

Abstract
BACKGROUND AND PURPOSE
Renal ischaemia/reperfusion (RI/R) injury is a major cause of acute kidney injury (AKI) and an important determinant of long-term kidney dysfunction. AMP-kinase and histone deacetylase sirtuin 1 (SIRT1) regulate cellular metabolism and are activated during hypoxia. We investigated whether AMP-kinase activator AICAR (5-amino-4-imidazolecarboxamide riboside-1-β-D-ribofuranoside) ameliorates RI/R injury and whether SIRT1 is involved in the pathogenesis.
EXPERIMENTAL APPROACH
Eight-week-old Sprague Dawley rats were divided into five groups: (i) sham-operated group; (ii) I/R group (40 min bilateral ischaemia followed by 24 h of reperfusion; (iii) I/R group + AICAR 50 mg·kg−1 i.v. given 60 min before operation; (iv). I/R group + AICAR 160 mg·kg−1 i.v; (v) I/R group + AICAR 500 mg·kg−1 i.v. Serum creatinine and urea levels were measured. Acute tubular necrosis (ATN), monocyte/macrophage infiltration and nitrotyrosine expression were scored. Kidney AMP-activated protein kinase (AMPK) and SIRT1 expressions were measured.
KEY RESULTS
Highest dose of AICAR decreased serum creatinine and urea levels, attenuated I/R injury-induced nitrosative stress and monocyte/macrophage infiltration, and ameliorated the development of ATN. Kidney I/R injury was associated with decreased AMPK phosphorylation and a fivefold increase in kidney SIRT1 expression. AICAR increased pAMPK/AMPK ratio and prevented the I/R-induced increase in renal SIRT1 expression.
CONCLUSIONS AND IMPLICATIONS
AICAR protects against the development of ATN after kidney I/R injury. Activators of kidney AMP kinase may thus represent a novel therapeutic approach to patients susceptible to AKI and to those undergoing kidney transplantation. The present study also suggests a role for SIRT1 in the pathogenesis of RI/R injury.
Keywords: acute kidney injury, ischaemia/reperfusion injury, acute tubular necrosis, AICAR, AMPK, SIRT1
Introduction
Acute kidney injury (AKI) occurs in 5% of all patients admitted to hospital and is associated with a 2- to 15-fold increase in mortality (Kunzendorf et al., 2010). AKI is the generic term for sudden decrease in renal function resulting in decreased urine output and retention of nitrogenous (creatinine and urea) and non-nitrogenous waste products in the blood (Lameire et al., 2005). Although the pathophysiology of AKI is complex and the aetiologies diverse, there is now accumulating evidence to indicate that hypotension, hypoperfusion, hypoxia, oxidative stress and renal vasoconstriction contribute to the pathogenesis (Kunzendorf et al., 2010). These detrimental changes in the renal haemodynamics and cellular metabolism have been shown to result in tubular and endothelial cell injury, mitochondrial damage and further renal vasoconstriction due to activation of the sympathetic nervous system and renin-angiotensin-aldosterone axis (Noiri et al., 2001; Devarajan, 2006; Hasegawa et al., 2010). Currently, there is no specific drug therapy for AKI, and recommendations for the prevention and non-dialysis treatment of AKI include mainly i.v. isotonic hydration, avoidance of nephrotoxins, maintenance of adequate systemic blood pressure, cardiac output and renal blood flow, as well as restoration and/or increase in urine flow (Lameire et al., 2005).
AMP-activated protein kinase (AMPK) is a ubiquitously expressed heterotrimeric kinase that acts as a highly conserved ultrasensitive energy sensor and participates in the regulation of energy-generating and energy-consuming pathways (Hallows et al., 2010). It is activated by an upstream kinase liver kinase B1 (LKB1) and when the intracellular AMP/ATP ratio is increased. It has been shown that AMPK plays an important role in the regulation of ion transport, podocyte function and diabetic renal hypertrophy in the kidneys (Hallows et al., 2010). AMPK is also a key regulator of lipid and glucose metabolism. Previous studies have provided evidence that AMPK is rapidly activated by acute renal ischaemia; however, the functional significance of AMPK activation remains undefined (Hallows et al., 2010). Mount et al. (2005) showed recently that although renal ischaemia activates AMPK within 1 min, the downstream target for AMPK, namely endothelial nitric oxide synthase (eNOS), was not phosphorylated. It is also unclear whether long-term pharmacological activation of AMPK provides protection against renal ischaemia/reperfusion (RI/R) injury.
Sirtuin 1 (SIRT1), a member of highly conserved nicotinamide adenine dinucleotide (NAD)-dependent class III histone deacetylases, is another ultrasensitive energy sensor. SIRT1 increases cellular stress resistance and genomic stability, and regulates cellular senescence via deacetylation of its target proteins such as p53, Forkhead box protein (FOXO) transcription factors and PPARγ-coactivator 1-α (PGC-1α) (Hao and Haase, 2010). Interestingly, SIRT1 activation has been shown to protect the kidney medulla from oxidative injury (He et al., 2010), and a close interplay between AMPK and SIRT1 in the regulation of cellular metabolism and inflammation has recently become apparent (Ruderman et al., 2010).
AICAR (5-amino-4-imidazolecarboxamide riboside-1-β-D-ribofuranoside) is an adenosine analogue, which binds directly to AMPK leading to allosteric modification and activation of AMPK. AICAR is taken up by adenosine transporters and subsequently phosphorylated to ZMP (5-aminoimidazole-4-carboxamide-1-β-D-furanosyl 5′-monophosphate) in the cell (Wong et al., 2009). ZMP in turn mimics AMP in AMPK signalling (Merrill et al., 1997). The aim of present study was to investigate whether AICAR ameliorates renal I/R injury. We were able to demonstrate that AICAR protected against the development of acute tubular necrosis (ATN) after kidney I/R injury. The beneficial effects of AICAR were mainly due to AMPK activation. However, AICAR also attenuated I/R injury-induced nitrosative stress, renal monocyte/macrophage infiltration and I/R injury-induced SIRT1 overexpression. Activators of kidney AMP kinase may thus represent a novel therapeutic approach to the treatment of AKI.
Methods
Experimental animals, RI/R, AICAR administration and sample preparation
Sixty-one 6-week-old male Sprague Dawley rats were purchased from Charles River Laboratories (Research Models and Services, Sulzfeld, Germany). The protocols were approved by the Animal Experimentation Committee of the University of Helsinki, Finland, and the Provincial State Office of Southern Finland (approval number STH059A), whose standards correspond to those of the American Physiological Society. Rats were kept under 12-h light/12-h dark cycle and they had free access to food and water. After a 1 week adaptation period, the rats were divided into five groups: (i) sham-operated controls receiving vehicle (0.9% NaCl 5 mL·kg−1 i.v; n= 17), (ii) I/R group receiving vehicle (n= 19), (iii) I/R group treated with AICAR (Bepharm Ltd, Shanghai, China) 50 mg·kg−1 i.v (n= 8), (iv) I/R group treated with AICAR 160 mg·kg−1 (n= 9), (v) I/R group treated with AICAR 500 mg·kg−1 i.v (n= 8). In a separate study, the effects of the highest dose of AICAR (500 mg·kg−1 i.v.) were examined in sham-operated rats (n= 6 in sham-operated rats receiving vehicle and n= 6 in sham-operated rats treated with AICAR). AICAR was dissolved in saline (0.9% NaCl) and given i.v. to the tail vein 60 min before the operation; the others received the same volume of vehicle.
