

Abstract
BACKGROUND AND PURPOSE
Angiotensin-(1–7) [Ang-(1–7)] has anti-inflammatory effects in models of cardiovascular disease and arthritis, but its effects in asthma are unknown. We investigated whether Ang-(1–7) has anti-inflammatory actions in a murine model of asthma.
EXPERIMENTAL APPROACH
The effects of Ang-(1–7) alone or in combination with the MAS1 receptor antagonist, A779, were evaluated over a 4 day period in an ovalbumin-challenged mouse model of allergic asthma. On day 5, bronchoalveolar lavage was performed, and lungs were sectioned and assessed histologically for quantification of goblet cells, perivascular and peribronchial inflammation and fibrosis. Biochemical analysis of the pro-inflammatory ERK1/2 and IκB-α was assessed. In addition, the effect of Ang-(1–7) on proliferation of human peripheral blood mononuclear cells (HPBMC) was investigated.
KEY RESULTS
Ang-(1–7) attenuated ovalbumin-induced increases in total cell counts, eosinophils, lymphocytes and neutrophils. Ang-(1–7) also decreased the ovalbumin-induced perivascular and peribronchial inflammation, fibrosis and goblet cell hyper/metaplasia. Additionally, Ang-(1–7) reduced the ovalbumin-induced increase in the phosphorylation of ERK1/2 and IκB-α. These effects of Ang-(1–7) were reversed by the MAS1 receptor antagonist A779. Furthermore, Ang-(1–7) inhibited phytohaemagglutinin (PHA)-induced HPBMC proliferation.
CONCLUSION AND IMPLICATIONS
Ang-(1–7), via its MAS1 receptor, acts as an anti-inflammatory pathway in allergic asthma, implying that activation of the MAS1 receptor may represent a novel approach to asthma therapy.
Keywords: asthma, inflammation, Ang-(1–7), MAS1 receptors, ERK1/2 and NF-κB
Introduction
Asthma is a chronic inflammatory disease of the airways that leads to airway remodelling and fibrosis and involves immune, inflammatory and structural cells, and is driven, in the majority of patients, by an allergic process (Holgate, 2011). Many cytokines, particularly Th2-derived, such as IL-4, IL5 IL-9 and IL-13, together with inflammatory mediators from activated mast cells, have been shown to be important in the disease pathogenesis (Finkelman et al., 2010; Hansbro et al., 2011). More recently, the discovery of additional cytokines such as the IL-17 (IL-17 A, E), a Th17-derived cytokine, and IL-33, which may play a role in sustaining the airway inflammation in asthma, has not only added further complexity to the disease but has also opened up further avenues for potential intervention (Finkelman et al., 2010; Hansbro et al., 2011). One postulated mechanism for the increased and sustained inflammatory response in asthma is due to an imbalance between pro- and anti-inflammatory mediators, the latter acting as a counter to the regulatory signalling pathways (Haworth and Levy, 2007).
Angiotensin-(1–7) [Ang-(1–7)] is a member of the renin–angiotensin system and has recently been shown to oppose the cardiovascular effects of pressor agents such as angiotensin II (Ang II), NA and endothelin-1 (Benter et al., 1995; 2006; Chappell, 2007). Ang-(1–7) has been identified as an endogenous ligand for the GPCR MAS1, which is a cell surface receptor that is highly expressed in the brain, heart, kidney, endothelium and leucocytes (Santos et al., 2003; Nie et al., 2009; Rabelo et al., 2011). Ang-(1–7) was shown to have anti-thrombotic and antiproliferative properties (Rabelo et al., 2011). Indeed, Ang-(1–7) inhibits Ang II-stimulated ERK1/2 and Rho kinase phosphorylation in the heart (Giani et al., 2007; 2008). Our studies have also shown that chronic treatment with Ang-(1–7) can inhibit hypertension- or diabetes-induced vascular, renal and cardiac dysfunction (Benter et al., 2006; 2007). Furthermore, Ang-(1–7) has been reported to have anti-fibrotic effects in the kidney via activation of the src homology-2 containing protein-tyrosine phosphatase-1 (SHP-1) and inhibition of high glucose-induced increase in p38-MAPK, cell protein synthesis and TGF-β production (Gava et al., 2009). There is also very recent evidence that Ang-(1–7) has anti-inflammatory actions. We have shown, in a model of combined hypertension and diabetes, that chronic treatment with Ang-(1–7) can inhibit renal NADPH oxidase (NOX) and cardiac NF-κB activity and inhibit the expression of C3, IL-6, IL-1β, Na 1p12 and Casp 1 in the heart (Benter et al., 2008; Al-Maghrebi et al., 2009). Furthermore, in an experimental model of arthritis, activation of the MAS1 receptor, by the MAS1 agonist AVE 0991 or Ang-(1–7), reduces the neutrophil accumulation, hypernociception and production of TNF-α, IL-1β, CXCL1 and histopathological changes evoked in this antigen-induced arthritis (AIA) (da Silveira et al., 2010). Moreover, MAS1−/− mice subjected to AIA had less pronounced neutrophil influx and cytokine release (da Silveira et al., 2010).
