

Abstract
BACKGROUND AND PURPOSE
The histamine H3 receptor was identified as the autoreceptor of brain histaminergic neurons. After its cloning, functional H3 receptor isoforms generated by a deletion in the third intracellular loop were found in the brain. Here, we determined if this autoreceptor was the long or the short isoform.
EXPERIMENTAL APPROACH
We hypothesized that the deletion would affect H3 receptor stereoselectivity. The effects of the enantiomers of two chiral ligands, Nα-methyl-α-chloromethylhistamine (NαMe-αClMeHA) and sopromidine, were investigated on cAMP formation at the H3(445) and H3(413) receptor isoforms, common to all species. They were further compared with their effects at autoreceptors. They were also compared on [35S]GTPγ[S] binding to membranes of rat cerebral cortex, striatum and hypothalamus, the richest area in autoreceptors.
KEY RESULTS
The stereoselectivity of NαMe-αClMeHA enantiomers as agonists was similar at the H3(413) receptor isoform and autoreceptors, but lower at the long isoform. While (S) sopromidine did not discriminate between the isoforms, (R) sopromidine was an antagonist at the H3(413) receptor isoform and autoreceptors, but a full agonist at the long isoform. In rat brain, stereoselectivity of NαMe-αClMeHA was higher in the hypothalamus than in cerebral cortex or striatum, whereas the opposite pattern was found for sopromidine.
CONCLUSIONS AND IMPLICATIONS
The pharmacological profiles of H3 receptor isoforms differed markedly, showing that the function of autoreceptors was fulfilled by a short isoform, such as the H3(413) receptor. Development of drugs selectively targeting autoreceptors might enhance their therapeutic efficacy and/or decrease incidence of side effects.
Keywords: histamine H3 receptor, autoreceptor, isoforms, enantiomers, stereoselectivity, rat brain, coupling properties
Introduction
The histamine H3 receptor was identified as an autoreceptor regulating histaminergic neuron activity in the brain (Arrang et al., 1983; 1987; receptor nomenclature follows Alexander et al., 2011). However, H3 receptors are also present on many other neuronal populations (Pillot et al., 2002), either as post-synaptic receptors or as heteroreceptors (Schlicker et al., 1994). The coupling of the H3 receptor to Gi/o proteins was confirmed by its cloning in humans (Lovenberg et al., 1999). Activation of recombinant H3 receptors inhibits adenylate cyclase, assayed as cAMP accumulation (Lovenberg et al., 1999), and activates phospholipase A2, assessed as arachidonic acid (AA) release (Morisset et al., 2000).
Various recombinant isoforms of the H3 receptor have been identified in different species including humans. These isoforms are generated by the deletion of a pseudo-intron, variable in length, located in the third intracellular loop of the receptor (Hancock et al., 2003). They are all expressed in the brain, but their respective functions therein remain unknown. Binding studies on these H3 receptor isoforms revealed only moderate pharmacological differences (Coge et al., 2001; Morisset et al., 2001; Rouleau et al., 2004). Functional studies with standard agonists revealed some differences in coupling between rat isoforms, which, however, were dependent on the response studied and cell type used (Morisset et al., 2000; Drutel et al., 2001). These agonists also displayed a higher potency at the human H3(365) receptor than at the human H3(445) receptor (Wellendorph et al., 2002; Bongers et al., 2007). Using various chiral histamine derivatives, we have previously shown a strong stereoselectivity of the rat H3 autoreceptor on inhibition of histamine release, with a preference of the H3 receptor for (+) enantiomers (i.e. corresponding to the S-configuration of L-histidine), such as R-(α)-methylhistamine (R-αMeHA) (Arrang et al., 1985a). In the present study, we tried to take advantage of this stereoselectivity to identify which of the long or short isoforms function as the autoreceptors. Our hypothesis was that the deletion within the third intracellular loop of the H3 receptor would generate three-dimensional modifications recognized by chiral compounds. We tested this hypothesis by characterizing the effects of Nα-methyl-α-chloromethylhistamine (NαMe-αClMeHA) and sopromidine, two chiral ligands for which the H3 receptor is stereoselective (Arrang et al., 1985a), at the rat long (non-deleted) H3(445) isoform and short H3(413) isoform. This short isoform was selected for the study because, in contrast to the others, it is the only one to be maintained in the brain from all species including rat (Morisset et al., 2001), mouse (Rouleau et al., 2004), guinea pig (Tardivel-Lacombe et al., 2000), monkey (Strakhova et al., 2008) and human (Coge et al., 2001; Tardivel-Lacombe et al., 2001). The stereoselectivity of each of the two isoforms was compared with the autoreceptor. Moreover, we used H3 receptor-mediated [35S]GTPγ[S] binding to rat brain membranes (Rouleau et al., 2002) in order to further compare the effects of NαMe-αClMeHA and sopromidine isomers in the hypothalamus, a region known to contain a very high density of histaminergic fibres and, hence, of H3 autoreceptors (Panula and Airaksinen, 1991; Tohyama et al., 1991; Wouterlood and Steinbusch, 1991), as well as in the cerebral cortex and striatum. All these approaches showed marked differences between isoforms and revealed that the autoreceptor is a short isoform.
Methods
Cloning and expression of the rH3(445) and rH3(413) receptor isoforms
The two rat H3 (rH3) receptor isoforms were cloned and expressed as previously described (Morisset et al., 2001). Briefly, cDNA inserts corresponding to the full-length coding sequence of the rat H3 isoforms, rH3(445) or rH3(413), were ligated into the mammalian expression vector pCIneo (Promega, Charbonnières, France). CHO-K1 cells were transfected with SuperFect Reagent (Qiagen, Courtaboeuf, France). Stable transfectants were selected with 2 mg·mL−1 of Geneticin, tested for their expression level of [125I]iodoproxyfan (IPX) binding sites and maintained in the presence of 1 mg·mL−1 of Geneticin.
[125I]IPX binding assays
CHO cells transfected with the two isoforms, CHO(rH3(445)R) and CHO(rH3(413)R) cells, and expressing 400–500 fmol·mg−1 protein were harvested, homogenized in ice-cold phosphate buffer (50 mM Na2HPO4/KH2PO4, pH 7.5) and centrifuged (140×g for 10 min at 4°C). The pellets were then suspended in 10 mL of phosphate buffer and homogenized with a Polytron homogenizer (Polytron, Inc., Norcross, GA, USA). After centrifugation at 23 000×g for 30 min at 4°C, the last pellets were washed superficially and sonicated for 30 s in fresh ice-cold buffer. Binding assays were performed as described previously (Morisset et al., 2001). Briefly, aliquots of membrane suspensions (≍20 µg of protein) were incubated for 60 min at 25°C with 30 pM [125I]IPX alone or together with drugs in increasing concentrations (200 µL final volume). Non-specific binding was determined using the selective H3 receptor agonist imetit at 1 µM. Incubations, performed in triplicate, were stopped by rapid filtration through glass microfibre filters (GF/B; Whatman, Clifton, NJ, USA) pre-soaked in 0.3% polyethylenimine. Radioactivity trapped on filters was counted with a gamma counter.
[3H]AA release
CHO(rH3(445)R) and CHO(rH3(413)R) cells expressing 400–500 fmol·mg−1 protein were seeded 24 h before the assay in 24 well-plates. After incubation for 2 h at 37°C with 0.5 µCi [3H]AA in DMEM-Nut mix F-12 (Invitrogen Life Technology, Cergy Pontoise, France) containing 0.2% BSA, the cells were washed twice and the drugs tested [histamine, R-αMeHA and for CHO(rH3(413)R) cells, S-(α)-methylhistamine] were added in increasing concentrations and incubated for 10 min. Cells were then incubated for 30 min with 2 µM of the Ca2+ ionophore A23187. [3H]AA release was determined by liquid scintillation counting.
cAMP accumulation
CHO(rH3(445)R) and CHO(rH3(413)R) cells were seeded 24 h before the assay in 96 well-plates. After incubation for 10 min at 37°C with 3 µM forskolin, drugs [histamine, R-αMeHA, R and S enantiomers of NαMe-αClMeHA and of sopromidine] were added, when required, at increasing concentrations in DMEM-Nut mix F-12 containing 100 µM isobutylmethyl xanthine. cAMP was extracted and measured by radioimmunoassay according to the instructions of the manufacturer (PerkinElmer Life Sciences, Boston, MA, USA).