An established model of renal I/R injury was used (Kennedy and Erlich, 2008). Rats were anaesthetized with isoflurane (anaesthesia induction in a chamber with 4–5% isoflurane at the flow rate 1.5 L·min−1), intubated and 1.5% isoflurane at rate 1.5 L·min−1 was used to maintain anaesthesia. Abdominal incisions were made and the renal pedicles were bluntly dissected. Bilateral renal ischaemia was induced by clamping renal pedicles for 40 min with microvascular clamps. Control animals were subjected to sham operation without renal pedicle clamping. The rats were hydrated with warm saline during the operation and the body temperature was maintained constantly at 37°C by using a heating pad until awake. The wounds were sutured after removal of the clips, and the animals were allowed to recover. Buprenorphine (0.1 mg·kg−1 s.c.) was used as post-operative analgesia; 24 h after the operation, the rats were again anaesthetized with isoflurane, and blood samples were collected from inferior vena cava with a 5 mL syringe and a 22G needle for biochemical measurements. The kidneys were excised, washed with ice-cold saline, blotted dry and weighed. Left kidney was used for Western Blot, qRT-PCR and histological examinations. Tissue samples for histology were fixed in 10% formalin and processed to paraffin with routine methodology. Samples for immunohistochemistry were snap-frozen in −38°C isopentane. For protein and gene expression studies, renal samples were snap-frozen in liquid nitrogen. Samples were stored at −80°C until assayed.
Kidney histology
For histological examination, 4 µm thick paraffin sections were cut and stained with haematoxylin eosin (n= 8–19 per group). Renal samples were visually examined by a pathologist (PF) with a Leica DMR microscope (Leica Microsystems AG, Heerbrugg, Switzerland) and morphological changes from the whole cross-sectional area of cortex and medulla were assessed according to the ATN scoring system adopted from Dragun et al. (2001; magnification × 200, ≥20 fields per kidney section quantified using the ATN-scoring system). Evaluation of histopathological changes included the loss of tubular brush border, tubular dilatation, cast formation and cell lysis. Tissue damage was quantified in a blinded manner and scored according to the percentage of damaged tubules in the sample: 0, no damage; 1, less than 25% damage; 2, 25–50% damage; 3, 50–75% damage; and 4, more than 75% damage.
Immunohistochemistry
For immunohistochemistry, frozen kidneys were processed and semi-quantitative scoring of inflammatory cells and kidney nitrotyrosine expression was performed (n= 8–19) as described in detail elsewhere (Helkamaa et al., 2003). The relative amount of antibody-positive signal in cortical and medullary areas per sample was determined with computerized densitometry (Leica IM500 and Leica QWIN software, Leica Microsystems AG, Heerbrugg, Switzerland). Primary monoclonal antibody against rat monocyte/macrophage ED-1 (Serotec Ltd, Oslo, Norway) and polyclonal antibody against nitrotyrosine (Upstate Biotechnology, Lake Placid, NY, USA), as well as peroxidase-conjugated rabbit anti-mouse and biotinylated anti-rabbit (Vector Laboratories Inc., Burlingame CA, USA) secondary antibodies (DAKO A/S, Glostrup, Denmark) were used.
Western blotting
Proteins from kidney samples (n= 5–8 per group) were isolated with lysis buffer (NaCl 136 mM, Na2HPO4 8 mM, KCl2 163 2.7 mM, KH2PO4 1.46 mM, Tween 20 0.001%, and complete protease inhibitors; Roche Diagnostics, Neuilly-Sur-Seine, France). Tissue samples were homogenized using a Bertin Precellys 24 homogenizer (Bertin Technologies, Aix en Provence, France), ceramic beads (Precellys CK14, Bertin Technologies) and a protocol consisting of 5000 rpm for 50 s repeated twice. Nuclear proteins for assessment of SIRT1 were extracted with Microcon's YM-10 filters and Centrifugal Filter Units (Millipore, Bedford, MA, USA) according to the instructions of the manufacturer. This protocol was first described by Schreiber et al. (1989). Samples were electrophoretically separated by 8% SDS-PAGE (20 µg total protein of the whole cell lysate per lane). Each lane corresponded to one rat and all five groups were run on one gel. Proteins were transferred to a PVDF membrane (Immobilon-P®, Millipore, Bedford, MA, USA) and blocked in 5% non-fat milk-TBS – 0.01% Tween-20® buffer. The membranes were probed with the following primary antibodies; AMPK (AMPKalpha, 1/1000, Cell Signaling, Beverly, MA, USA), pAMPK (pAMPKalpha, 1/1000, Cell Signaling) and SIRT1 (Anti-Sir2, 1/1000, Upstate, Millipore, Temecula, CA, USA). Tubulin was used as the loading control (Antialpha tubulin, 1/3000; Abcam, Cambridge, MA, USA). Horseradish peroxidase-conjugated anti-rabbit secondary antibody (Chemicon, Temecula, CA, USA) was subjected to enhanced chemiluminescence solution (ECLplus, Amersham Biosciences, Buckinghamshire, UK). The relative protein expressions in separate samples from the membranes were quantified with a Fluorescent Image Analyzer (FUJIFILM Corp, Tokyo, Japan).
Quantitative real-time RT-PCR
Quantitative real-time RT-PCR was performed using the LightCycler instrument (Roche Diagnostics, Neuilly sur Seine, France) for detection of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (Nox1), mitochondrial superoxidedismutase (MnSOD), catalase and ribosomal 18S mRNA, as described elsewhere (Louhelainen et al., 2009). Briefly, total RNA from the rat kidneys (n= 7–9 per group) was collected with TRIzol (Gibco, Invitrogen, Carlsbad, CA, USA), treated with deoxyribonuclease 1 (Sigma Chemicals, St. Louis, MO, USA) and reverse transcribed to cDNA by reverse-transcription enzyme (Im-Prom-II reverse transcription system, Promega, Madison, WI, USA). One microlitre of cDNA was subjected to quantitative real-time PCR for the detection of Nox1, as well as MnSOD, catalase and ribosomal 18S mRNA. The following primers were used: Nox1-2 sequence (5′-3′) down TTCTGCCGGGAGCGATAA, up GGAGTTGCAGGAGTCCTCATTTT; MnSOD down CCTCGGTGACGTTCAGATTGT, up TTAACGCGCAGATCATGCA; catalase down GGGCTGGGCTCAATGC, up TCAGCGACCGAGGGAT; and 18 s down TTTTCGTCACTACCTCCCCG, up ACATCCAAGGAAGGCAGCAG. The samples were amplified using FastStart DNA Master SYBR Green 1 (Roche Diagnostics, Neuilly sur Seine, France) according to the protocol of the manufacturer. The quantities of the PCR products were quantified with an external standard curve amplified from purified PCR product.