Based on the evidence that Ang-(1–7) has anti-inflammatory effects in cardiovascular disease and arthritis, and inhibits NF-κB- and ERK1/2-dependent pathways (Benter et al., 2008; Al-Maghrebi et al., 2009; da Silveira et al., 2010), which have important roles in asthma, we have employed a murine model of asthma to investigate (i) whether Ang-(1–7) has anti-inflammatory effects in this model; (ii) the role of the MAS1 receptor in mediating any Ang-(1–7)-induced effects; (iii) the effect of Ang-(1–7) on ovalbumin induced NF-κB- and ERK1/2-dependent signalling; and (iv) the effect of Ang-(1–7) on phytohaemagglutinin (PHA)-induced proliferation of human peripheral blood mononuclear cells (HPBMCs).
Methods
Reagents
Chemicals used included ovalbumin (grade V), halothane, absolute ethanol (Merck KGaA, Darmstadt, Germany), formaldehyde (Surechem Products LTD, Suffolk, UK), Alu-Gel-S (SERVA Electrophoresis GmbH, Heidelberg, Germany), isotone II diluent solution (Beckman Coulter Inc., Krefeld, Germany), Zap-OGLOBIN (Coulter Electronics LTD, Buckinghamshire, UK), Diff-Quik (Baxter Dade AG, Dudingen, Switzerland), PBS (Sigma-Aldrich, St Louis, MO, USA), Ang-(1–7), A779, Tris-base, NaCl, Na4P2O7, NaF, CaCl2, MgCl2, Glycerol, NP4O, Na3VO4, PMSF, protease inhibitor cocktail, Ponceau 2R (Sigma-Aldrich), phosphatase inhibitor cocktail (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) xylene, ethanol (Honeywell Riedel-de Haën®, Seelze, Germany), haematoxylin, eosin, glacial acetic acid, ferric chloride, HCl, periodic acid, DMSO (BDH Laboratory Supplies, Poole, UK), Dpx mountant, picric acid, mercuric chloride, phosphomolybdic acid, light green SF yellowish, Schiff's reagent (Sigma-Aldrich), acid fuchsin (Gurr Microscopy Materials; BDH Ltd., Poole, UK), rabbit monoclonal antibodies for total and phosphorylated IκB-α and rabbit polyclonal antibody for phosphorylated ERK1/2 (Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit monoclonal anti-actin antibody, phenanthroline (Sigma-Aldrich), nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany).
Animals
Male BALB/c mice (6–8 weeks old) used in this study were maintained under temperature-controlled conditions with an artificial 12 h light/dark cycle and were allowed standard chow and water ad libitum. The total number of mice used was 105. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). All experimental protocols were approved by the Animal Welfare Committee and complied with regulations for the use of Laboratory Animals in the Health Sciences Centre, Kuwait University.
Immunization, challenge and drug treatment
BALB/c mice were immunized once by i.p. injection of 10 µg ovalbumin in 0.2 mL of alu-Gel-S on day 0. Ten days later, mice were challenged intranasally, once a day, over four consecutive days, with 20 µg ovalbumin in 50 µL PBS. Control animals were similarly immunized with ovalbumin and challenged intranasally with 50 µL PBS.
Two studies were carried out. In the first study [dose–response curve (DRC) study], five treatment groups (n = 6–16) were established. Groups 1 and 2 were treated with the vehicle (water) and challenged intranasally with PBS and ovalbumin, respectively. All groups 3 to 5 were challenged with ovalbumin and were pretreated 30 min before and 1 h after challenge with Ang-(1–7) at 0.03, 0.1 and 0.3 mg·kg−1; i.p., respectively. The drug/vehicle treatment was continued for four consecutive days. The reason for choosing to administer Ang-(1–7) twice per day, and close to the allergen challenge, was mainly due to the short half-life of Ang-(1–7).
In the second study, four treatment groups (of n = 11–18) were established. Groups 1 and 2 were treated (i.p.) with the vehicle (water) for A779 and then 1 h later treated with the vehicle (water) for Ang-(1–7) and 30 min; thereafter, mice were challenged intranasally with PBS and ovalbumin respectively. One hour subsequently, these mice were treated with the vehicle (water) for Ang-(1–7). Group 3 was treated (i.p.) with the vehicle (water) for A779 and 1 h later treated with Ang-(1–7) (0.3 mg·kg−1; i.p.). Thirty minutes thereafter, these mice were challenged intranasally with ovalbumin and 1 h subsequently were treated with Ang-(1–7) (0.3 mg·kg−1; i.p.). Group 4 was treated with A779 (1 mg·kg−1; i.p.), and 1 h later, the mice were treated with Ang-(1–7) (0.3 mg·kg−1; i.p.). Thirty minutes thereafter, mice were challenged intranasally with ovalbumin, and 1 h subsequently, they were treated with Ang-(1–7) 0.3 mg·kg−1; i.p.). The drug/vehicle treatment and PBS/ovalbumin intranasal challenges were continued for four consecutive days. The dose and route of administration of A779 were chosen based on a previous study by our group (Al-Maghrebi et al., 2009).
Bronchoalveolar lavage fluid (BALF) cell counts and differentiation
On day 5, mice were killed with an overdose of halothane. BALF was collected by cannulating the trachea and washing the lungs with saline solution (4 × 0.3 mL each). BALF cells were counted using a particle size counter (Z1 series, Beckman Coulter), and cytospins (Shandon Scientific Ltd, Cheshire, UK) were prepared. Cells were stained with Diff-Quik, and a differential count of 200 cells was performed using standard morphological criteria. Results are expressed as total cell count mL−1 in BALF and differential cells as absolute cell count mL−1.