[35S]GTPγ[S] binding assays
[35S]GTPγ[S] binding to brain membranes was performed as previously described (Rouleau et al., 2002). The cerebral cortex, striatum and hypothalamus were dissected out from brains of male Wistar rats (160–200 g, Janvier, Le Genest-Saint-Isle, France), homogenized in ice-cold buffer (50 mM Tris/HCl, pH 7.4) and centrifuged (140×g for 10 min at 4°C). The supernatants were centrifuged twice at 23 000×g for 15 min at 4°C. The final pellets were suspended in 50 volumes of buffer. Membranes (8–22 µg) were pretreated for 30 min at 25°C with adenosine deaminase (1 U·mL−1; Roche, Meylan, France) and incubated for 60 min at 25°C with 0.1 nM [35S]GTPγ[S] and, when required, the various drugs were tested (R and S enantiomers of NαMe-αClMeHA and of sopromidine), in 1 mL of assay buffer (50 mM Tris/HCl, 50 mM NaCl, 5 mM MgCl2, 10 µM GDP, 0.02% BSA, pH 7.4). In order to prevent any interaction of the drugs with histamine H1 and H2 receptors, all incubations were performed in the presence of 100 nM mepyramine and 10 µM cimetidine. The non-specific binding was determined using GTPγS (10 µM). Incubations were stopped by rapid filtration under vacuum through Whatman GF/B filters. Filters were washed twice with 4 mL of ice-cold water and the radioactivity retained on the filters was measured by liquid scintillation spectrometry.
Analysis of data
The curves were analysed with an iterative least-squares method by non-linear regression using a one-site cooperative model (Gbahou et al., 2006). The method provided estimates for EC50 values, IC50 values and their SEM. The apparent affinity constants (Ki values) of NαMe-αClMeHA enantiomers on inhibition of [125I]IPX binding were calculated from their IC50 values by using the relationship (Cheng and Prussoff, 1973): Ki= IC50/1 + (S/X), where S represents the concentration (30 pM) and X is the apparent dissociation constant (KD) of [125I]IPX at the rH3(445) receptor isoform (85 pM) and at the rH3(413) receptor isoform (82 pM). The same relationship was used to calculate Ki values of sopromidine isomers tested against histamine (100 nM) on cAMP formation at recombinant isoforms. In that case, the total curves were analysed with X representing the EC50 values of histamine at the rH3(445) (9 nM) and rH3(413) receptor isoforms (15 nM). Their Ki values on specific [35S]GTPγ[S] binding induced by 1 µM imetit to brain membranes were also calculated with the same relationship, taking into account only the antagonistic part of the curves (above 100%) and an EC50 value of imetit of 2 ± 1 nM (not shown).
Statistical evaluation of the results was performed by one-way anova, followed by Student's Newman–Keuls post hoc test. For statistical comparison of the EC50 values, IC50 values or maximal effects of two compounds (i.e. two enantiomers or R-αMeHA vs. histamine), the two corresponding curves were analysed by two-way anova, followed by Bonferroni post hoc test.
The activity ratio of two enantiomers (termed S/R ratio when the S enantiomer was preferred and R/S ratio when the R enantiomer was preferred) was determined as follows: [EC50 (or Ki) value of the preferred (R or S) enantiomer/EC50 (or Ki) value of the non-preferred (R or S) enantiomer] and was used as an index of the stereoselectivity (Arrang et al., 1985a).
Materials
[125I]IPX (2000 Ci·mmol−1) was prepared as described (Krause et al., 1997). [3H]AA (211 Ci·mmol−1) and [35S]GTPγ[S] (1250 Ci·mmol−1) were from PerkinElmer Life Sciences. Histamine, imetit, thioperamide, isobutylmethyl xanthine, forskolin and BSA were purchased from Sigma-Aldrich (Saint Quentin Fallavier, France). Ionophore A23187 and adenosine deaminase were obtained from Roche. The enantiomers of α-MeHA, NαMe-αClMeHA and sopromidine were provided by W. Schunack (Freie Universität Berlin, Germany).
Results
Effect of histamine and R-α-methylhistamine on forskolin-induced cAMP accumulation and A23187-evoked [3H]AA release in CHO(rH3(445)R) and CHO(rH3(413)R) cells
The cloning of the rat receptor isoforms rH3(445) or rH3(413) in CHO cells yielded numerous clones stably expressing various densities of these isoforms. As expected from the negative coupling of the H3 receptor to adenylate cyclase (Lovenberg et al., 1999; Morisset et al., 2000), histamine used at a maximal concentration (1 µM) induced an inhibition of forskolin-induced cAMP accumulation, which was already significant at a density as low as ∼20 fmol·mg−1 protein of rH3(445) receptor (Figure 1). The magnitude of this inhibition dramatically increased with the density of each isoform, to become almost total at a density of 1000 fmol·mg−1 protein of rH3(413) receptor (cAMP rate of 299 ± 59 fmol vs. 2788 ± 198 fmol with forskolin alone, Figure 1). The influence of the receptor density on the histamine effect was similar for both isoforms. At a density of 300 fmol·mg−1 protein, histamine inhibited forskolin-induced cAMP accumulation by 45 ± 5% and 45 ± 10% with the receptor isoforms rH3(445) and rH3(413) respectively. At a density of 400–500 fmol·mg−1 protein, the histamine-induced inhibition was 70 ± 3% and 74 ± 3% respectively (Figure 1).
Figure 1.
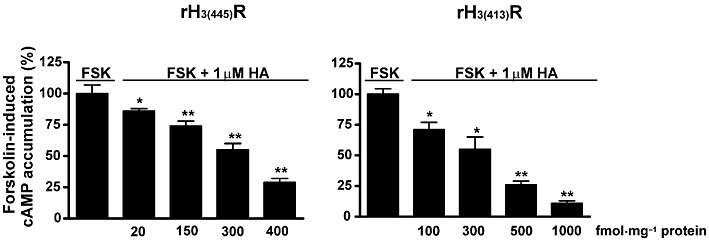
Effect of histamine (HA) on forskolin (FSK)-induced cAMP accumulation in CHO cells expressing various densities of the rH3(445) and rH3(413) receptor isoforms. cAMP accumulation was induced by forskolin (3 µM) alone or in the presence of histamine (1 µM) in CHO(rH3(445)R) and CHO(rH3(413)R) cells expressing increasing isoform densities (up to 1000 fmol·mg−1 protein, as determined using [125I]IPX binding assay). The results are means ± SEM of values from two to six different experiments with five determinations each. *P < 0.01, **P < 0.001, significantly different from forskolin alone.
These cell lines expressing 400–500 fmol·mg−1 protein of each isoform were selected in subsequent experiments. The agonist effect of histamine displayed a similar profile at rH3(445) and rH3(413) isoforms, with a maximal inhibition of cAMP formation of 75–80% and EC50 values of 9 ± 2 and 15 ± 4 nM respectively (Figure 2A and Table 1). The H3 receptor agonist R-αMeHA behaved as a full agonist with EC50 values of 3.6 ± 3.4 nM at rH3(445) and 1.8 ± 0.2 nM at rH3(413) receptors (Figure 2A), leading to potencies of R-αMeHA relative to histamine at the two isoforms of 266% and 833% respectively. Two-way anova indicated that the difference between the curves of R-α-MeHA and histamine was significant for rH3(413) receptors [F(1,36) = 19.74, P < 0.0001], but not for rH3(445) receptors [F(1,14) = 2.54, P= 0.13]. Post hoc analysis confirmed that R-α-MeHA was significantly more potent than histamine at rH3(413) receptors (P < 0.01 at 3 nM and P < 0.05 at 10 nM, Figure 2A).
Figure 2.