Analysis of protein carbonyls
For detection of protein carbonyls in the kidney (n= 7–9 samples per group), we used commercially available OxiSelect Protein Carbonyl elisa Kit (Cell Biolabs, Inc., San Diego, CA, USA) according to the manufacturer's instructions.
Biochemical determinations
Creatinine, urea and electrolytes from serum samples, as well as serum lipids and liver enzymes were measured by routine laboratory techniques (ADVIA 1650 Chemistry System, Siemens Healthcare Diagnostics Inc., Deerfield, IL, USA).
Statistical analysis
Data are presented as the mean + SEM. Statistically significant differences in mean values were tested by anova and Bonferroni's post hoc test, or by Student's t-test, when appropriate. The differences were considered significant when P < 0.05.
Results
Effects of AICAR on serum creatinine and urea levels in I/R injury
Acute kidney I/R injury was associated with a 7.4-fold increase in serum creatinine concentration and a 7.5-fold increase in serum urea level as compared to sham-operated controls (Figure 1A and B). In rats with I/R injury, the highest dose of AICAR decreased serum creatinine and urea concentrations by 35% and 25%, respectively, whereas the lower AICAR doses did not significantly influence serum creatinine and urea levels (Figure 1). In sham-operated rats, AICAR treatment did not influence serum urea level; however, AICAR slightly but significantly increased serum creatinine concentration by 10% (Supporting Information Table S1).
Figure 1.
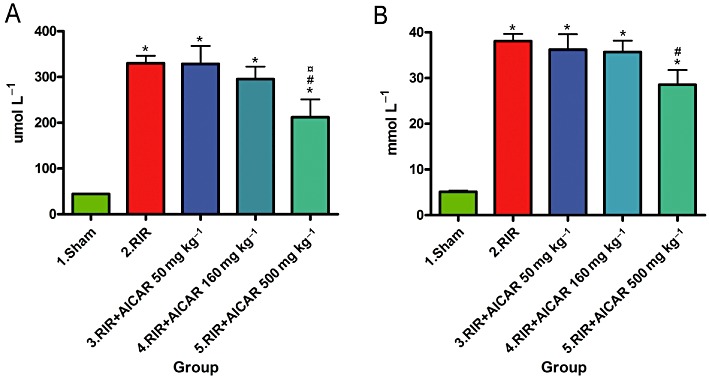
Effects of AICAR on serum creatinine (A) and serum urea concentrations (B) in rats with RI/R injury. Sham denotes sham-operated rats; RIR, rats with renal I/R injury; RIR + AICAR50, rats with I/R injury treated with AICAR at the dose 50 mg·kg−1 i.v.; RIR + AICAR160, rats with I/R injury treated with AICAR at the dose 160 mg·kg−1 i.v.; RIR + AICAR500, rats with I/R injury treated with AICAR at the dose 500 mg·kg−1 i.v. AICAR, 5-amino-4-imidazolecarboxamide riboside-1-β-D-ribofuranoside. Means ± SEM are given, n= 8–19 in each group. *P < 0.05 versus sham; #P < 0.05 versus RIR; P < 0.05 versus RIR+AICAR50.
Effects of AICAR on kidney morphology in I/R injury
Histopathological analysis of the kidneys harvested 24 h after I/R showed marked injury of the renal parenchyma comprising vast necrosis of the tubuloepithelial cells, tubular dilatation and cast formation (Figure 2). The highest dose of AICAR significantly ameliorated I/R injury-induced ATN.
Figure 2.
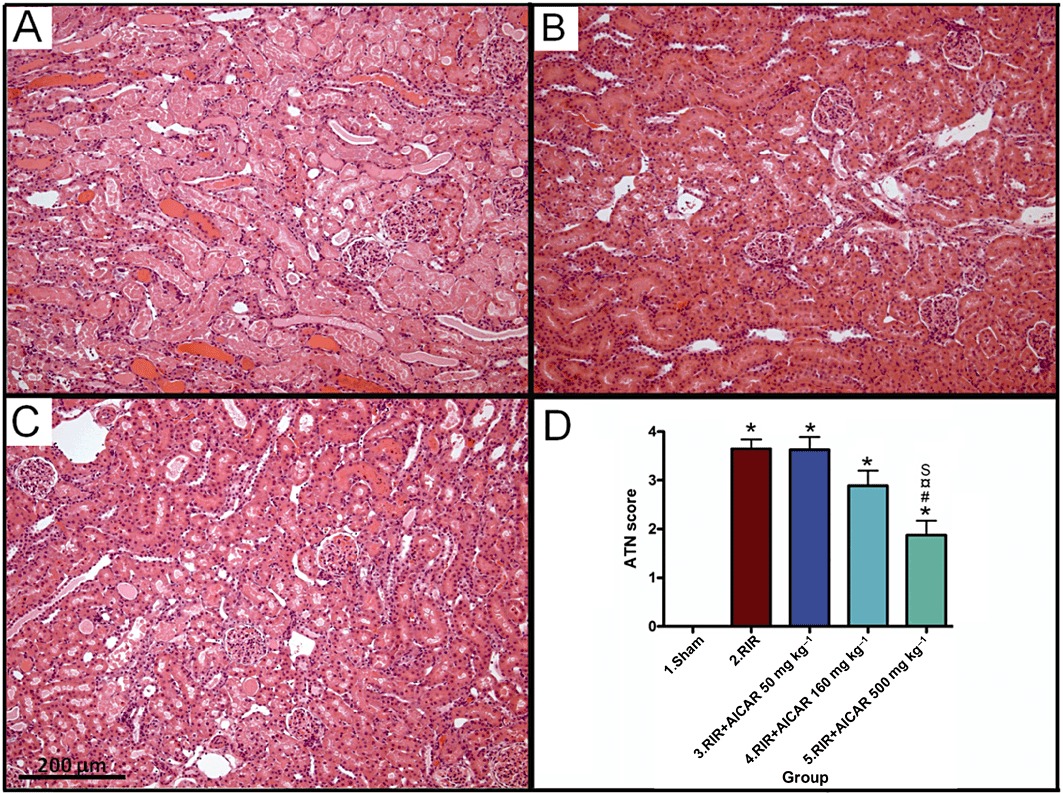
Effects of AICAR on ATN in rats with RI/R injury. Representative photomicrographs from untreated rats with I/R injury (A), sham-operated rats (B) and rats with I/R injury treated with 500 mg·kg−1 AICAR (C) are given. Magnification × 100, scale bar 200 µm. Quantification of ATN with a scoring system is presented in (D). Means ± SEM are given, n= 8–19 in each group. *P < 0.05 versus sham; #P < 0.05 versus RIR; P < 0.05 versus RIR + AICAR50; S P < 0.05 versus RIR + AICAR160. For abbreviations, see Figure legend 1.