Lung tissue preparation for histopathology
Lungs from half of the animals from all treatment groups were removed, immersed in 10% formalin, embedded in paraffin wax, routinely processed, sectioned 5 µm thick and stained with haematoxylin and eosin (H&E) and examined for pathological changes under light microscopy as described previously (El-Hashim et al., 2011). Another set of sections was also stained with Masson's Trichrome stains to evaluate fibrillar collagen and connective tissue matrix dispositions. Mucus and mucus-containing goblet cells in the bronchial epithelium were stained with a periodic acid–Schiff (PAS). Three serial sections were mounted on each slide. All the slides were independently scored by three different observers, and the average of these three scores was used in this study to reduce intra-observer variation. Ten sections from each mouse were assessed in this study using a Zeiss 40 microscope. A semiquantitative four-level lung pathology score was used to grade the extent of abnormalities in each microscopic field at 200×. The grading scale is shown below.
Grade degree description:
0 Normal histology.
1 Minimal changes.
2 Moderate changes.
3 Severe changes.
Western blot analyses of IκB−α and ERK1/2
SDS-PAGE gel electrophoresis and Western blotting for total and phosphorylated forms of IκB-α and ERK1/2 were performed. Briefly, after the animals had been killed, lungs from half of the animals from all treatment groups were removed, washed with PBS (pH 7.4) at 4°C and snap-frozen in liquid nitrogen and stored at −80°C. The tissue samples were then pulverized whilst frozen and transferred to lysis buffer (pH 7.6) containing 10 mM Tris-base, 140 mM NaCl, 10 mM Na4P2O7, 1 mM NaF, 1 mM CaCl2, 1 mM MgCl2, 10% glycerol, 1% NP40, 2 mM Na3VO4, 1 mM PMSF, protease inhibitor cocktail and phosphatase inhibitor cocktail. The tissues were then homogenized using Polytron PT 4000 (Kinematica, Switzerland), and the samples were left to lyse completely by incubation on ice-cold shaker for 30 min, centrifuged at 13.4× g for 20 min at 4°C to collect supernatants. Protein concentrations were then measured by Lowry's protein assay. Aliquots containing equal amounts of protein were subjected to SDS-PAGE gel electrophoresis and transferred onto nitrocellulose membranes. Membranes were then incubated with the indicated antibodies and subsequently with appropriate secondary antibodies conjugated to horseradish peroxidase. Immunoreactive bands were detected with SuperSignal chemiluminescent substrate (GE Healthcare, Buckinghamshire, UK) using Kodak autoradiography film (G.R.I., Rayne, UK). To ensure equal loading of proteins, β-actin levels were detected using primary rabbit anti-human β-actin antibody followed by the secondary anti-rabbit IgG horseradish peroxidase-conjugated antibody. Images were analysed and quantified by densitometry (Bio-Rad, Philadelphia, PA, USA).
HPBMC proliferation
Proliferation and cytokine assays were performed as previously described (El-Hashim et al., 2010). Briefly, HPBMCs were separated from peripheral blood of normal control (n = 6) individuals, with no history of allergic disease, by Ficoll–Hypaque density gradient centrifugation. Cells were suspended at 1 × 106 mL−1 in complete culture medium (RPMI 1640 supplemented with 2 mM L-glutamine, 200 IU·mL−1 penicillin, 100 µg·mL−1 streptomycin and 10% FBS; all from Life Technologies, Basel, Switzerland), phenanthroline (1 µM) (peptidase inhibitor) and stimulated with PHA (10 µg mL−1) in the presence and absence of 10−3–10−7 µM Ang-(1–7). Cell proliferation was measured by pulsing cultures with [3H]-thymidine for a period of 18 h after an initial culture of 24 h.
Statistics
All numerical values are expressed as mean ± SEM. Total cell counts represent the number of BALF cells mL−1. Absolute cell counts mL−1 represents the number of each cell type mL−1 of BALF. Data for the HPBMC proliferation experiments are expressed as % of [3H]-thymidine incorporation in control cultures (cultures, which received PHA). The data for these experiments are presented as counts min-1. For the histopathology, a semiquantitative four-level lung pathology score was used to grade the extent of abnormalities in each microscopic field at 200×. Kruskal–Wallis analysis of variance was used to compare mean differences between individual groups, total and differential cell data, histopathological grading and Western blots and a post hoc analysis (Dunn's method) was used to determine if there were differences between individual groups. For the effects of Ang-(1–7) and A779 on PHA-induced proliferation of HPBMCs, Student's paired t-test was used to compare differences. The mean difference was considered as significant at a probability level of less than 0.05. Analysis was performed with Sigma plot/Stat for Windows version 11 (Systat Software Inc., San Jose, CA, USA). For the histolopathology data, the average scores from each group were statistically analysed using SPSS V. 17 software (Evanston, IL, USA).
Results
Effect of Ang-(1–7) on total and differential cell count
Ovalbumin challenge induced a significant (P < 0.05) increase in BALF total cell count (Figure 1A), eosinophils (Figure 1B), lymphocytes (Figure 1C), neutrophils (Figure 1D) and macrophages (Figure 1E). Treatment of mice with Ang-(1–7) (0.03, 0.1 and 0.3 mg·kg−1; i.p.) resulted in a dose-dependent decrease in the total cell count compared with vehicle-treated mice (P < 0.05; Figure 1A). Treatment with Ang-(1–7) also caused a significant (P < 0.05) dose-dependent decrease in eosinophil influx compared with ovalbumin-challenged mice (Figure 1B), lymphocytes (Figure 1C) and neutrophils (Figure 1D).