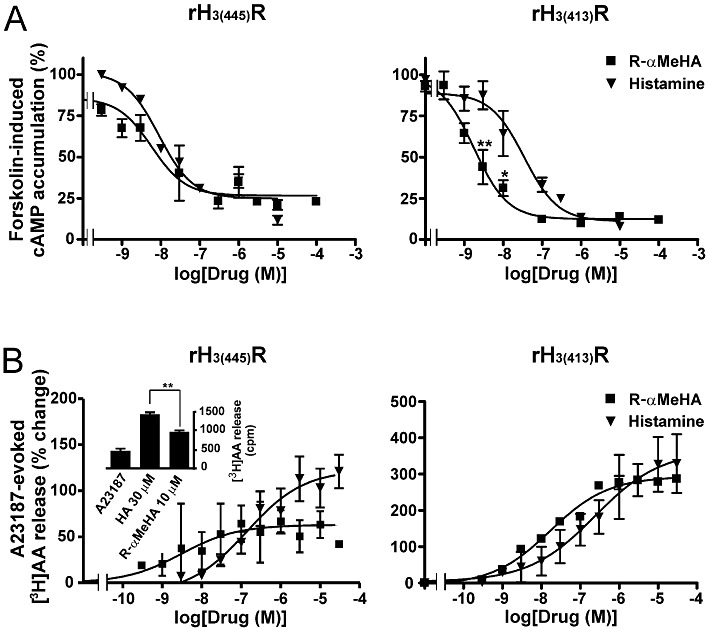
Effects of R-αMeHA and histamine on forskolin-induced cAMP accumulation (A) and A23187-evoked [3H]AA release (B) in CHO(rH3(445)R) and CHO(rH3(413)R) cells expressing 350–500 fmol·mg−1 protein of receptor. Results are means ± SEM of values from two to eight separate experiments with three to five determinations each. In (B), the inset represents the effects of both compounds at a maximal concentration within one of the experiments. *P < 0.05, **P < 0.01, significantly different from histamine.
Table 1.
Compared agonist potencies (EC50, µM) of R(−) and S(+) NαMe-αClMeHA at the recombinant rH3(445) and rH3(413) receptor isoforms and at the rat H3 autoreceptor
| Compounds | rH3(445) receptor | rH3(413) receptor | H3 autoreceptora |
|---|---|---|---|
| Histamine | 0.009 ± 0.002 (100) | 0.015 ± 0.004 (100) | 0.062 ± 0.014 (100) |
| R(−) NαMe-αClMeHA | 0.27 ± 0.04 (3.3) | 81 ± 24 (0.02) | 1100 ± 0.5 (0.006) |
| S(+) NαMe-αClMeHA | 0.084 ± 0.014 (11) | 1.5 ± 0.8 (1) | 5.5 ± 1.9 (1.1) |
| Ratio S/R | 3.2 | 54 | 200 |
Values from Arrang et al. (1985a). The values at the two isoforms are derived from the data shown in Figure 3. The potencies relative to histamine (=100) are indicated between parentheses and were calculated as the ratio: EC50 value of histamine/EC50 value of agonist × 100.
On [3H]AA release, a signalling pathway positively coupled to recombinant H3 receptors (Morisset et al., 2000; Gbahou et al., 2003), histamine increased A23187-evoked [3H]AA release with EC50 values of 177 ± 68 and 213 ± 50 nM at rH3(445) and rH3(413) receptor isoforms respectively (Figure 2B). R-αMeHA increased [3H]AA release with EC50 values of 1.6 ± 0.9 nM at rH3(445) and 19 ± 5 nM at rH3(413) receptors (Figure 2B), leading to potencies of R-αMeHA relative to histamine at the two isoforms of 11063% and 1121% respectively. Two-way anova failed to show any significant difference between the curves of R-α-MeHA and histamine not only at rH3(413) receptors but also at rH3(445) receptors [F(1,41) = 0.94, P= 0.33]. This finding probably resulted from the variability of the response between experiments coupled to the low magnitude of the increase induced by R-α-MeHA at rH3(445) receptors. However, the curve fitting showed that R-αMeHA behaved as a full agonist at rH3(413) receptors, but as a partial agonist at rH3(445) receptors with an intrinsic activity of 50% that of histamine (Figure 2B). In agreement, when the maximal effect of R-α-MeHA was compared with that of histamine within each of the eight experiments performed at rH3(445) receptors with three to five determinations, it was found to be significantly lower (P < 0.01) in all the experiments (inset of Figure 2B), with an intrinsic activity of R-αMeHA ranging from 33% to 61% that of histamine, confirming the partial agonism by R-αMeHA at the long isoform (Figure 2B). Due to the limited availability of the drug, the S enantiomer of αMeHA could be tested only at the shorter isoform on [3H]AA release and increased this release with an EC50 of 900 ± 450 nM (data not shown). At this rH3(413) isoform, the comparison with histamine yielded therefore a relative potency for S-αMeHA of 24% and the comparison with R-αMeHA yielded a marked stereoselectivity with an activity ratio R/S = 47.4.
Effects of (R) and (S) NαMe-αClMeHA on specific [125I]IPX binding and forskolin-induced cAMP accumulation in CHO(rH3(445)R) and CHO(rH3(413)R) cells
In addition to αMeHA, the effect of NαMe-αClMeHA, another chiral analogue of histamine previously studied at autoreceptors (Arrang et al., 1985a), was investigated at rH3(445) and rH3(413) receptor isoforms.
Its two enantiomers were first studied on the [125I]IPX specific binding assay using membranes of CHO cells expressing a similar density of rH3(445) and rH3(413) receptors. The R and S enantiomers of NαMe-αClMeHA inhibited [125I]IPX binding with deduced Ki values at rH3(445) receptors of 480 ± 60 and 220 ± 40 nM respectively (S/R ratio = 2.2). The corresponding Ki values of the R and S enantiomers at rH3(413) receptors were 1.5 ± 0.3 and 1.1 ± 0.2 µM (S/R ratio = 1.4) (Figure 3A).
Figure 3.
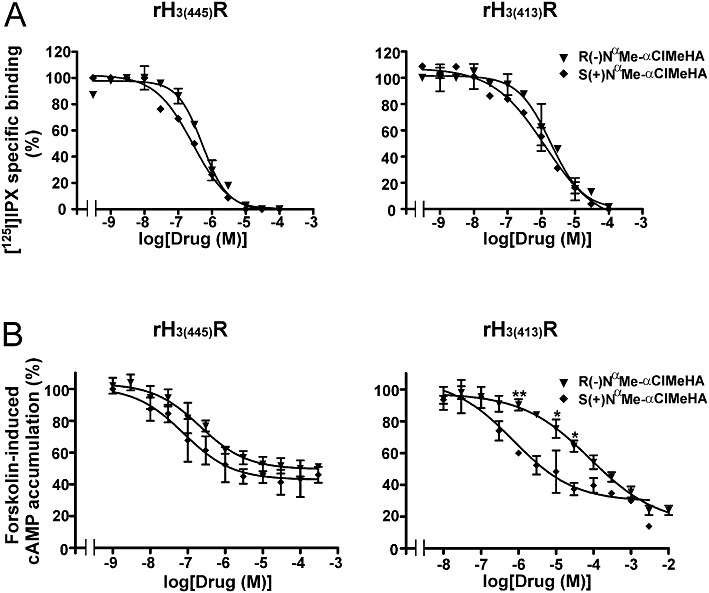
Effects of the R(−) and S(+) enantiomers of NαMe-αClMeHA on specific [125I]IPX binding (A) and forskolin-induced cAMP accumulation (B) in CHO(rH3(445)R) and CHO(rH3(413)R) cells expressing 500 fmol·mg−1 protein of receptor. Each point represents the mean ± SEM of values from two to three different experiments with three to five determinations each. *P < 0.05, **P < 0.01, significantly different from S(+) NαMe-αClMeHA.
On cAMP accumulation induced by forskolin, the comparison of their plateau with that of histamine showed that R(−) and S(+) NαMe-αClMeHA behaved as partial agonists at rH3(445) receptors with an intrinsic activity of ∼60% that of histamine, but as full agonists at rH3(413) receptors (Figure 3B). At the rH3(445) receptor isoform, their respective EC50 values were 270 ± 40 and 84 ± 14 nM, leading to respective potencies relative to histamine of 3.3% and 11% and to an S/R ratio of 3.2 (Figure 3B and Table 1). At the rH3(413) receptor isoform, the respective EC50 values of R(−) and S(+) NαMe-αClMeHA were 81 ± 24 and 1.5 ± 0.8 µM, leading to respective potencies relative to histamine of 0.02% and 1% and to an S/R ratio of 54 (Figure 3B and Table 1). The statistical analysis using two-way anova showed a significant difference between the curves of the two enantiomers both at rH3(445) receptors [F(1,37) = 7.68, P < 0.01] and at rH3(413) receptors [F(1,37) = 31.54, P < 0.0001]. However, post hoc analysis led to significant differences at several concentrations only at rH3(413) receptors (P < 0.01 at 1 µM; P < 0.05 at 10 and 30 µM, Figure 3B). These data show that if the S(+) enantiomer was preferred to the R(−) enantiomer at both isoforms, the stereoselectivity was more pronounced at the rH3(413) receptors isoform (S/R ratio of 54 vs. 3.2) (Table 1). Moreover, the comparison of the potencies relative to histamine obtained at the two recombinant isoforms with those previously reported at autoreceptors (Arrang et al., 1985a) shows that the relative potency of S(+) NαMe-αClMeHA at rH3(445) receptors (11%) was only 10-fold lower at rH3(413) receptors and at autoreceptors (1% and 1.1%, respectively), whereas the relative potency of R(−) NαMe-αClMeHA at rH3(445) receptors (3.3%) was 200- to 500-fold lower at rH3(413) receptors and autoreceptors (0.02% and 0.006%, respectively), yielding a similar stereoselectivity on these two systems (S/R ratio = 54 vs. 200) (Table 1).