Effects of AICAR on monocyte/macrophage recruitment and kidney nitrotyrosine expression in I/R injury
Acute kidney I/R injury was associated with a 23-fold increase in the number of ED-1-positive inflammatory cells in the kidney (Figure 3), and a 12.5-fold increase in renal nitrotyrosine expression (Figure 4) as compared to sham-operated controls. The difference in the number of renal ED-1-positive cells between sham-operated group and the groups with two highest AICAR doses did not reach statistical significance (Figure 3). AICAR at a dose of 160 mg·kg−1 also significantly ameliorated renal nitrosative stress (Figure 4).
Figure 3.
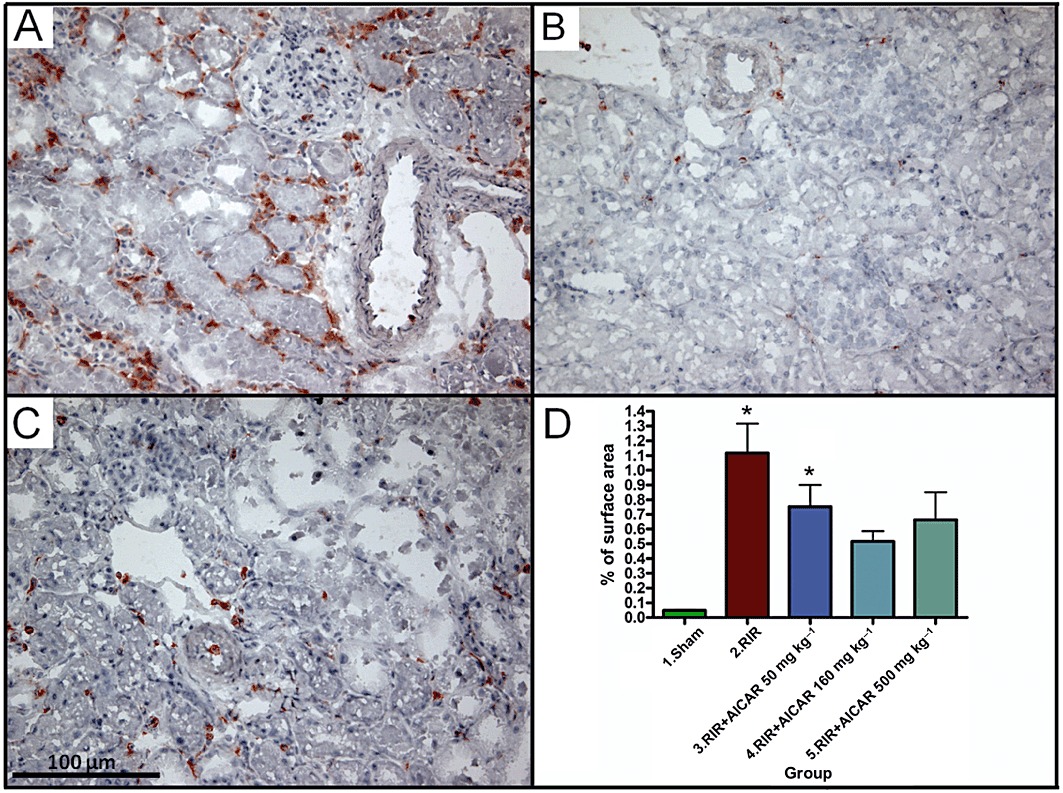
Effects of AICAR on monocyte/macrophage infiltration (ED1-immunopositive cells) in rats with RI/R injury. Representative photomicrographs from untreated rats with I/R injury (A), sham-operated rats (B) and rats with I/R injury treated with 500 mg·kg−1 AICAR (C) are given. Magnification × 200, scale bar 100µm. Quantification of ED1-positive cells in the kidney is presented in (D). Means ± SEM are given, n= 8–19 in each group. *P < 0.05 versus sham; #P < 0.05 versus RIR. For abbreviations, see Figure 1 legend.
Figure 4.
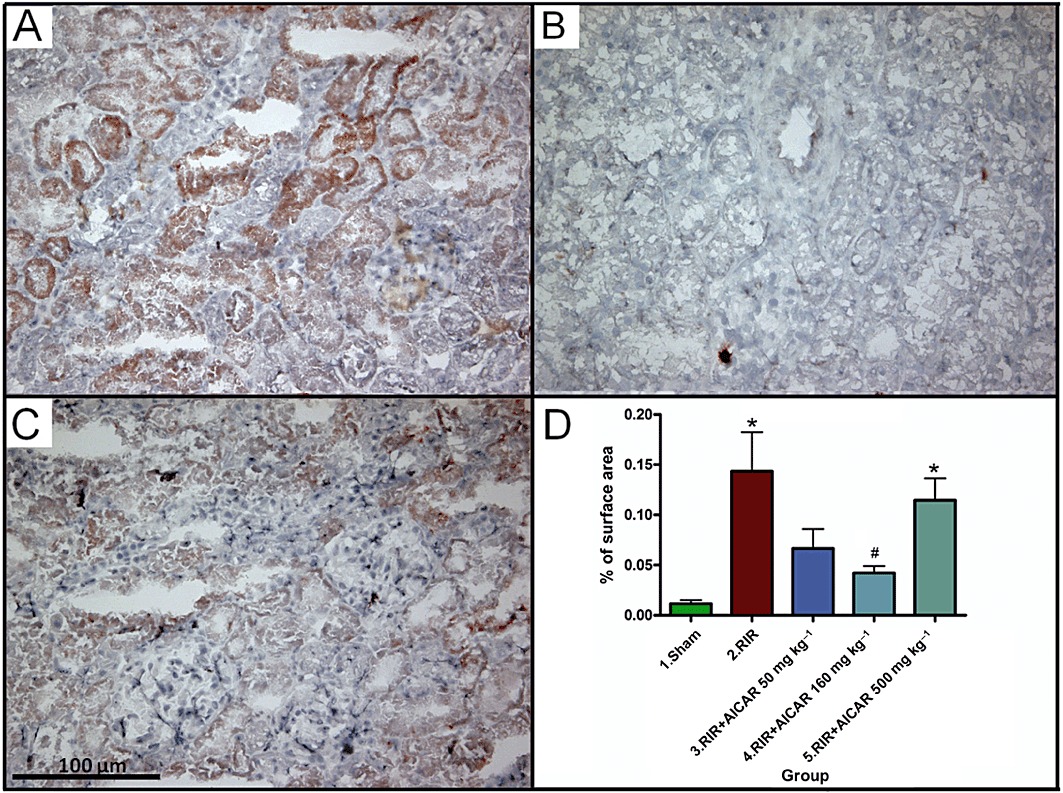
Effects of AICAR on nitrosative stress measured as kidney nitrotyrosine expression in rats with RI/R injury. Representative photomicrographs from untreated rats with I/R injury (A), sham-operated rats (B) and rats with I/R injury treated with 500 mg·kg−1 AICAR (C) are given. Magnification × 200, scale bar 100 µm. Quantification of nitrotyrosine expression in the kidney is presented in (D). Means ± SEM are given, n= 8–19 in each group. *P < 0.05 versus sham; #P < 0.05 versus RIR. For abbreviations, see Figure 1 legend.