Figure 1.
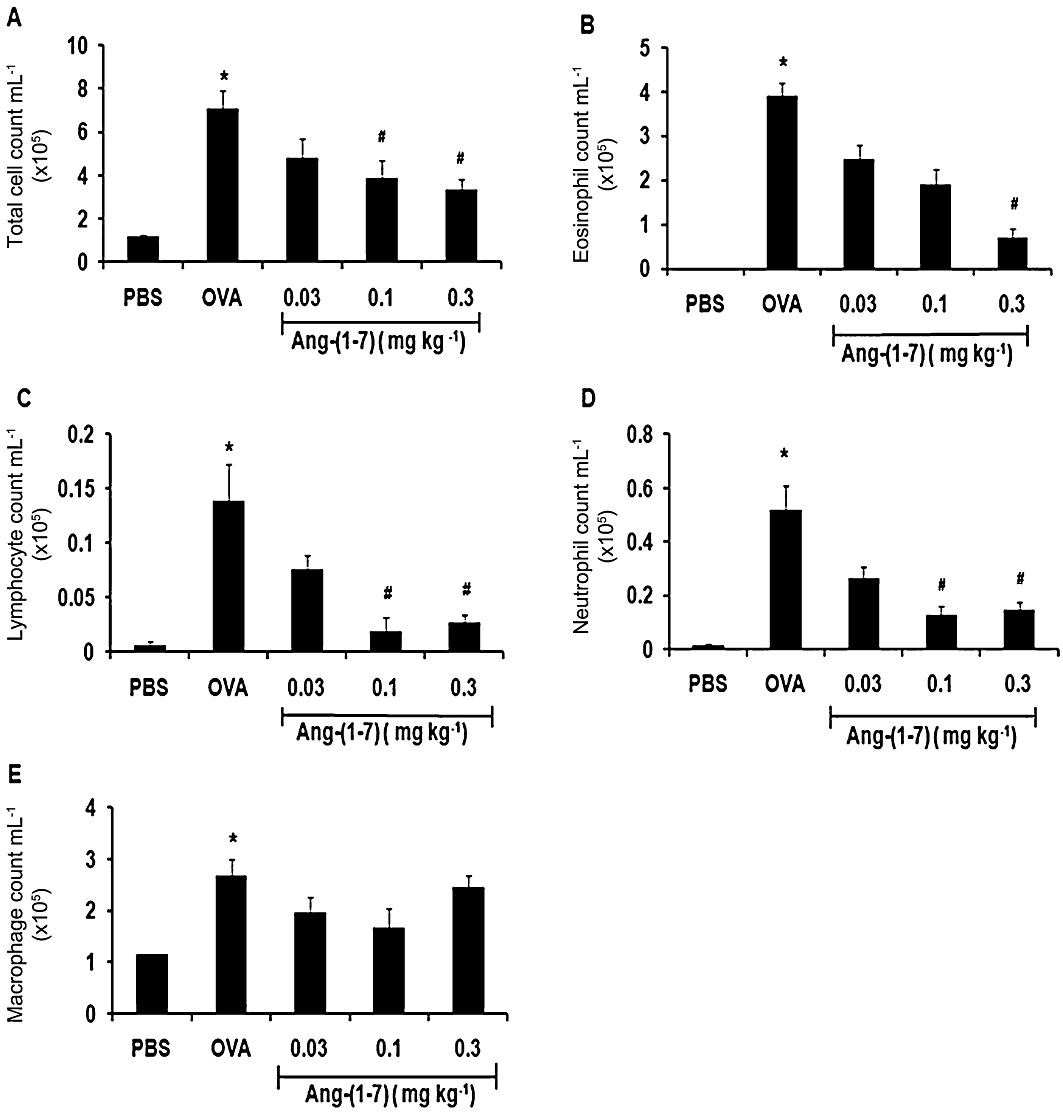
Effect of Ang-(1–7) (0.03, 0.1 and 0.3 mg·kg−1; i.p.) on ovalbumin-induced change in total BALF cell count (A), eosinophils (B), lymphocytes (C), neutrophils (D) and macrophage (E). Treatment with Ang-(1–7) inhibited the ovalbumin-induced increase in total cell influx, eosinophils, lymphocytes and neutrophils in the airways. Data are expressed as mean ± SEM (n = 6–16). *P < 0.05 versus time-matched PBS-challenged mice. #P < 0.05 versus time-matched ovalbumin-challenged mice.
Effect of A779 on Ang-(1–7) -induced decrease in total and differential cell counts
Again, ovalbumin challenge induced a significant (P < 0.05) increase in BALF total cell count (Figure 2A), eosinophils (Figure 2B), lymphocytes (Figure 2C), neutrophils (Figure 2D) and macrophages (Figure 2E). Treatment of mice with Ang-(1–7) (0.3 mg·kg−1; i.p.) resulted in a significant (P < 0.05) decrease in the total cell count (Figure 2A), eosinophil (Figure 2B), lymphocytes (Figure 2C) and neutrophils (Figure 2D) compared with vehicle-treated mice. Treatment with A779 (1 mg·kg−1; i.p.) significantly (P < 0.05) inhibited the Ang-(1–7)-induced decrease in total cell count (Figure 2A), eosinophils (Figure 2B), lymphocytes (Figure 2C) and neutrophils (Figure 2D) compared with vehicle-treated mice. In the ovalbumin-challenged mice treated with A779 alone, there was no significant difference (P > 0.05) between this group and the vehicle-treated ovalbumin-challenged group in either total cell count (5.2 ± 1.2 vs. 5.9 ± 1.2 × 105 mL−1 cells) or differential cell counts (data not shown for differential counts). Additionally, A779 treatment did not significantly affect basal total (2.0 ± 0.2 vs. 1.3 ± 0.1 × 105 mL−1 cells) or differential cell count (data not shown for differential counts) when compared with vehicle-pretreated PBS-challenged mice.