Effects of the R(−) and S(+) enantiomers of sopromidine on forskolin-induced cAMP accumulation in CHO(rH3(445)R) and CHO(rH3(413)R) cells
R(−) and S(+) sopromidine were first investigated alone on forskolin-induced cAMP accumulation (Figure 4A). At the rH3(445) receptor isoform, R(−) sopromidine decreased the response in a concentration-dependent manner (EC50= 79 ± 34 nM), with a maximal effect similar to that of histamine and was therefore behaving as a full agonist at this isoform. In contrast, S(+) sopromidine alone mimicked the effect of thioperamide and increased the response with an EC50 of 180 ± 90 nM, thereby acting as an inverse agonist (Figure 4A and Table 2). At the rH3(413) receptor isoform, R(−) sopromidine behaved again as an agonist (EC50= 91 ± 78 nM), but the comparison of its maximal effect with that of histamine indicated that it acted as a very partial agonist with an intrinsic activity of ∼20% (Figure 4A). S(+) sopromidine alone behaved also as an inverse agonist at this isoform, with an EC50 of 340 ± 92 nM and a maximal effect similar to that of thioperamide.
Figure 4.
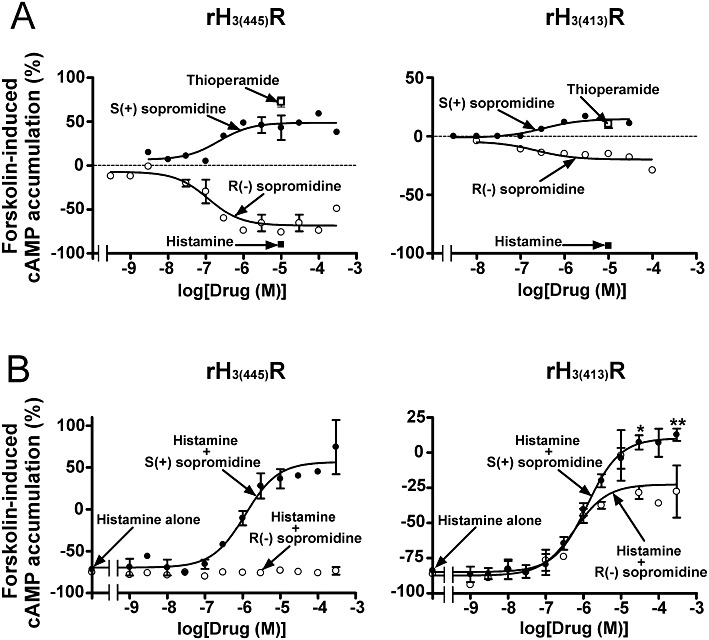
Effects of the R(−) and S(+) enantiomers of sopromidine on forskolin-induced cAMP accumulation in CHO(rH3(445)R) and CHO(rH3(413)R) cells. Cells were incubated with 3 µM forskolin and, when required, increasing concentrations of R(−) or S(+) sopromidine added alone (A) or in the presence of 100 nM histamine (B). The data are the means ± SEM of values from two to four separate experiments with five determinations each. *P < 0.05, **P < 0.01, significantly different from histamine + R(−) sopromidine.
Table 2.
Compared properties of R(−) and S(+) sopromidine at the recombinant rH3(445) and rH3(413) receptor isoforms and at the rat H3 autoreceptor
| R(−) sopromidine | S(+) sopromidine | |||||
|---|---|---|---|---|---|---|
| Condition | rH3(445) receptor | rH3(413) receptor | H3 autoreceptora | rH3(445) receptor | rH3(413) receptor | H3 autoreceptora |
| Alone | Full agonist (EC50= 79 ± 34 nM) | Partial agonist (EC50= 91 ± 78 nM) | nd | Inverse agonist (EC50= 180 ± 90 nM) | Inverse agonist (EC50= 340 ± 92 nM) | nd |
| With histamine | No effect (Ki > 100 µM) | Antagonist (Ki= 63 ± 40 nM) | Antagonist (Ki= 56 ± 22 nM) | Antagonist (Ki= 250 ± 120 nM) | Antagonist (Ki= 220 ± 100 nM) | Antagonist (Ki= 45 ± 11 nM) |
Values from Arrang et al. (1985a). The values at the two isoforms are derived from data shown in Figure 4.
nd, not determined.
The antagonist potency of the two enantiomers was then evaluated against a sub-maximal concentration of histamine (100 nM) (Figure 4B). At the rH3(445) receptor isoform, R(−) sopromidine tested up to 300 µM had no apparent effect against histamine. In contrast, the S enantiomer completely reversed the inhibition of forskolin-induced cAMP formation induced by histamine and an enhancement of the response was even observed at the highest concentration due to its own inverse agonist effect. Analysis of the total curve yielded a Ki value for S(+) sopromidine of 250 ± 120 nM (Figure 4B and Table 2). At the rH3(413) receptor isoform, R(−) sopromidine partially reversed the effect of histamine with a Ki of 63 ± 40 nM and a plateau similar to that obtained with the compound added alone (Figure 4). The S enantiomer completely reversed the effect of histamine with a Ki value of 220 ± 100 nM. Again, its own inverse agonist effect enhanced the response at the highest concentration (Figure 4B and Table 2). Two-way anova confirmed the statistical significance of the difference between the two curves [F(1,34) = 12.39, P < 0.001] and post hoc analysis confirmed the significant difference of the plateau (P < 0.01 at 300 µM; P < 0.05 at 30 µM, Figure 4B). The comparison of the properties of R(−) and S(+) sopromidine at the two recombinant isoforms with those previously reported at autoreceptors (Arrang et al., 1985a) shows that R(−) sopromidine behaves as a full agonist at the rH3(445) receptor isoform, but as an antagonist with a similar potency at the rH3(413) receptor isoform and at autoreceptors (Ki of 63 ± 40 and 56 ± 22 nM, respectively, Table 2). In contrast, S(+) sopromidine behaved as an antagonist/inverse agonist in the three systems (Table 2).
Effects of the R(−) and S(+) enantiomers of NαMe-αClMeHA and sopromidine on [35S]GTPγ[S] binding to rat brain membranes
The effects of the enantiomers of NαMe-αClMeHA and sopromidine were studied at native H3 receptors mediating [35S]GTPγ[S] binding to rat brain membranes (Rouleau et al., 2002). In [35S]GTPγ[S] binding assays, the H3 selectivity of their effects was ensured by blockade of H1 and H2 receptors with corresponding antagonists, that is, mepyramine and cimetidine, used at maximal concentrations. R(−) and S(+) NαMe-αClMeHA increased specific [35S]GTPγ[S] binding in the cerebral cortex, striatum and hypothalamus in a concentration-dependent and saturable manner. The EC50 value of the S enantiomer was similar in the three regions (3.8 ± 0.4, 9.2 ± 3.1 and 6.1 ± 5.8 µM in the cerebral cortex, striatum and hypothalamus respectively). The EC50 value of the R enantiomer increased from 9.7 ± 1.9 µM in the striatum to 14 ± 7 µM in the cerebral cortex and to 77 ± 34 µM in the hypothalamus, leading S/R ratios to increase from 1.0 in the striatum to 3.7 in the cerebral cortex and 13 in the hypothalamus (Figure 5A). The plateau reached by the two enantiomers was not significantly different from the plateau reached by imetit in the cerebral cortex and striatum (116 ± 1.6% and 118 ± 3.5%, respectively), indicating that the compounds were acting as full agonists in these two regions. However, in the hypothalamus, their maximal effect was half of that of imetit (3 ± 1% and 4 ± 1% vs. 8 ± 2%, respectively), showing that they behaved as partial agonists in this region. In this area, two-way anova indicated a difference between the two curves close to statistical significance [F(1,52) = 3.47, P= 0.06].
