachidonic acid derivatives and direct phosphorylation via protein kinases A, C and G and tyrosine kinase (Premkumar and Ahern, 2000; Vellani et al., 2001; Kwong and Lee, 2002; Gu et al., 2003). Moreover, there is also evidence that various inflammatory mediators such as ATP (Szallasi et al., 2007), bradykinin (Hwang and Oh, 2002), nerve growth factor (Ganju et al., 1998; Chuang et al., 2001) or PGE2 (Kwong and Lee, 2002; Lee et al., 2002) may indirectly lead to activation of the TRPV1 channel via activation of their respective receptors. There is strong experimental evidence that the combination of direct and indirect mechanisms finely tune the TRPV1 activity. Each of the different known modes of direct TRPV1 activation (protons, heat and vanilloids) is capable of sensitizing the channel to other agonists. Similarly, inflammatory mediators from the external milieu found in disease conditions can indirectly sensitize the receptor. It is this sensitization of the TRPV1 receptor in inflammatory disease that could hold the key and contribute to the transduction of noxious signalling for normally innocuous stimuli causing AHR/hypertussive responses in patients with chronic respiratory disease (Adcock, 2009). It seems reasonable to suggest that the various mechanisms for sensitization provide a scenario for TRPV1 to be tonically active, and this activity may contribute to the underlying pathology in airway disease. Although the evidence in our study points to a peripheral effect of the TRPV1 antagonists – indeed, PF-04065463 was designed to be peripherally restricted (brain/plasma ratio = 0.04) – we cannot completely rule out a central effect as we do not have the equivalent data for SB-705498. However, neither compound induced any behavioural effects at the doses examined.
In conclusion, the data from this study show that TPV1 antagonists inhibit the increased responsiveness to a low dose of histamine evoked by OA challenge in sensitized guinea pigs. This suggests that TRPV1 on airway sensory nerves may play an important role in the development of non-specific AHR that occurs in inflammatory diseases of the airways. To date, much importance has been focused on the development of TRPV1 antagonists for the treatment of pain. Our data, together with the already existing data for a role of TRPV1 channels in the airways in cough, provide evidence that TRPV1 antagonists may have potential for development as therapeutics for the treatment of non-specific AHR in respiratory disease.
Acknowledgments
The authors acknowledge David Collins for statistical advice.
Glossary
- AHR
airway hyper-responsiveness
- AUC
area under the curve
- COPD
chronic obstructive pulmonary disease
- dT
Δ time
- GERD
gastro-oesophageal reflux disease
- LAR
late asthmatic response
- OA
ovalbumin
- sGaw
specific airway conductance
- sRaw
specific air resistance
- TRPA1
transient receptor potential ankyrin 1
- TRPM8
transient receptor potential melastatin
- TRPV1
transient receptor potential vanilloid 1
- TRPV4
transient receptor potential vanilloid 4
Conflict of interest
None.
References
- Adcock JJ. TRPV1 receptors in sensitisation of cough and pain reflexes. Pulm Pharmacol Ther. 2009;22:65–70. doi: 10.1016/j.pupt.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Scott BP, Nasser N, Ao H, Maher MP, Dubin AE, et al. Pharmacology and antitussive efficacy of 4-(3-trifluoromethylpyridin-2-yl)-piperazine-1-carboxylic acid (5-trifluromethyl-pyridin-2-yl)-amide (JNJ17203212), a transient receptor potential vanilloid 1 antagonist in guinea pigs. J Pharmacol Exp Ther. 2007;323:665–674. doi: 10.1124/jpet.107.127258. [DOI] [PubMed] [Google Scholar]
- Carr MJ, Undem BJ. Inflammation-induced plasticity of the afferent innervation of the airways. Environ Health Perspect. 2001;109:567–571. doi: 10.1289/ehp.01109s4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Ganju P, O'Bryan JP, Der C, Winter J, James IF. Differential regulation of SHC proteins by nerve growth factor in sensory neurones PC12 cells. Eur J Neurosci. 1998;10:1995–2008. doi: 10.1046/j.1460-9568.1998.00209.x. [DOI] [PubMed] [Google Scholar]
- Geppetti P, Materazzi S, Nicoletti P. The transient receptor potential vanilloid 1: role in airway inflammation and disease. Eur J Pharmacol. 2006;533:207–214. doi: 10.1016/j.ejphar.2005.12.063. [DOI] [PubMed] [Google Scholar]
- Gu Q, Kwong K, Lee LY. Ca2+ transient evoked by chemical stimulation is enhanced by PGE2 in vagal sensory neurones: role of cAMP/PKA signalling pathway. J Neurophysiol. 2003;89:1985–1993. doi: 10.1152/jn.00748.2002. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Benham CD, Randall A, Davis JB. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol Sci. 2002;23:183–191. doi: 10.1016/s0165-6147(02)01999-5. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Hannan SL, Jerman JC, Egerton J, Smart D, Rami HK, et al. 2005. SB-705498: a novel potent and selective TRPV1 antagonist which inhibits the capsaicin-, acid- and heat-mediated activation of the receptor. Society for Neuroscience, Washington, DC: 2005, online. Abstract No. 36.7.