Figure 2.
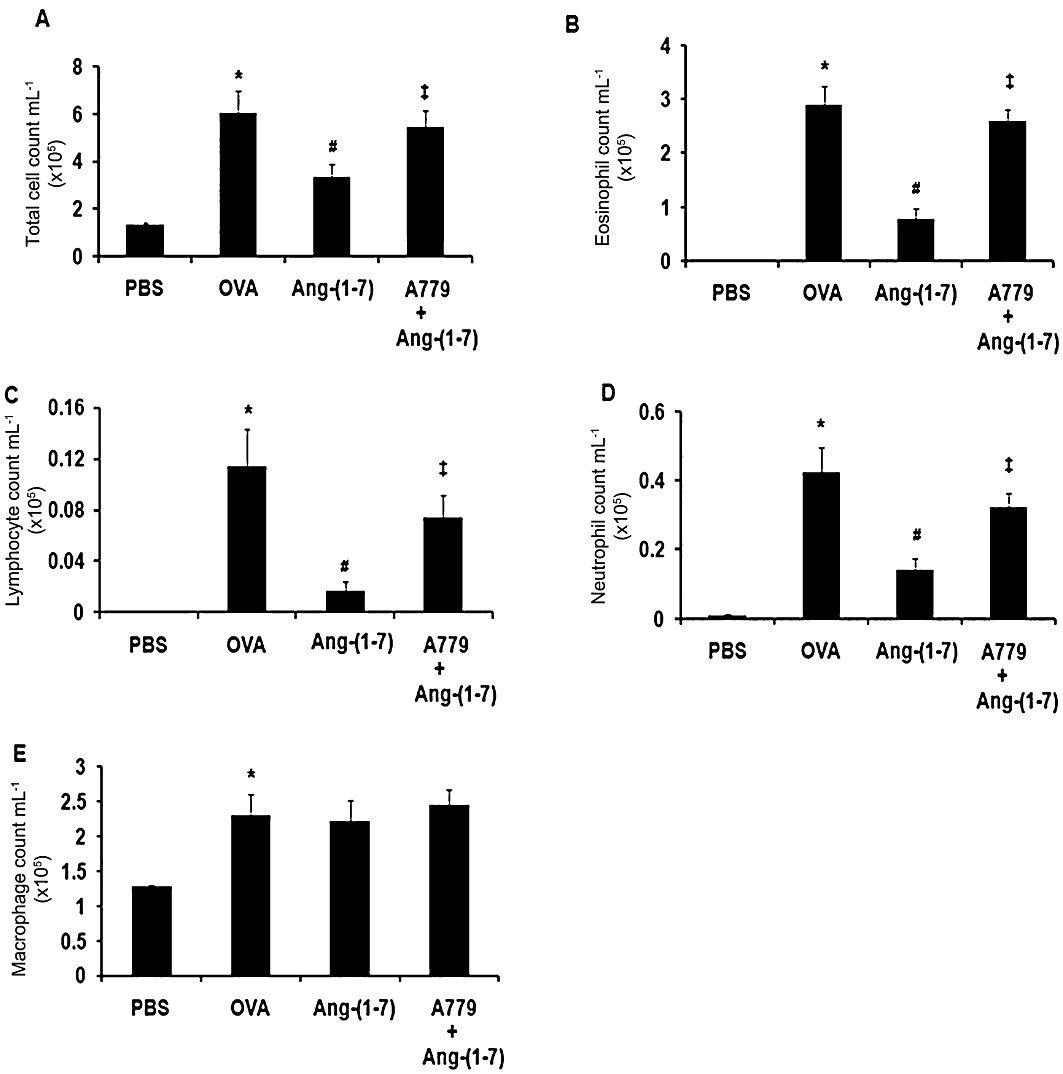
Effect of A779 (1 mg·kg−1; i.p.) on Ang-(1–7) (0.3 mg·kg−1; i.p)-mediated effects on ovalbumin-induced change in total BALF cell count (A), eosinophils (B), lymphocytes (C), neutrophils (D) and macrophage (E). Treatment with A779 significantly attenuated the Ang-(1–7)-mediated inhibition of the ovalbumin-induced increase in total cell influx, eosinophils, lymphocytes and neutrophils in the airways. Data are expressed as mean ± SEM (n = 11–18). *P < 0.05 versus time-matched PBS-challenged mice. #P < 0.05 versus time-matched ovalbumin-challenged mice. ‡P < 0.05 versus time-matched Ang-(1–7)-treated ovalbumin-challenged mice.