Figure 5.
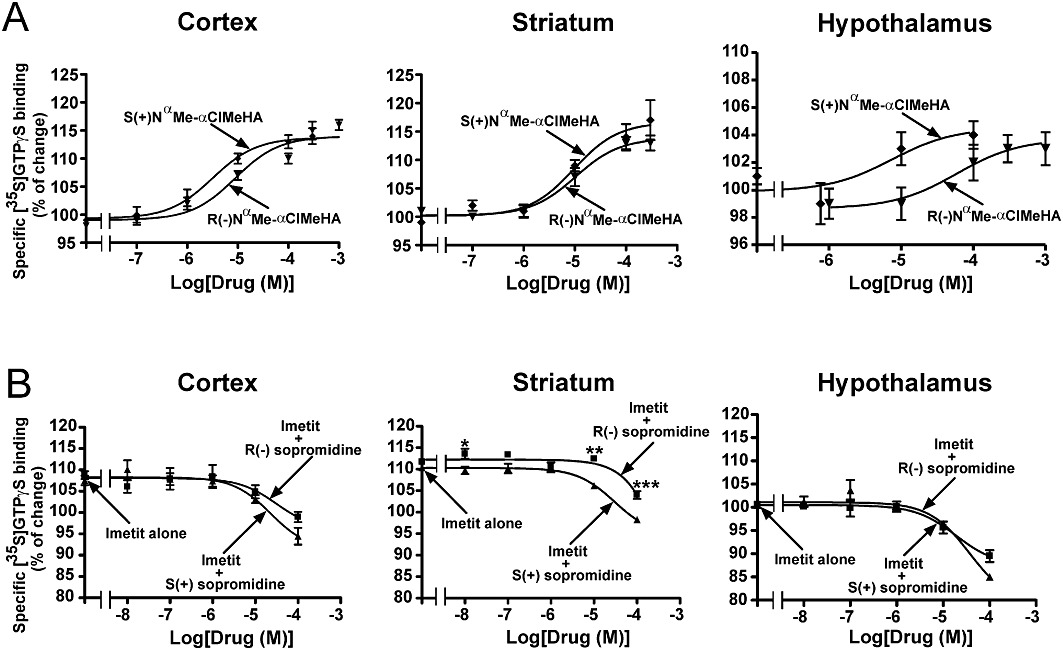
Effects of the R(−) and S(+) enantiomers of NαMe-αClMeHA and sopromidine on specific [35S]GTPγ[S] binding to membranes from various rat brain regions. Membranes were incubated with 0.1 nM [35S]GTPγ[S] in the presence of increasing concentrations of NαMe-αClMeHA enantiomers alone (A), or sopromidine enantiomers in the presence of 1 µM imetit (B). In order to prevent any interaction of the drugs with H1 and H2 receptors, all incubations were performed in the presence of 100 nM mepyramine and 10 µM cimetidine. Data are means ± SEM of 4–16 determinations from two to four separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from imetit + S(+) sopromidine.
In the three regions, the two isomers of sopromidine antagonized the increase in specific [35S]GTPγ[S] binding induced by 1 µM imetit (Figure 5B). In the presence of 100 µM (highest concentration tested) of each of the two enantiomers, [35S]GTPγ[S] binding had returned to control values (100%) in the cerebral cortex and striatum, and was even further decreased (by up to 15%) in the hypothalamus, thereby revealing the inverse agonist properties of the compounds (Figure 5B). Taking into account only the antagonistic part of the curves (i.e. above 100%) and an EC50 value of imetit of 2 ± 1 nM (not shown), the apparent Ki values found for the S enantiomer were roughly similar in the three regions (47 ± 14, 14 ± 0.6 and 23 ± 9 nM in the hypothalamus, cerebral cortex and striatum, respectively), whereas the apparent Ki values found for the R enantiomer increased from 21 ± 14 nM in the hypothalamus to 37 ± 35 nM in the cerebral cortex and 163 ± 92 nM in the striatum. The resulting S/R ratios increased from 2 in the hypothalamus, to 3 in the cerebral cortex and 8 in the striatum (Figure 5B). In agreement, two-way anova revealed that the difference observed between the effects of the R and S enantiomers of sopromidine reached significance only in the striatum [F(1,79) = 35.48, P < 0.0001], post hoc analysis indicating a significant difference between the two enantiomers at 10 µM (P < 0.01) and 100 µM (P < 0.001) (Figure 5B).
Discussion
All the findings of this study support the assumption that the function of the autoreceptors modulating histaminergic neurons was not fulfilled by the long isoform of the H3 receptor, but by a short form, such as the H3(413)R isoform. These findings included: (i) the partial agonist effect of R-αMeHA on [3H]AA release mediated by the long isoform has never been found either at native H3 autoreceptors or at short isoforms; (ii) the stereoselectivity ratio of enantiomers of NαMe-αClMeHA at autoreceptors was only fourfold lower at the functional H3(413) receptor isoform, but was much lower at the functional long isoform; (iii) this stereoselectivity of NαMe-αClMeHA on [35S]GTPγ[S] binding was higher in the hypothalamus, the region of origin of histaminergic neurons, than in the cerebral cortex or striatum; (iv) (R) sopromidine behaved as an antagonist with a similar potency at functional H3(413) receptor isoforms and autoreceptors, but as a full agonist at the long isoform; (v) sopromidine isomers exhibited no stereoselectivity at autoreceptors and on [35S]GTPγ[S] binding in the hypothalamus but showed some stereoselectivity in the cerebral cortex or striatum.
The observation that autoreceptors are the short receptor isoforms is consistent with their expression level in the brain. Functional (Schlicker et al., 1994; Haas et al., 2008), localization (Goodchild et al., 1999; Anichtchik et al., 2001; Pillot et al., 2002) and lesion (Cumming et al., 1991; Pollard et al., 1993; Anichtchik et al., 2000) studies showed that the majority of H3 receptors in the brain are not autoreceptors on histaminergic neurons, but are post-synaptic receptors or heteroreceptors present on other neuronal populations. In addition, in most brain areas from different species including human, the expression of the long isoform is largely predominant compared with shorter functional isoforms, with deletions in the third intracellular loop. such as the H3(413) receptor isoform (Tardivel-Lacombe et al., 2000; Coge et al., 2001; Drutel et al., 2001; Morisset et al., 2001; Rouleau et al., 2004). Although the long isoform can be excluded, our study does not identify which short isoform(s) fulfill(s) the autoreceptor function. However, whereas all the other splice variants differ in composition between species (Hancock et al., 2003), the expression of the H3(413) receptor isoform is the only one to be maintained in the brain from all species including rat (Morisset et al., 2001), mouse (Rouleau et al., 2004), guinea pig (Tardivel-Lacombe et al., 2000), monkey (Strakhova et al., 2008) and human (Coge et al., 2001; Tardivel-Lacombe et al., 2001). This may therefore indicate that this short isoform does play the role of autoreceptor including in human brains. The involvement of shorter variants such as the H3(397) receptor in rodent (Drutel et al., 2001; Morisset et al., 2001; Rouleau et al., 2004), or the H3(365) receptor in human (Coge et al., 2001; Wellendorph et al., 2002; Bongers et al., 2007), alone or with the H3(413) receptor isoform, cannot, however, be entirely ruled out. Unfortunately, the stereoselectivity of the human H3(365) receptor could not be evaluated in this study because no response was produced by this isoform in CHO cells (data not shown), as also reported in one of the earlier studies (Coge et al., 2001).