- Gunthorpe MJ, Hannan SL, Smart D, Jerman JC, Arpino S, Smith GD, et al. Characterisation of SB-705498, a potent and selective vanilloid receptor-1 (VR1/TRPV1) antagonist that inhibits the capsaicin-, acid-, and heat-induced activation of the receptor. J Pharmacol Exp Ther. 2007;321:1183–1192. doi: 10.1124/jpet.106.116657. [DOI] [PubMed] [Google Scholar]
- Ho CY, Gu Q, Lin YS, Lee LY. Sensitivity of vagal afferent endings to chemical irritants in the rat lung. Respir Physiol. 2001;127:113–124. doi: 10.1016/s0034-5687(01)00241-9. [DOI] [PubMed] [Google Scholar]
- Hwang SW, Oh U. Hot channels in airways: pharmacology of the vanilloid receptor. Curr Opin Pharmacol. 2002;3:235–242. doi: 10.1016/s1471-4892(02)00149-2. [DOI] [PubMed] [Google Scholar]
- Jia Y, McLeod RL, Hey JA. TRPV1 receptor: a target for the treatment of pain, cough, airway disease and urinary incontinence. Drug News Perspect. 2005;18:165–171. doi: 10.1358/dnp.2005.18.3.892761. [DOI] [PubMed] [Google Scholar]
- Kwong K, Lee LY. PGE2 sensitises cultured pulmonary vagal sensory neurones to chemical and electrical stimuli. J Appl Physiol. 2002;93:1419–1428. doi: 10.1152/japplphysiol.00382.2002. [DOI] [PubMed] [Google Scholar]
- Lee LY, Widdicombe JG. Modulation of airway sensitivity to inhaled irritants: role of inflammatory mediators. Environ Health Perspect. 2001;109:585–589. doi: 10.1289/ehp.01109s4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY, Kwong K, Lin YS, Gu Q. Hypersensitivity of bronchopulmonary C-fibres induced by airway mucosal inflammation: cellular mechanisms. Pulm Pharmacol Ther. 2002;15:199–204. doi: 10.1006/pupt.2002.0338. [DOI] [PubMed] [Google Scholar]
- Matthews PJ, Aziz Q, Facer P, Davis JB, Thompson DG, Anand P. Increased capsaicin receptor TRPV1 nerve fibres in the inflamed human oesophagus. Eur J Gastroenterol Hepatol. 2004;16:897–902. doi: 10.1097/00042737-200409000-00014. [DOI] [PubMed] [Google Scholar]
- McLeod RL, Fernandez X, Correll CC, Phelps TP, Jia Y, Wang X, et al. TRPV1 antagonists attenuate antigen-provoked cough in ovalbumin sensitized guinea pigs. Cough. 2006;2:10. doi: 10.1186/1745-9974-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod RL, Jia Y, McHugh NA, Fernandez X, Mingo GG, Wang X, et al. Sulfur-dioxide exposure increases TRPV1-mediated responses in nodose ganglia cells and augments cough in guinea pigs. Pulm Pharmacol Ther. 2007;20:750–757. doi: 10.1016/j.pupt.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Nasra J, Belvisi MG. Modulation of sensory nerve function and the cough reflex: understanding disease pathogenesis. Pharmacol Ther. 2009;124:354–375. doi: 10.1016/j.pharmthera.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase cepsilon and identification of two target serine series. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Pare PD, Hogg JC. Mechanics of airway narrowing. Am Rev Respir Dis. 1989;139:242–246. doi: 10.1164/ajrccm/139.1.242. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Raemdonck K, de Alba J, Birrell MA, Grace M, Maher SA, Irvin CG, et al. A role for sensory nerves in the late asthmatic response. Thorax. 2011;67:19–25. doi: 10.1136/thoraxjnl-2011-200365. [DOI] [PubMed] [Google Scholar]
- Rami HK, Thompson M, Wyman P, Jerman JC, Egerton J, Brough S, et al. Discovery of small molecule antagonists of TRPV1. Bioorg Med Chem Lett. 2004;14:3631–3634. doi: 10.1016/j.bmcl.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Rami HK, Thompson M, Stemp G, Fell S, Jerman JC, Stevens AJ, et al. Discovery of SB-705498: a potent, selective and orally bioavailable TRPV1 antagonist suitable for clinical development. Bioorg Med Chem Lett. 2006;16:3287–3291. doi: 10.1016/j.bmcl.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Schuiling M, Zuidhof AB, Meurs H, Zaagsma J. Role of tachykinin NK2-receptor activation in the allergen-induced late asthmatic reaction, airway hyperreactivity and airway inflammatory cell influx in conscious, unrestrained guinea-pigs. Br J Pharmacol. 1999;127:1030–1038. doi: 10.1038/sj.bjp.0702628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof of concept. Nat Rev Drug Discov. 2007;6:357–372. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Tominaga T. Structure and function of TRPV1. Pflugers Arch. 2005;451:143–150. doi: 10.1007/s00424-005-1457-8. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, et al. The cloned capsaicin receptor integrates multiple pain producing stimuli. Neuron. 1998;21:532–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Toward TJ, Broadley KJ. Early and late bronchoconstriction, airway hyper-reactivity, leucocyte influx and lung histamine and nitric oxide after inhaled antigen: effects of dexamethasone and rolipram. Clin Exp Allergy. 2004;34:91–102. doi: 10.1111/j.1365-2222.2004.01833.x. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Milan A, Gatti R, Zanasi A, Harrison S, Fontana G, et al. Antitussive activity of iodo-resiniferatoxin in guinea pigs. Thorax. 2004;59:769–772. doi: 10.1136/thx.2003.012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Stelt M, Di Marzo V. Endovallinoids. Putative endogenous ligands of transient receptor potential vallinoid 1 channels. Eur J Biochem. 2004;271:1827–1834. doi: 10.1111/j.1432-1033.2004.04081.x. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Horie S, Michael GJ, Keir S, Spina D, Page CP, et al. Immunohistochemical co-localization of transient receptor potential vanilloid (TRPV)1 and sensory neuropeptides in the guinea-pig respiratory system. Neuroscience. 2006;141:1533–1543. doi: 10.1016/j.neuroscience.2006.04.073. [DOI] [PubMed] [Google Scholar]