Histological changes
In general, H&E-stained lung sections from control mice (immunized with ovalbumin and challenged intranasally with PBS) showed consistently normal histology with the different stains used (Figures 3A, 4A and 5A). However, lung sections from mice that were challenged intranasally with ovalbumin showed severe and marked perivascular and peribronchial inflammatory cell infiltration (Figures 3B, 4B and 5B), severe perivascular and peribronchial fibrosis (Figure 4B) and marked goblet cell hyper/metaplasia (Figure 5B), suggesting airway remodelling. In contrast, lung sections from ovalbumin-challenged mice treated with Ang-(1–7) (0.3 mg·kg−1; i.p.) showed a marked improvement in all the histopathological parameters assessed (Figures 3C, 4C and 5C); there was a significant (P < 0.05) decrease in the perivascular and peribronchial inflammatory cell infiltration (Figure 6A), perivascular and peribronchial fibrosis (Figures 4C and 6B) and goblet cell hyper/metaplasia (Figures 5C and 6C). However, lung sections from mice that were treated with Ang-(1–7) and A779 (1 mg·kg−1; i.p.) had an overall pronounced and severe degree of airway inflammation (Figures 3D, 4D and 5D). There was significant (P < 0.05) perivascular and peribronchial inflammatory cell infiltration (Figures 3D and 6A). There was also evidence of prominent and significant (P < 0.05) perivascular and peribronchial fibrosis (Figures 4D and 6B), and mice also had significant (P < 0.05) goblet cell hyper/metaplasia (Figures 5D and 6C). Changes in this group were very similar to the histological changes noted in the ovalbumin-challenged, vehicle-treated group.
Figure 3.
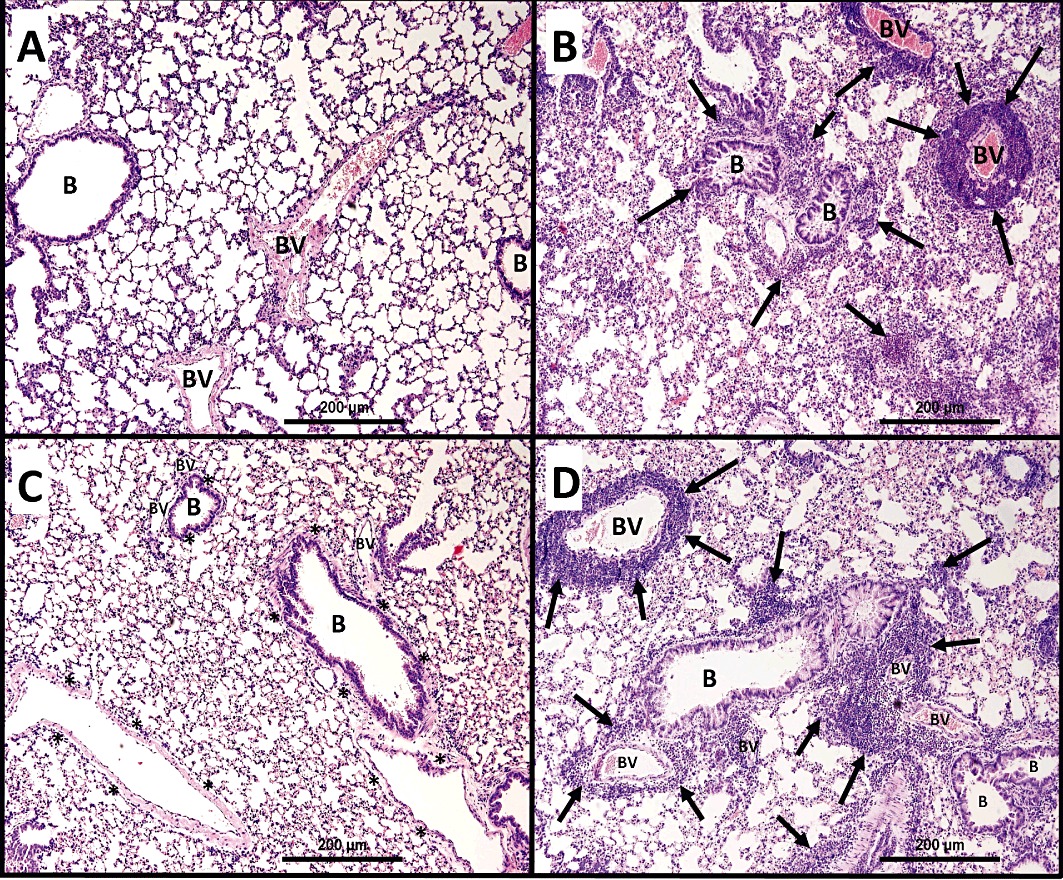
Representative low-magnification light photomicrographs display H&E staining of whole lung samples from (A) PBS vehicle (n = 6), (B) ovalbumin (OVA)-challenged (n = 6), (C) OVA-challenged, Ang-(1–7) (0.3 mg·kg−1; i.p.) treated (n = 6), (D) OVA-challenged, Ang-(1–7) + A779 (1 mg·kg−1; i.p.) (n = 6) treated groups. OVA-challenged/vehicle-treated mice showed marked peribronchial and perivascular inflammatory cell infiltrations (B) (n = 6) compared with PBS-challenged mice (A) (n = 6). Treatment with Ang-(1–7) (0.3 mg·kg−1; i.p.) (C) (n = 6) resulted in significant reduction in the peribronchial and perivascular dark-staining inflammatory cell infiltration and was comparable with PBS vehicle group (A). Treatment with A779 (1 mg·kg−1; i.p.) (D) (n = 6) significantly blocked the Ang-(1–7)-mediated inhibition of the peribronchial and perivascular inflammatory cell infiltrations. B, bronchioles; BV, blood vessels; ( ), marked peribronchial and perivascular inflammatory cell infiltrations.