The mechanisms leading a short, rather than the long, isoform to play the role of autoreceptor remain unknown. It is worth noting that our observations on the H3 receptor resemble those on the D2 dopamine receptor. The short isoform of the D2 receptor also differs from the D2 long isoform by a 29-amino acid deletion in the third cytoplasmic loop and also functions as an autoreceptor (Usiello et al., 2000; Centonze et al., 2002; Lindgren et al., 2003). This functional selectivity was suggested on the basis of a predominant location of the short isoform in dopaminergic neurons (Khan et al., 1998; Jomphe et al., 2006) and/or from a differential coupling leading the short isoform to be preferred for the autoreceptor function (Senogles, 1994; Guiramand et al., 1995; Liu et al., 1996; Wolfe and Morris, 1999; Van et al., 2007). These two suggestions may apply to the functional selectivity of H3 receptors. However, whether histaminergic neurons selectively express the short isoforms of H3 receptors is not yet known, inasmuch as transcripts of both long and short isoforms have been observed in the hypothalamus, their region of origin (Morisset et al., 2001). Alternatively, the autoreceptor function of short isoforms may result from their selective signalling properties. More interestingly, both H3 (Takeshita et al., 1998; Stevens et al., 2001; Moreno-Delgado et al., 2009) and D2 (Wolfe and Morris, 1999) receptors inhibit high voltage-activated Ca2+ channels, which is likely to underlie their autoreceptor function on histamine and dopamine release respectively. However, the same coupling, that is, inhibition of the cAMP pathway, can be used by native H3 autoreceptors to inhibit histamine synthesis in the brain (Moreno-Delgado et al., 2009) and by both long and short isoforms. Therefore, whether H3 and D2 receptor short isoforms selectively couple to a common effector to inhibit histaminergic and dopaminergic neuron activity, respectively, remains to be shown.
This study confirms our studies on autoreceptors indicating that H3 receptors prefer (+) enantiomers corresponding to the S-configuration of L-histidine, such as R-αMeHA or S-NαMe-αClMeHA (Arrang et al., 1985a). However, we show for the first time that H3 receptor isoforms differ markedly in their pharmacological profiles, indicating that they correspond to distinct H3 receptor conformations. These different conformations resulting from differences in the third intracellular loop are not expected to generate significant differences in the affinity of the ligands, known to interact with the transmembrane domains. In agreement, differences between isoforms in binding studies were always limited (Coge et al., 2001; Morisset et al., 2001; Rouleau et al., 2004). It is worth noting, however, that the affinity of betahistine was ∼6-fold higher at the rH3(413) than at the rH3(445) receptor isoform, but very similar to its antagonist potency at rat autoreceptors (Arrang et al., 1985b), which already led us to suggest that the function of autoreceptor was fulfilled by a short, rather than long, H3 receptor isoform (Gbahou et al., 2010). Moreover, it is well-established that H3 receptors exist in multiple conformations displaying different pharmacological profiles (Gbahou et al., 2003) and that H3 receptor binding sites do not represent, at least solely, functional receptors. For example, inverse agonist radioligands label a much larger population of H3 receptors than agonist radioligands (Witte et al., 2006; Yao et al., 2006; Mezzomo et al., 2007), and agonist radioligands bind to both uncoupled and coupled states of the H3 receptor (Arrang et al., 1990). It explains that, whereas (R)- and (S)-αMeHA conserve their stereoselectivity in binding and functional studies (Arrang et al., 1987; 1990), the stereoselectivity of the R and S enantiomers of NαMe-αClMeHA, observed here in functional responses, was not observed in binding assays. All these considerations confirm that binding assays are not appropriate for the screening of compounds for a particular functional or therapeutic use.
Rather than having differences in affinity, H3 receptor isoforms differ markedly at the level of their coupling to G proteins. Indeed, all of our data on the three studied responses can be explained by differences in agonist potency and/or intrinsic activity between the two isoforms. Firstly, R-αMeHA acted surprisingly as an intrinsic partial agonist at the long isoform. This effect was observed only for [3H]AA release, a response with a low coupling efficiency of the H3 receptor (Morisset et al., 2000). For cAMP accumulation, where the H3 receptor displays a higher coupling efficiency, a full agonist is expected to reach its maximal activity with only a partial occupancy of these receptors. A partial but potent agonist such as R-αMeHA is then expected to behave as an apparent full agonist by occupying more, if not all, functional receptors. In agreement, the relative potency of R-αMeHA was higher on [3H]AA release than on cAMP accumulation.
Secondly, differences in coupling of isoforms also accounts for their strong differences in stereoselectivity observed with NαMe-αClMeHA enantiomers, both on cAMP formation mediated by the recombinant isoforms and on [35S]GTPγ[S] binding to brain membranes. These differences were, in fact, generated by a lower potency of the non-preferred isomer, that is, the R(−), at the short isoform, whereas the potency of the preferred isomer, that is, the S(+), remained unchanged.
Thirdly, at both isoforms, the two enantiomers of sopromidine displayed opposite intrinsic properties, with the R(−) isomer behaving as agonist (full or partial) and the S(+) isomer acting as an inverse agonist. Their potency as agonist, antagonist or inverse agonist remained roughly similar, indicating that they stabilized conformations with similar binding properties, but different coupling properties. At autoreceptors inhibiting histamine release, we previously failed to detect both the partial agonist effect of R(−) sopromidine and the inverse agonist effect of S(+) sopromidine, and concluded that they were both behaving as full antagonists (Arrang et al., 1985a), presumably because our system involved a lower density of receptors with no apparent constitutive activity (Morisset et al., 2000). Therefore, in contrast to what we concluded from histamine release experiments, the H3 receptor also displays a strong stereoselectivity for sopromidine enantiomers. Moreover, as found with NαMe-αClMeHA enantiomers, this stereoselectivity differed between the two isoforms, with the inverse agonist effect of S(+) sopromidine remaining unchanged, in contrast to the agonist effect of the R(−) isomer, which was full at the long isoform but very partial at the short isoform. [35S]GTPγ[S] binding showed that this difference in stereoselectivity of isoforms also occurred in the brain. The absence of stereoselectivity of sopromidine tested as antagonist against imetit in the hypothalamus is consistent with the high density of autoreceptors in this region, both isomers being expected to act as apparent antagonists with similar potencies. In the cerebral cortex and striatum, the presumably higher density of long isoforms is likely to account for the stereoselectivity observed in these two regions, the agonist property of the R(−) isomer at these long isoforms counteracting its apparent antagonist properties at short isoforms. In agreement, this stereoselectivity was mainly generated by a decrease in the potency of the R(−) isomer in the cerebral cortex and striatum compared with the hypothalamus, whereas the potency of the S(+) isomer remained roughly the same in all three regions.
It is worth noting that these differences in coupling of H3 receptor isoforms may be ligand-dependent because, in contrast to the compounds used in the present study, the two enantiomers of αMeHA revealed a similar stereoselectivity of rodent and human H3 receptor isoforms (Arrang et al., 1987; 1990; Wulff et al., 2002; Hancock et al., 2003).
In conclusion, the present findings show that short, but not long, isoforms fulfil the function of autoreceptor and thereby confirm the hypothesis that H3 receptor isoforms have distinct functional roles in the brain. The roles played by the long isoform remain to be explored, but its transcripts, being predominant in most brain regions, may encode for the numerous receptors present on neurons other than histaminergic neurons, either at the post-synaptic level (somatodendritic receptors) or pre-synaptic level (heteroreceptors) (Pillot et al., 2002). The pharmacological differences that we report here between isoforms indicate that it should be possible to identify ligands selective for each of them. Such ligands should then be helpful in discriminating between the selective functions of isoforms at the pre- and post-synaptic levels. Numerous pharmaceutical companies have invested considerable efforts in the clinical development of inverse agonists at the H3 receptor, as possible treatments for wakefulness and cognition disorders (narcolepsy, Alzheimer's disease and attention deficit hyperactivity disorder) (Hancock and Fox, 2004; Passani et al., 2004; Celanire et al., 2005; Leurs et al., 2005; Esbenshade et al., 2006; Arrang et al., 2007; Parmentier et al., 2007; Lin et al., 2008), but these drugs are not selective for any isoform. The clinical development of compounds that selectively target autoreceptors may enhance their therapeutic efficacy, in the above-mentioned disorders, as well as in other more controversial indications including food intake disorders (Hancock, 2003), seizures (Kamei, 2001) or schizophrenia (Southam et al., 2009; Burban et al., 2010; Motawaj and Arrang, 2011). Moreover, because no compound has yet been successfully developed into clinical use, the possible side effects of H3 receptor inverse agonists remain unknown but may be decreased with compounds selective for short isoforms.
Acknowledgments
This study was supported by INSERM. The enantiomers of NαMe-αClMeHA and sopromidine were provided by W. Schunack (Freie Universität Berlin, Germany).