Effects of AICAR on kidney AMPK expression in I/R injury
To investigate the cellular mechanisms mediating the effects of AICAR in I/R injury, we measured the total expression and phosphorylation level of AMPK in the kidneys by Western blot. Kidneys harvested 24 h after I/R injury showed a 40% decrease in the ratio of phosphorylated/total AMPK expression as compared to sham-operated rats, whereas the total expression of AMPK in the kidney remained unaltered (Figure 5). AICAR at the highest treatment dose increased significantly the phosphorylation level of AMPK in the kidneys (Figure 5).
Figure 5.
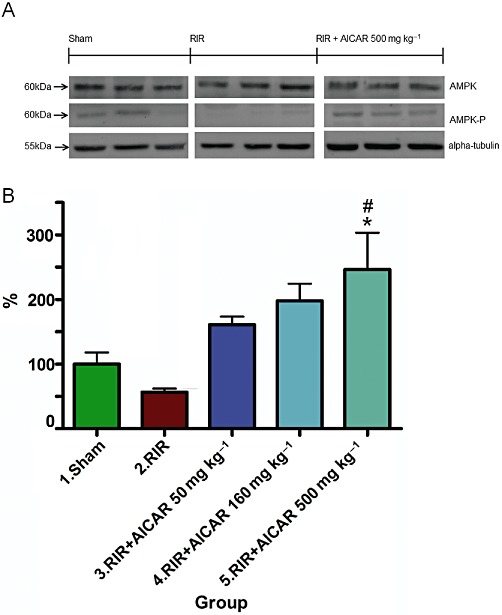
Effects of AICAR on kidney AMPK expression in rats with RI/R injury. The expressions of total AMPK, phosphorylated AMPK and housekeeping protein α-tubulin, measured by Western blot (20 µg total protein of the whole cell lysate per lane), are given in (A). The phosphorylation level of AMPK measured as p-AMPK/AMPK ratio is given in (B). Means ± SEM from three different runs are given, n= 4–5 in each group. *P < 0.05 versus sham; #P < 0.05 versus RIR. For abbreviations, see Figure 1 legend.
Effects of AICAR on kidney SIRT1 expression in I/R injury
Acute kidney I/R injury was associated with a fourfold increase in kidney SIRT1 expression measured from the nuclear protein fraction as compared to sham-operated controls (Figure 6). AICAR treatment prevented the I/R injury-induced increase in kidney SIRT1 expression in a dose-dependent manner (Figure 6).
Figure 6.
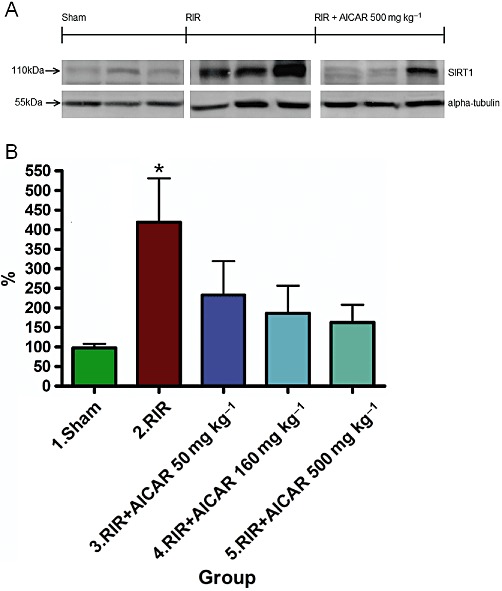
Effects of AICAR on kidney SIRT1 expression in rats with RI/R injury. The expression of SIRT1 and housekeeping protein α-tubulin, measured by Western blot (20 µg nuclear protein fraction per lane), are given in (A). Kidney SIRT1 expression measured as SIRT1/α-tubulin ratio is given in (B). Means ± SEM from three different runs are given, n= 4–5 per group in each gel. *P < 0.05 versus sham. For abbreviations, see Figure 1 legend.
Effects of AICAR on oxidative stress markers in I/R injury
To further investigate the role of oxidative stress in the pathophysiology of renal I/R injury, we measured the renal mRNA expression of pro-oxidative Nox1, and the mRNA expressions of the anti-oxidative catalase and MnSOD. I/R injury was associated with approximately 90% decrease in renal catalase mRNA expression (Figure 7A), and a 2.3-fold increase in renal Nox1 mRNA expression (Figure 7B), whereas no change was found in MnSOD mRNA expression in the kidneys (Figure 7C). The highest dose of AICAR decreased renal Nox1 mRNA expression, whereas the renal mRNA expressions of catalase or MnSOD were not influenced by AICAR treatment (Figure 7A–C).
Figure 7.
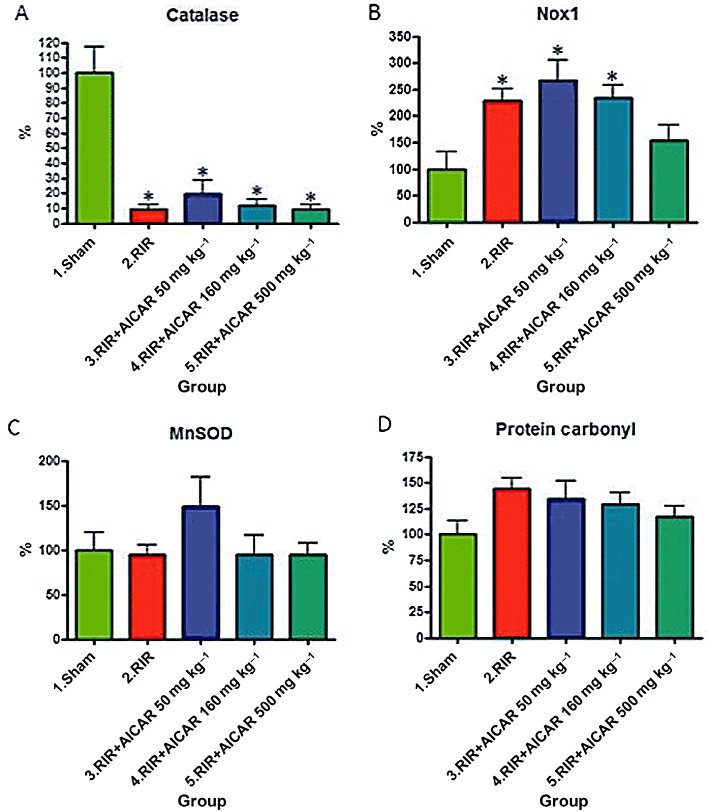
Effects of AICAR on oxidative stress markers in rats with RI/R injury. Renal mRNA expression of catalase (A), Nox1 (B) and mitochondrial superoxide dismutase (MnSOD; C) are given. The levels of protein carbonyls in the kidneys are shown in (D). Means ± SEM are given, n= 6–9 in each group. *P < 0.05 versus sham. For abbreviations, see Figure 1 legend.
I/R injury in the kidneys was associated with a non-significant 50% increase in the level of renal protein carbonyls (Figure 7D). AICAR treatment did not influence the concentration of protein carbonyls in the kidneys (Figure 7D).