), marked peribronchial and perivascular inflammatory cell infiltrations.
Figure 4.
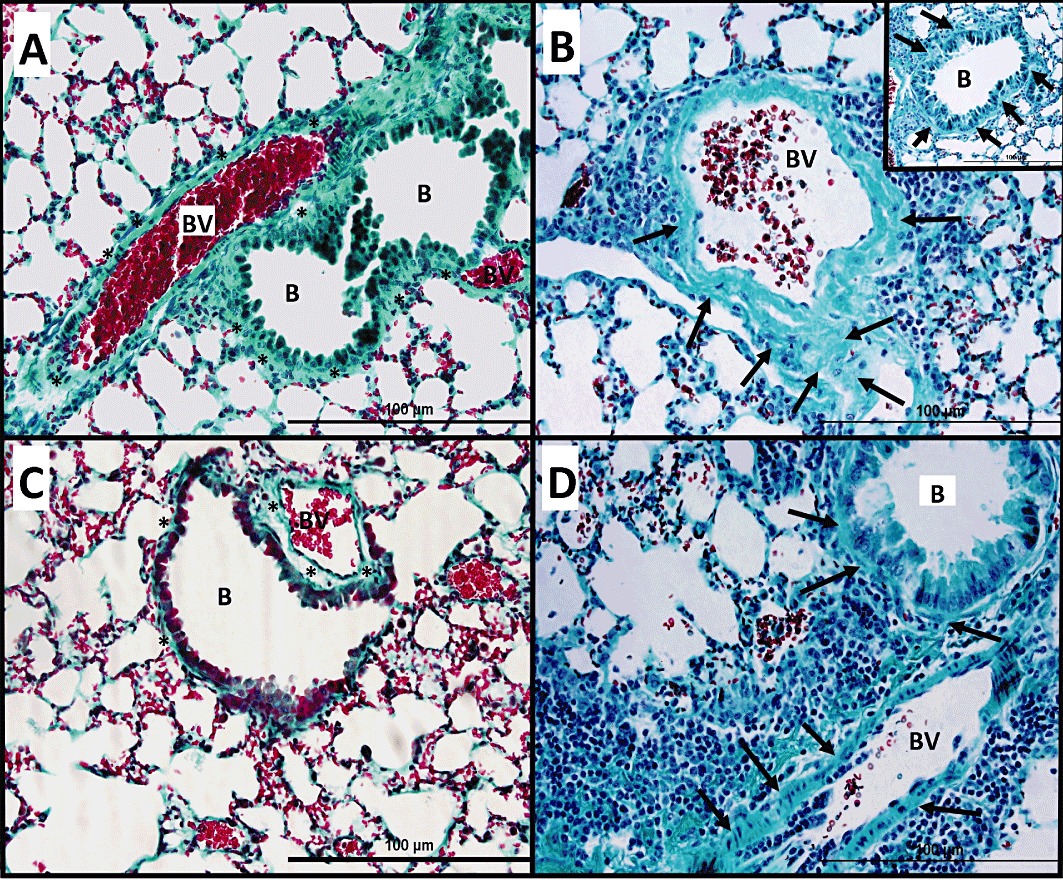
Masson's Trichrome staining of lung samples from ovalbumin-challenged/vehicle-treated mice show significant peribronchial and perivascular fibrosis (B) (n = 6) compared with PBS-challenged mice (A) (n = 6). Treatment with Ang-(1–7) (0.3 mg·kg−1) (C) (n = 6) resulted in a significant reduction in peribronchial and perivascular fibrosis and was similar to in appearance to PBS vehicle group (A). Treatment with A779 (1 mg·kg−1; i.p.) (D) (n = 6) significantly blocked the Ang-(1–7)-induced inhibition of peribronchial and perivascular fibrosis; ( ), significant and remarkable peribronchial and perivascular fibrosis.
), significant and remarkable peribronchial and perivascular fibrosis.
Figure 5.