Glossary
- αMeHA
α-methylhistamine
- AA
arachidonic acid
- IPX
iodoproxyfan
- NαMe-αClMeHA
Nα-methyl-α-chloromethylhistamine
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5th edn.) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichtchik OV, Huotari M, Peitsaro N, Haycock JW, Mannisto PT, Panula P. Modulation of histamine H3 receptors in the brain of 6-hydroxydopamine-lesioned rats. Eur J Neurosci. 2000;12:3823–3832. doi: 10.1046/j.1460-9568.2000.00267.x. [DOI] [PubMed] [Google Scholar]
- Anichtchik OV, Peitsaro N, Rinne JO, Kalimo H, Panula P. Distribution and modulation of histamine H3 receptors in basal ganglia and frontal cortex of healthy controls and patients with Parkinson's disease. Neurobiol Dis. 2001;8:707–716. doi: 10.1006/nbdi.2001.0413. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Schwartz JC, Schunack W. Stereoselectivity of the histamine H3-presynaptic autoreceptor. Eur J Pharmacol. 1985a;117:109–114. doi: 10.1016/0014-2999(85)90478-9. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Quach TT, Dam Trung Tuong M, Yeramian E, Schwartz JC. Actions of betahistine at histamine receptors in the brain. Eur J Pharmacol. 1985b;111:73–84. doi: 10.1016/0014-2999(85)90115-3. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Lancelot JC, Lecomte JM, Pollard H, Robba M, et al. Highly potent and selective ligands for histamine H3-receptors. Nature. 1987;327:117–123. doi: 10.1038/327117a0. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Roy J, Morgat JL, Schunack W, Schwartz JC. Histamine H3 receptor binding sites in rat brain membranes: modulations by guanine nucleotides and divalent cations. Eur J Pharmacol. 1990;188:219–227. doi: 10.1016/0922-4106(90)90005-i. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Morisset S, Gbahou F. Constitutive activity of the histamine H3 receptor. Trends Pharmacol Sci. 2007;28:350–357. doi: 10.1016/j.tips.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Bongers G, Krueger KM, Miller TR, Baranowski JL, Estvander BR, Witte DG, et al. An 80-amino acid deletion in the third intracellular loop of a naturally occurring human histamine H3 isoform confers pharmacological differences and constitutive activity. J Pharmacol Exp Ther. 2007;323:888–898. doi: 10.1124/jpet.107.127639. [DOI] [PubMed] [Google Scholar]
- Burban A, Sadakhom C, Dumoulin D, Rose C, Le Pen G, Frances H, et al. Modulation of prepulse inhibition and stereotypies in rodents: no evidence for antipsychotic-like properties of histamine H3-receptor inverse agonists. Psychopharmacology (Berl) 2010;210:591–604. doi: 10.1007/s00213-010-1863-2. [DOI] [PubMed] [Google Scholar]
- Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJ. Histamine H3 receptor antagonists reach out for the clinic. Drug Discov Today. 2005;10:1613–1627. doi: 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Gubellini P, Pisani A, Borrelli E, Bernardi G, et al. Dopamine D2 receptor-mediated inhibition of dopaminergic neurons in mice lacking D2 L receptors. Neuropsychopharmacology. 2002;27:723–726. doi: 10.1016/S0893-133X(02)00367-6. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prussoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Coge F, Guenin SP, Audinot V, Renouard-Try A, Beauverger P, Macia C, et al. Genomic organization and characterization of splice variants of the human histamine H3 receptor. Biochem J. 2001;355:279–288. doi: 10.1042/0264-6021:3550279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming P, Shaw C, Vincent SR. High affinity histamine binding site is the H3 receptor: characterization and autoradiographic localization in rat brain. Synapse. 1991;8:144–151. doi: 10.1002/syn.890080208. [DOI] [PubMed] [Google Scholar]
- Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, et al. Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol. 2001;59:1–8. [PubMed] [Google Scholar]
- Esbenshade TA, Fox GB, Cowart MD. Histamine H3 receptor antagonists: preclinical promise for treating obesity and cognitive disorders. Mol Interv. 2006;6:77–88. doi: 10.1124/mi.6.2.5. 59. [DOI] [PubMed] [Google Scholar]
- Gbahou F, Rouleau A, Morisset S, Parmentier R, Crochet S, Lin JS, et al. Protean agonism at histamine H3 receptors in vitro and in vivo. Proc Natl Acad Sci U S A. 2003;100:11086–11091. doi: 10.1073/pnas.1932276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbahou F, Vincent L, Humbert-Claude M, Tardivel-Lacombe J, Chabret C, Arrang JM. Compared pharmacology of human histamine H3 and H4 receptors: structure-activity relationships of histamine derivatives. Br J Pharmacol. 2006;147:744–754. doi: 10.1038/sj.bjp.0706666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbahou F, Davenas E, Morisset S, Arrang JM. Effects of betahistine at histamine H3 receptors: mixed inverse agonism/agonism in vitro and partial inverse agonism in vivo. J Pharmacol Exp Ther. 2010;334:945–954. doi: 10.1124/jpet.110.168633. [DOI] [PubMed] [Google Scholar]
- Goodchild RE, Court JA, Hobson I, Piggott MA, Perry RH, Ince P, et al. Distribution of histamine H3-receptor binding in the normal human basal ganglia: comparison with Huntington's and Parkinson's disease cases. Eur J Neurosci. 1999;11:449–456. doi: 10.1046/j.1460-9568.1999.00453.x. [DOI] [PubMed] [Google Scholar]
- Guiramand J, Montmayeur JP, Ceraline J, Bhatia M, Borrelli E. Alternative splicing of the dopamine D2 receptor directs specificity of coupling to G-proteins. J Biol Chem. 1995;270:7354–7358. doi: 10.1074/jbc.270.13.7354. [DOI] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- Hancock AA. H3 receptor antagonists/inverse agonists as anti-obesity agents. Curr Opin Investig Drugs. 2003;4:1190–1197. [PubMed] [Google Scholar]
- Hancock AA, Fox GB. Perspectives on cognitive domains, H3 receptor ligands and neurological disease. Expert Opin Investig Drugs. 2004;13:1237–1248. doi: 10.1517/13543784.13.10.1237. [DOI] [PubMed] [Google Scholar]
- Hancock AA, Esbenshade TA, Krueger KM, Yao BB. Genetic and pharmacological aspects of histamine H3 receptor heterogeneity. Life Sci. 2003;73:3043–3072. doi: 10.1016/j.lfs.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Jomphe C, Tiberi M, Trudeau LE. Expression of D2 receptor isoforms in cultured neurons reveals equipotent autoreceptor function. Neuropharmacology. 2006;50:595–605. doi: 10.1016/j.neuropharm.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Kamei C. Involvement of central histamine in amygdaloid kindled seizures in rats. Behav Brain Res. 2001;124:243–250. doi: 10.1016/s0166-4328(01)00218-2. [DOI] [PubMed] [Google Scholar]
- Khan ZU, Mrzljak L, Gutierrez A, de la Calle A, Goldman-Rakic PS. Prominence of the dopamine D2 short isoform in dopaminergic pathways. Proc Natl Acad Sci U S A. 1998;95:7731–7736. doi: 10.1073/pnas.95.13.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Stark H, Schunack W. Iododestannylation: an improved synthesis of [125I]iodoproxyfan, a specific radioligand of the histamine H3 receptor. J Labelled Comp Radiopharm. 1997;39:601–606. [Google Scholar]
- Leurs R, Bakker RA, Timmerman H, de Esch IJ. The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat Rev Drug Discov. 2005;4:107–120. doi: 10.1038/nrd1631. [DOI] [PubMed] [Google Scholar]
- Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, et al. An inverse agonist of the histamine H3 receptor improves wakefulness in narcolepsy: studies in orexin-/- mice and patients. Neurobiol Dis. 2008;30:74–83. doi: 10.1016/j.nbd.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, et al. Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci U S A. 2003;100:4305–4309. doi: 10.1073/pnas.0730708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LX, Monsma FJ, Jr, Sibley DR, Chiodo LA. D2L, D2S, and D3 dopamine receptors stably transfected into NG108-15 cells couple to a voltage-dependent potassium current via distinct G protein mechanisms. Synapse. 1996;24:156–164. doi: 10.1002/(SICI)1098-2396(199610)24:2<156::AID-SYN7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol. 1999;55:1101–1107. [PubMed] [Google Scholar]