Effects of AICAR on serum biochemistry in I/R injury
Acute kidney I/R injury was associated with 98%, 26% and 509% increases in the serum concentrations of alanine aminotransferase (ALAT), alkaline phosphatase (AFOS) and aspartate aminotransferase (ASAT), respectively, as compared to sham-operated controls, indicating AKI-induced hepatic injury (Table 1). AKI also markedly increased the serum levels of serum γ-glutamyl transferase and serum glutamate dehydrogenase (Table 1). The highest dose of AICAR decreased serum ASAT level but did not restore the other markers of I/R injury-induced hepatic damage/dysfunction (Table 1).
Table 1.
Effects of AICAR treatment on serum biochemistry in rats with kidney I/R injury
| Variable | Sham (n= 17) | RIR (n= 19) | RIR + AICAR50 (n= 8) | RIR + AICAR160 (n= 9) | RIR + AICAR500 (n= 8) | anova P-value |
|---|---|---|---|---|---|---|
| s-ALAT (U·L−1) | 39.3 ± 2.8 | 77.7 ± 4.2* | 67 ± 7.1* | 59 ± 4.0* | 62 ± 6.2* | 0.2413 |
| s-AFOS (U·L−1) | 257.9 ± 15.3 | 324.6 ± 17.7 | 293 ± 17.1 | 292 ± 23.1 | 321 ± 37.8 | 0.2840 |
| s-ASAT (U·L−1) | 120.4 ± 5.9 | 612.5 ± 42.1* | 648 ± 96.3* | 505 ± 52.1* | 374 ± 49.2*# | <0.0001 |
| s-GGT (U·L−1) | 0.0 ± 0 | 27.3 ± 2.6 | 26.1 ± 6.0 | 19.2 ± 2.8 | 13.2 ± 2.5 | <0.0001 |
| s-GLDH (U·L−1) | 10.0 ± 0.8 | 198.8 ± 19.8* | 195 ± 28.0* | 201 ± 24.4* | 129 ± 31.7* | <0.0001 |
| s-Protein(g·L−1) | 52.1 ± 0.8 | 52.5 ± 0.8 | 53.6 ± 1.3 | 52.6 ± 1.8 | 53.3 ± 1.2 | 0.4843 |
| s-Albumin (g·L−1) | 27.9 ± 0.4 | 27.6 ± 0.5 | 28.4 ± 0.7 | 27.7 ± 0.8 | 28.0 ± 0.5 | 0.4838 |
| s-K (mmol·L−1) | 4.7 ± 0.1 | 6.9 ± 0.3* | 6.4 ± 0.3* | 6.1 ± 0.2* | 5.6 ± 0.3# | <0.0001 |
| s-Na (mmol·L−1) | 143.3 ± 1.7 | 142.6 ± 2.2 | 149 ± 1.0 | 145 ± 1.9 | 150 ± 0.9 | 0.0267 |
| s-Cl (mmol·L−1) | 99.6 ± 0.9 | 91.5 ± 1.2* | 98 ± 2.7# | 95 ± 1.9 | 100 ± 2.0# | 0.1513 |
| s-Calsium (mmol·L−1) | 2.4 ± 0.0 | 2.4 ± 0.0 | 2.55 ± 0.1 | 2.47 ± 0.1 | 2.50 ± 0.0 | 0.0002 |
| s-Pi (mmol·L−1) | 2.9 ± 0.1 | 4.7 ± 0.3* | 4.3 ± 0.3* | 4.2 ± 0.3* | 3.7 ± 0.3 | <0.0001 |
| s-Chol (mmol·L−1) | 1.6 ± 0.1 | 1.6 ± 0.1 | 1.50 ± 0.1 | 1.45 ± 0.1 | 1.67 ± 0.1 | 0.8392 |
| s-Trigly (mmol·L−1) | 0.8 ± 0.0 | 0.9 ± 0.1 | 0.89 ± 0.1 | 0.76 ± 0.1 | 0.71 ± 0.1 | 0.0406 |
Sham denotes sham-operated rats; RIR, rats with renal I/R injury; RIR+AICAR50, rats with I/R injury treated with AICAR at the dose 50 mg·kg−1 i.v.; RIR+AICAR160, rats with I/R injury treated with AICAR at the dose 160 mg·kg−1 i.v.; RIR+AICAR500, rats with I/R injury treated with AICAR at the dose 500 mg·kg−1 i.v. Means ± SEM are given, n= 8–19 in each group.
P < 0.05 versus sham;
P < 0.05 versus RIR.
Serum potassium and phosphate concentrations were increased and the level of serum chloride decreased in rats with I/R injury (Table 1). These changes in serum electrolytes were attenuated by highest AICAR dose.
Neither I/R injury nor drug treatment influenced serum sodium, calcium, albumin, protein, cholesterol or triglyceride concentration.
In sham-operated rats, AICAR treatment increased the serum concentration of ALAT by 1.3-fold and that of ASAT by 1.4-fold (Supporting Information Table S1).
Discussion
The acute response initiated by ischaemia-reperfusion injury in the kidneys is characterized by increased production of reactive oxygen species (ROS) and induction of pro-inflammatory cytokines, leading to inflammatory response and ATN (Bonventre and Yang, 2011). The important finding of the present study was that AICAR ameliorated the development of ATN in a rat I/R injury. AICAR attenuated I/R injury-induced nitrosative stress and monocyte/macrophage infiltration in the kidneys. We here also reported that kidney I/R injury is associated with decreased AMPK phosphorylation and a fivefold increase in the expression SIRT1 in the kidneys. AICAR increased AMPK phosphorylation and prevented the I/R-induced increase in renal SIRT1 expression.
Previous studies have provided evidence that AICAR protects against I/R injury in several tissues, such as heart and liver (Bullough et al., 1994; Alkhulaifi and Pugsley, 1995; Galinanes et al., 1995; Mathew et al., 1995; Peralta et al., 2001). Lin et al. (2004) were the first to demonstrate that the combination therapy with AICAR and N-acetyl cysteine attenuates renal I/R injury and improves the outcome of the transplanted kidney after prolonged cold preservation. Consistent with the present study, Lee et al. (2009) have reported recently that preconditioning of the kidneys by AICAR, given as a single low-dose treatment 10 min before ischaemia, ameliorates renal I/R injury. Furthermore, in the same study, the authors demonstrated that acute AICAR treatment phosphorylated AMPK in the sham-operated rats. In contrast to the study by Lee et al. (2009), here we investigated the effects of AICAR in rats with kidney I/R injury by using three different AICAR doses. Furthermore, we examined the effects of I/R injury as well as the long-term effects of AICAR treatment on two major metabolic sensors, namely AMPK and SIRT1, from kidney samples harvested 24 h after the reperfusion. We assessed the renal effects of AICAR by combining functional data with the widely used histological scoring for ATN. Furthermore, renal nitrotyrosine expression and the number of infiltrated monocytes/macrophages were used in the quantification of local nitrosative stress and leucocyte recruitment, respectively. To further investigate the role of oxidative stress in the pathophysiology of AKI, we measured the renal mRNA expression of pro-oxidative Nox1 and of anti-oxidative catalase and MnSOD. We also quantified protein oxidation from the kidney samples by using a commercially available protein carbonyl elisa kit. It was found that renal I/R injury is associated with significant leucocyte infiltration, renal nitrosative stress, up-regulation of Nox1 and down-regulation of catalase mRNA expression, as well as a non-significant increase in the level of protein carbonyls in the kidneys. These findings strongly suggest the involvement of oxidative stress in the pathogenesis of renal I/R injury. Interestingly, the renoprotective effects of AICAR were associated with a decrease in nitrosative stress and amelioration of monocyte/macrophage infiltration in the kidneys. AICAR also ameliorated I/R-induced increase in renal Nox1 expression. Our study is thus in very good accordance with the previous study by Kim et al. (2008) reporting suppression of ROS formation by AICAR and studies by Lin et al. (2004) and Gaskin et al. (2009) demonstrating the anti-inflammatory actions of AICAR. It should also be noted that the anti-oxidative and anti-inflammatory effects of AICAR could also explain, at least in part, improvement of urea and creatinine clearance, as AICAR has been shown to increase the bioavailability of NO (Morrow et al., 2003; Gaskin et al., 2007), and could thereby improve post-ischaemic blood flow in the kidneys (Versteilen et al., 2006).
AMPK is a heterotrimeric serine/threonine kinase that plays a central role in the regulation of cellular energy metabolism. AMPK consists of a catalytic α subunit (α1, α2) and regulatory, non-catalytic β (β1, β2) and γ (γ1, γ2, γ3) subunits. In the kidney, the α1 subunit is highly expressed, whereas the expression of the α2 subunit is barely detectable (Kim and Tian, 2010). Mount et al. (2005) demonstrated recently that acute renal ischaemia leading to energy depletion and falling levels of ATP with a simultaneous rise in AMP activates AMPK within 1 min and that the activation of AMPK remains for a least 30 min. At present, data on AMPK activity at later phases after reperfusion are sparse. In the present study, we demonstrated that kidneys harvested 24 h after I/R injury showed a 40% decrease in AMPK activity when measured as the ratio of phosphorylated/total AMPK expression. These findings suggest an acute compensatory/regulatory role for AMPK in kidney I/R injury. In contrast, AICAR treatment increased the phosphorylation level of AMPK in the kidney. Although AICAR has been shown to exert several mechanisms of action, we believe that renoprotection found in the present study was largely due to AMPK activation. Previous studies have provided evidence that AICAR suppresses the production of ROS (Kim et al., 2008), inhibits pro-inflammatory NF-kB signalling (Katerelos et al., 2010), and inhibits inflammation in MRL/lpr mouse mesangial cells (Peairs et al., 2009) through AMPK activation. Further studies are warranted to examine the upstream AMPK kinases as well as the substrates that are phosphorylated by AMPK in the kidney.
SIRT1 is a member of the sirtuin family that are highly conserved NAD-dependent enzymes regulating life span in lower organisms and act as metabolic sensors of the cell (Finkel et al., 2009). SIRT1 increases cellular stress resistance, genomic stability and regulates cellular senescence and energy metabolism via deacetylation of the target proteins such as P53, FOXO transcription factors and PGC-1. In the kidney, SIRT1 has been shown to be cytoprotective and to participate in the regulation of blood pressure and sodium homeostasis (Hao and Haase, 2010). SIRT1 activation protects the kidney medulla from oxidative injury (He et al., 2010). Furthermore, kidney-specific overexpression of SIRT1 (Hasegawa et al., 2010) and SIRT1 activation by resveratrol (Kim et al. (2008) have been shown to ameliorate cisplatin-induced AKI. Interestingly, SIRT1 has also been shown to protect the heart from I/R injury (Hsu et al., 2010; Nadtochiy et al., 2011a,b). In the present study, renal SIRT1 expression was increased by fourfold in the kidney when measured 24 h after I/R injury. Our findings thus suggest sustained SIRT1 activation after kidney I/R injury whereas AMPK activation seems to occur only during the acute kidney ischaemia. We also reported here that AICAR treatment prevented the I/R injury-induced increase in kidney SIRT1 expression. It could therefore be postulated that amelioration of the renal nitrosative stress and monocyte/macrophage infiltration by AICAR leading to improved excretion of nitrogenous waste and tissue morphology diminish the need for SIRT1 overexpression after I/R injury.
The morbidity and mortality from AKI is high in part due to extra-renal complications such as hepatic dysfunction. Development of liver injury may also lead to other extra-renal complications such as respiratory failure, intestinal barrier destruction and systemic inflammatory response syndrome (Elapavaluru and Kellum, 2007). Park et al. showed recently rapid peri-portal hepatocyte necrosis, vacuolization, neutrophil infiltration and up-regulation of pro-inflammatory genes (TNF-a, IL-17A, IL-6) in the liver after AKI, suggesting an intestinal source of hepatic injury (Park et al., 2011). Interestingly, the highest dose of AICAR decreased the serum ASAT level in rats with renal I/R injury. In contrast, AICAR treatment produced a slight (13–36%) but significant increase in liver enzymes in sham-operated rats. Further studies are thus warranted to investigate the molecular mechanisms mediating the effects of AICAR on liver function.
In conclusion, using a rat model of kidney I/R injury, we here reported that AICAR protects against the development of ATN via mechanisms linked to decreased nitrosative stress and monocyte/macrophage infiltration and activation of AMPK in the kidneys. The present study thus suggests a therapeutic role for AMPK activators in patients susceptible to AKI and to those undergoing kidney transplantation.
Acknowledgments
The present study was supported by the Sigrid Juselius Foundation and the Academy of Finland. We are grateful to Mrs Päivi Leinikka, Mrs Nada Bechara-Hirvonen and Mrs Anneli von Behr for excellent technical assistance.