- Mezzomo K, Cumming P, Minuzzi L. Comparison of the binding distribution of agonist and antagonist ligands for histamine H3 receptors in pig brain by quantitative autoradiography. Eur J Pharmacol. 2007;564:75–79. doi: 10.1016/j.ejphar.2007.01.087. [DOI] [PubMed] [Google Scholar]
- Moreno-Delgado D, Gomez-Ramirez J, Torrent-Moreno A, Gonzalez-Sepulveda M, Blanco I, Ortiz J. Different role of cAMP dependent protein kinase and CaMKII in H3 receptor regulation of histamine synthesis and release. Neuroscience. 2009;164:1244–1251. doi: 10.1016/j.neuroscience.2009.08.068. [DOI] [PubMed] [Google Scholar]
- Morisset S, Rouleau A, Ligneau X, Gbahou F, Tardivel-Lacombe J, Stark H, et al. High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature. 2000;408:860–864. doi: 10.1038/35048583. [DOI] [PubMed] [Google Scholar]
- Morisset S, Sasse A, Gbahou F, Héron A, Ligneau X, Tardivel-Lacombe J, et al. The rat H3 receptor: gene organization and multiple isoforms. Biochem Biophys Res Commun. 2001;280:75–80. doi: 10.1006/bbrc.2000.4073. [DOI] [PubMed] [Google Scholar]
- Motawaj M, Arrang JM. Ciproxifan, a histamine H3-receptor antagonist/inverse agonist, modulates methamphetamine-induced sensitization in mice. Eur J Neurosci. 2011;33:1197–1204. doi: 10.1111/j.1460-9568.2011.07618.x. [DOI] [PubMed] [Google Scholar]
- Panula P, Airaksinen M. The histaminergic neuronal system as revealed with antisera against histamine. In: Watanabe T, Wada H, editors. Histaminergic Neurons: Morphology and Function. Boca Raton, FL: CRC; 1991. pp. 127–144. [Google Scholar]
- Parmentier R, Anaclet C, Guhennec C, Brousseau E, Bricout D, Giboulot T, et al. The brain H3-receptor as a novel therapeutic target for vigilance and sleep-wake disorders. Biochem Pharmacol. 2007;73:1157–1171. doi: 10.1016/j.bcp.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Passani MB, Lin JS, Hancock A, Crochet S, Blandina P. The histamine H3 receptor as a novel therapeutic target for cognitive and sleep disorders. Trends Pharmacol Sci. 2004;25:618–625. doi: 10.1016/j.tips.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Pillot C, Héron A, Cochois V, Tardivel-Lacombe J, Ligneau X, Schwartz JC, et al. A detailed mapping of the histamine H3 receptor and its gene transcripts in rat brain. Neuroscience. 2002;114:173–193. doi: 10.1016/s0306-4522(02)00135-5. [DOI] [PubMed] [Google Scholar]
- Pollard H, Moreau J, Arrang JM, Schwartz JC. A detailed autoradiographic mapping of histamine H3 receptors in rat brain areas. Neuroscience. 1993;52:169–189. doi: 10.1016/0306-4522(93)90191-h. [DOI] [PubMed] [Google Scholar]
- Rouleau A, Ligneau X, Tardivel-Lacombe J, Morisset S, Gbahou F, Schwartz JC, et al. Histamine H3-receptor-mediated [35S]GTP gamma[S] binding: evidence for constitutive activity of the recombinant and native rat and human H3 receptors. Br J Pharmacol. 2002;135:383–392. doi: 10.1038/sj.bjp.0704490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau A, Héron A, Cochois V, Pillot C, Schwartz JC, Arrang JM. Cloning and expression of the mouse histamine H3 receptor: evidence for multiple isoforms. J Neurochem. 2004;90:1331–1338. doi: 10.1111/j.1471-4159.2004.02606.x. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Malinowska B, Kathmann M, Gothert M. Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundam Clin Pharmacol. 1994;8:128–137. doi: 10.1111/j.1472-8206.1994.tb00789.x. [DOI] [PubMed] [Google Scholar]
- Senogles SE. The D2 dopamine receptor isoforms signal through distinct Gi alpha proteins to inhibit adenylyl cyclase. A study with site-directed mutant Gi alpha proteins. J Biol Chem. 1994;269:23120–23127. [PubMed] [Google Scholar]
- Southam E, Cilia J, Gartlon JE, Woolley ML, Lacroix LP, Jennings CA, et al. Preclinical investigations into the antipsychotic potential of the novel histamine H3 receptor antagonist GSK207040. Psychopharmacology (Berl) 2009;201:483–494. doi: 10.1007/s00213-008-1310-9. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Eriksson KS, Brown RE, Haas HL. The mechanism of spontaneous firing in histamine neurons. Behav Brain Res. 2001;124:105–112. doi: 10.1016/s0166-4328(01)00219-4. [DOI] [PubMed] [Google Scholar]
- Strakhova MI, Fox GB, Carr TL, Witte DG, Vortherms TA, Manelli AM, et al. Cloning and characterization of the monkey histamine H3 receptor isoforms. Eur J Pharmacol. 2008;601:8–15. doi: 10.1016/j.ejphar.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Takeshita Y, Watanabe T, Sakata T, Munakata M, Ishibashi H, Akaike N. Histamine modulates high-voltage-activated calcium channels in neurons dissociated from the rat tuberomammillary nucleus. Neuroscience. 1998;87:797–805. doi: 10.1016/s0306-4522(98)00152-3. [DOI] [PubMed] [Google Scholar]
- Tardivel-Lacombe J, Rouleau A, Héron A, Morisset S, Pillot C, Cochois V, et al. Cloning and cerebral expression of the guinea pig histamine H3 receptor: evidence for two isoforms. Neuroreport. 2000;11:755–759. doi: 10.1097/00001756-200003200-00020. [DOI] [PubMed] [Google Scholar]
- Tardivel-Lacombe J, Morisset S, Gbahou F, Schwartz JC, Arrang JM. Chromosomal mapping and organization of the human histamine H3 receptor gene. Neuroreport. 2001;12:321–324. doi: 10.1097/00001756-200102120-00028. [DOI] [PubMed] [Google Scholar]
- Tohyama M, Tamiya R, Inagaki N, Takagi H. Morphology of histaminergic neurons with histidine decarboxylase as a marker. In: Watanabe T, Wada H, editors. Histaminergic Neurons: Morphology and Function. Boca Raton, FL: CRC; 1991. pp. 107–126. [Google Scholar]
- Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, et al. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408:199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Van H, II, Banihashemi B, Wilson AM, Jacobsen KX, Czesak M, Albert PR. Differential signaling of dopamine-D2S and -D2L receptors to inhibit ERK1/2 phosphorylation. J Neurochem. 2007;102:1796–1804. doi: 10.1111/j.1471-4159.2007.04650.x. [DOI] [PubMed] [Google Scholar]
- Wellendorph P, Goodman MW, Burstein ES, Nash NR, Brann MR, Weiner DM. Molecular cloning and pharmacology of functionally distinct isoforms of the human histamine H3 receptor. Neuropharmacology. 2002;42:929–940. doi: 10.1016/s0028-3908(02)00041-2. [DOI] [PubMed] [Google Scholar]
- Witte DG, Yao BB, Miller TR, Carr TL, Cassar S, Sharma R, et al. Detection of multiple H3 receptor affinity states utilizing [3H]A-349821, a novel, selective, non-imidazole histamine H3 receptor inverse agonist radioligand. Br J Pharmacol. 2006;148:657–670. doi: 10.1038/sj.bjp.0706752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe SE, Morris SJ. Dopamine D2 receptor isoforms expressed in AtT20 cells differentially couple to G proteins to acutely inhibit high voltage-activated calcium channels. J Neurochem. 1999;73:2375–2382. doi: 10.1046/j.1471-4159.1999.0732375.x. [DOI] [PubMed] [Google Scholar]
- Wouterlood F, Steinbusch H. Afferent and efferent fiber connections of histaminergic neurons in the rat brain: comparison with dopaminergic, noradrenergic and serotonergic systems. In: Watanabe T, Wada H, editors. Histaminergic Neurons: Morphology and Function. Boca Raton, FL: CRC; 1991. pp. 145–162. [Google Scholar]
- Wulff BS, Hastrup S, Rimvall K. Characteristics of recombinantly expressed rat and human histamine H3 receptors. Eur J Pharmacol. 2002;453:33–41. doi: 10.1016/s0014-2999(02)02382-8. [DOI] [PubMed] [Google Scholar]
- Yao BB, Witte DG, Miller TR, Carr TL, Kang CH, Cassar S, et al. Use of an inverse agonist radioligand [3H]A-317920 reveals distinct pharmacological profiles of the rat histamine H3 receptor. Neuropharmacology. 2006;50:468–478. doi: 10.1016/j.neuropharm.2005.10.008. [DOI] [PubMed] [Google Scholar]