Glossary
- AFOS
alkaline phosphatase
- AICAR
5-amino-4-imidazolecarboxamide riboside-1-β-D-ribofuranoside
- AKI
acute kidney injury
- ALAT
alanine aminotransferase
- AMPK
AMP-activated protein kinase
- ASAT
aspartate aminotransferase
- ATN
acute tubular necrosis
- eNOS
endothelial nitric oxide synthase
- FOXO
Forkhead box protein
- LKB1
liver kinase B1
- MnSOD
manganese superoxide dismutase
- NAD
nicotinamide adenine dinucleotide
- Nox1
nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1
- PGC-1α
peroxisome proliferator-activated receptor γ-coactivator 1-α
- RI/R
renal ischaemia/reperfusion
- SIRT1
sirtuin 1 (silent information regulator 1)
- ZMP
5-aminoimidazole-4-carboxamide-1-β-D-furanosyl 5′-monophosphate
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1 Effects of AICAR treatment on serum biochemistry in sham-operated rats. Sham denotes sham-operated rats; Sham+AICAR500, sham operated-rats treated with AICAR at the dose 500 mg·kg−1 i.v. Means ± SEM are given, n = 6 in both groups
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alkhulaifi AM, Pugsley WB. Role of acadesine in clinical myocardial protection. Br Heart J. 1995;73:304–305. doi: 10.1136/hrt.73.4.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough DA, Zhang C, Montag A, Mullane KM, Young MA. Adenosine-mediated inhibition of platelet aggregation by acadesine. A novel antithrombotic mechanism in vitro and in vivo. J Clin Invest. 1994;94:1524–1532. doi: 10.1172/JCI117493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- Elapavaluru S, Kellum JA. Why do patients die of acute kidney injury? Acta Clin Belg Suppl. 2007;2:326–331. doi: 10.1179/acb.2007.074. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinanes M, Zhai X, Bullough D, Mullane KM, Hearse DJ. Protection against injury during ischemia and reperfusion by acadesine derivatives GP-1-468 and GP-1-668. Studies in the transplanted rat heart. J Thorac Cardiovasc Surg. 1995;110:752–761. doi: 10.1016/S0022-5223(95)70108-7. [DOI] [PubMed] [Google Scholar]
- Gaskin FS, Kamada K, Yusof M, Korthuis RJ. 5′-AMP-activated protein kinase activation prevents postischemic leukocyte-endothelial cell adhesive interactions. Am J Physiol Heart Circ Physiol. 2007;292:H326–H332. doi: 10.1152/ajpheart.00744.2006. [DOI] [PubMed] [Google Scholar]
- Gaskin FS, Kamada K, Yusof M, Durante W, Gross G, Korthuis RJ. AICAR preconditioning prevents postischemic leukocyte rolling and adhesion: role of K(ATP) channels and heme oxygenase. Microcirculation. 2009;16:167–176. doi: 10.1080/10739680802355897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows KR, Mount PF, Pastor-Soler NM, Power DA. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol. 2010;298:F1067–F1077. doi: 10.1152/ajprenal.00005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao CM, Haase VH. Sirtuins and their relevance to the kidney. J Am Soc Nephrol. 2010;21:1620–1627. doi: 10.1681/ASN.2010010046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem. 2010;285:13045–13056. doi: 10.1074/jbc.M109.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest. 2010;120:1056–1068. doi: 10.1172/JCI41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helkamaa T, Finckeberg P, Louhelainen M, Merasto S, Rauhala P, Lapatto R, et al. Entacapone protects from angiotensin II-induced inflammation and renal injury. J Hypertens. 2003;21:2353–2363. doi: 10.1097/00004872-200312000-00025. [DOI] [PubMed] [Google Scholar]
- Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–2182. doi: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katerelos M, Mudge SJ, Stapleton D, Auwardt RB, Fraser SA, Chen CG, et al. 5-aminoimidazole-4-carboxamide ribonucleoside and AMP-activated protein kinase inhibit signalling through NF-kappaB. Immunol Cell Biol. 2010;88:754–760. doi: 10.1038/icb.2010.44. [DOI] [PubMed] [Google Scholar]
- Kennedy SE, Erlich JH. Murine renal ischaemia-reperfusion injury. Nephrology (Carlton) 2008;13:390–396. doi: 10.1111/j.1440-1797.2008.00979.x. [DOI] [PubMed] [Google Scholar]
- Kim JE, Kim YW, Lee IK, Kim JY, Kang YJ, Park SY. AMP-activated protein kinase activation by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) inhibits palmitate-induced endothelial cell apoptosis through reactive oxygen species suppression. J Pharmacol Sci. 2008;106:394–403. doi: 10.1254/jphs.fp0071857. [DOI] [PubMed] [Google Scholar]
- Kim M, Tian R. Targeting AMPK for cardiac protection: opportunities and challenges. J Mol Cell Cardiol. 2010;51:548–553. doi: 10.1016/j.yjmcc.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzendorf U, Haase M, Rolver L, Haase-Fielitz A. Novel aspects of pharmacological therapies for acute renal failure. Drugs. 2010;70:1099–1114. doi: 10.2165/11535890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Lameire N, Van Biesen W, Vanholder R. Acute renal failure. Lancet. 2005;365:417–430. doi: 10.1016/S0140-6736(05)17831-3. [DOI] [PubMed] [Google Scholar]
- Lee S, Chang Y, Na K, Lee K, Suh K, Kim S, et al. The preconditioning with AICAR protects against subsequent renal ischemia reperfusion injury. Korean J Nephrol. 2009;28:96–102. [Google Scholar]
- Lin A, Sekhon C, Sekhon B, Smith A, Chavin K, Orak J, et al. Attenuation of ischemia-reperfusion injury in a canine model of autologous renal transplantation. Transplantation. 2004;78:654–659. doi: 10.1097/01.tp.0000131664.18670.17. [DOI] [PubMed] [Google Scholar]
- Louhelainen M, Vahtola E, Forsten H, Merasto S, Kyto V, Finckenberg P, et al. Oral levosimendan prevents postinfarct heart failure and cardiac remodeling in diabetic Goto-Kakizaki rats. J Hypertens. 2009;27:2094–2107. doi: 10.1097/HJH.0b013e32832f0ce4. [DOI] [PubMed] [Google Scholar]
- Mathew JP, Rinder CS, Tracey JB, Auszura LA, O'Connor T, Davis E, et al. Acadesine inhibits neutrophil CD11b up-regulation in vitro and during in vivo cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1995;109:448–456. doi: 10.1016/S0022-5223(95)70275-X. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- Mount PF, Hill RE, Fraser SA, Levidiotis V, Katsis F, Kemp BE, et al. Acute renal ischemia rapidly activated the energy sensor AMPK but does not increase phosphorylation of eNOS-Ser1177. Am J Physiol Renal Physiol. 2005;289:F1103–F1115. doi: 10.1152/ajprenal.00458.2004. [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. 2011a;89:643–649. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, et al. SIRT1-mediated acute cardioprotection. Am J Physiol Heart Circ Physiol. 2011b;301:H1506–H1512. doi: 10.1152/ajpheart.00587.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noiri E, Nakao A, Uchida K, Tsukahara H, Ohno M, Fujita T, et al. Oxidative and nitrosative stress in acute renal ischemia. Am J Physiol Renal Physiol. 2001;281:F948–F957. doi: 10.1152/ajprenal.2001.281.5.F948. [DOI] [PubMed] [Google Scholar]
- Park SW, Chen SW, Kim M, Brown KM, Kolls JK, D'Agati VD, et al. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab Invest. 2011;91:63–84. doi: 10.1038/labinvest.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, et al. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol. 2009;156:542–551. doi: 10.1111/j.1365-2249.2009.03924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta C, Bartrons R, Serafin A, Blazquez C, Guzman M, Prats N, et al. Adenosine monophosphate-activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat. Hepatology. 2001;34:1164–1173. doi: 10.1053/jhep.2001.29197. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298:E751–E760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding-proteins with mini-extracts, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteilen AM, Korstjens IJ, Musters RJ, Groeneveld AB, Sipkema P. Rho kinase regulates renal blood flow by modulating eNOS activity in ischemia-reperfusion of the rat kidney. Am J Physiol Renal Physiol. 2006;291:F606–F611. doi: 10.1152/ajprenal.00434.2005. [DOI] [PubMed] [Google Scholar]
- Wong AKF, Howie J, Petrie JR, Lang CC. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin Sci. 2009;116:607–620. doi: 10.1042/CS20080066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.