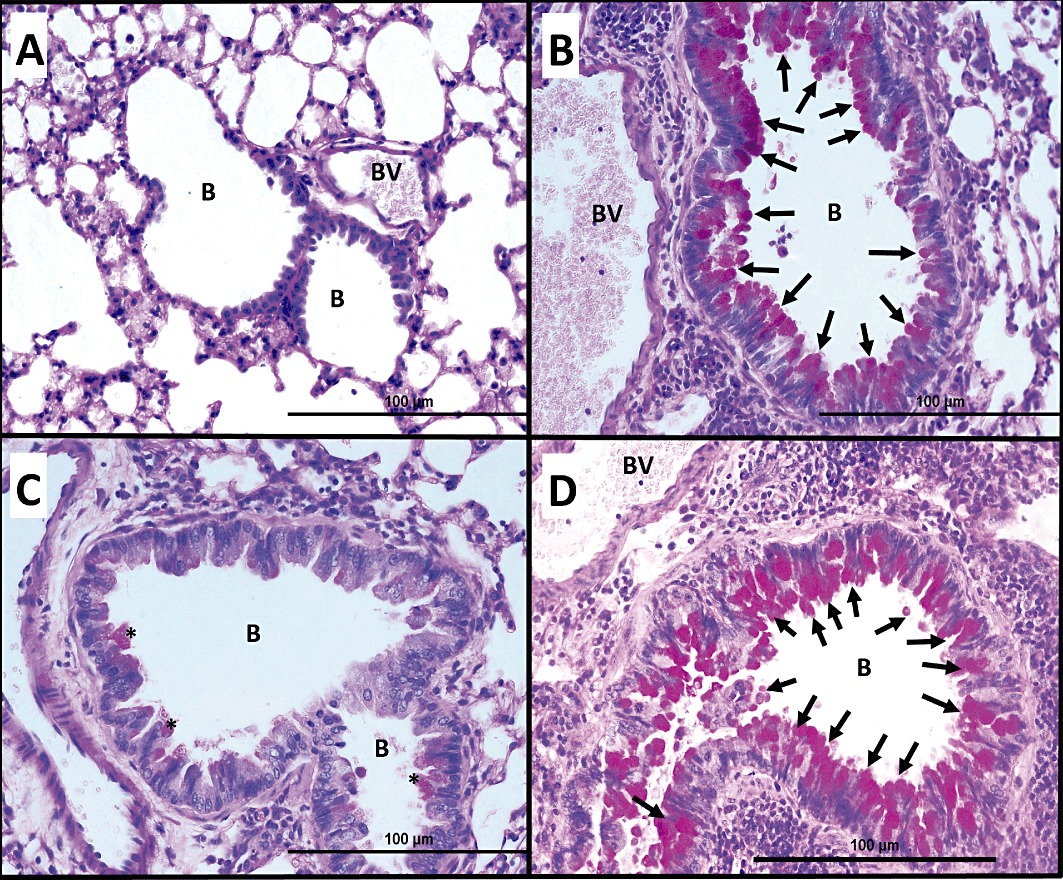
Histological examination of PAS stain of ovalbumin-challenged/vehicle-pretreated mice shows significant bronchial mucus production and goblet cell hyper/metaplasia in mice (B) (n = 6) compared with PBS-challenged mice (A) (n = 6). Treatment with Ang-(1–7) (0.3 mg·kg−1; i.p.) (C) (n = 6) resulted in a significant reduction (asterisks) in bronchial mucus production and goblet cell hyper/metaplasia and was similar to PBS vehicle group (A). Treatment with A779 (1 mg·kg−1; i.p.) (D) (n = 6) significantly blocked the Ang-(1–7)-mediated inhibition of bronchial mucus production and goblet cell hyper/metaplasia in mice; ( ), significant and remarkable bronchial mucus production and goblet cell hyper/metaplasia.
), significant and remarkable bronchial mucus production and goblet cell hyper/metaplasia.
Figure 6.
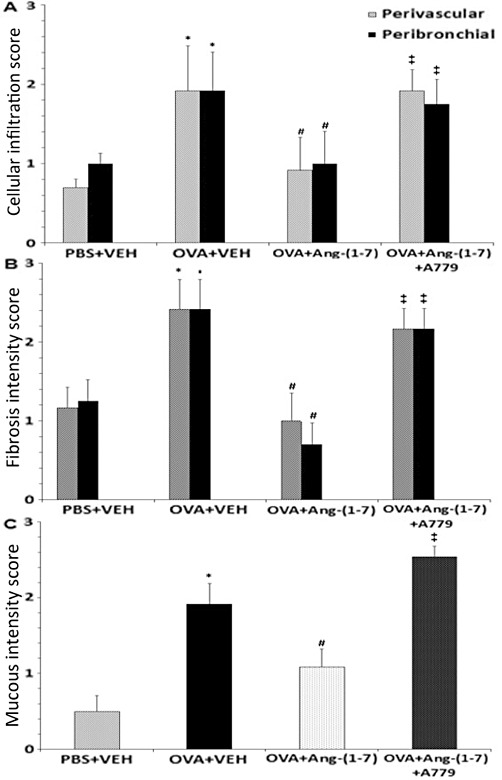
Effect of Ang-(1–7) (0.3 mg·kg−1; i.p), Ang-(1–7) + A779 (1 mg·kg−1; i.p.) on: (A) ovalbumin-induced increase in histological cellular infiltration, (B) ovalbumin-induced increase in peribronchial and perivascular fibrosis and (C) ovalbumin-induced increase in mucus production and goblet cell hyper/metaplasia. Data are expressed as mean ± SEM (n = 6). *P < 0.05 versus time-matched PBS-challenged mice. #P < 0.05 versus time-matched ovalbumin-challenged mice. ‡P < 0.05 versus time-matched Ang-(1–7)-treated ovalbumin-challenged mice.
Effect of Ang-(1–7) and A779 on the ovalbumin-induced increase in p-IκB-α and p-ERK1/2 in the lungs
Ovalbumin challenge significantly increased the levels of p-ERK1/2 compared with the PBS-challenged control animals (Figure 7A,B). Treatment with Ang-(1–7) significantly (P < 0.05) reduced the level of ERK1/2 phosphorylation compared with the ovalbumin-challenged animals. However, treatment with A779 significantly (P < 0.05) prevented the Ang-(1–7)-induced reduction in the level of p-ERK1/2 (Figure 7A,7B). Phosphorylation of serine-32 in p-IκB-α is required for activation of NF-κB activation. Ovalbumin challenge also significantly increased the levels of p-IκB-α compared with the PBS-challenged control animals (Figure 7A,C). Treatment with Ang-(1–7) reduced the level of p-IκB-α (Figure 7A,C). However, treatment with A779 significantly prevented the Ang-(1–7)-induced reduction in the level of p-IκB-α (Figure 7A,C).
Figure 7.
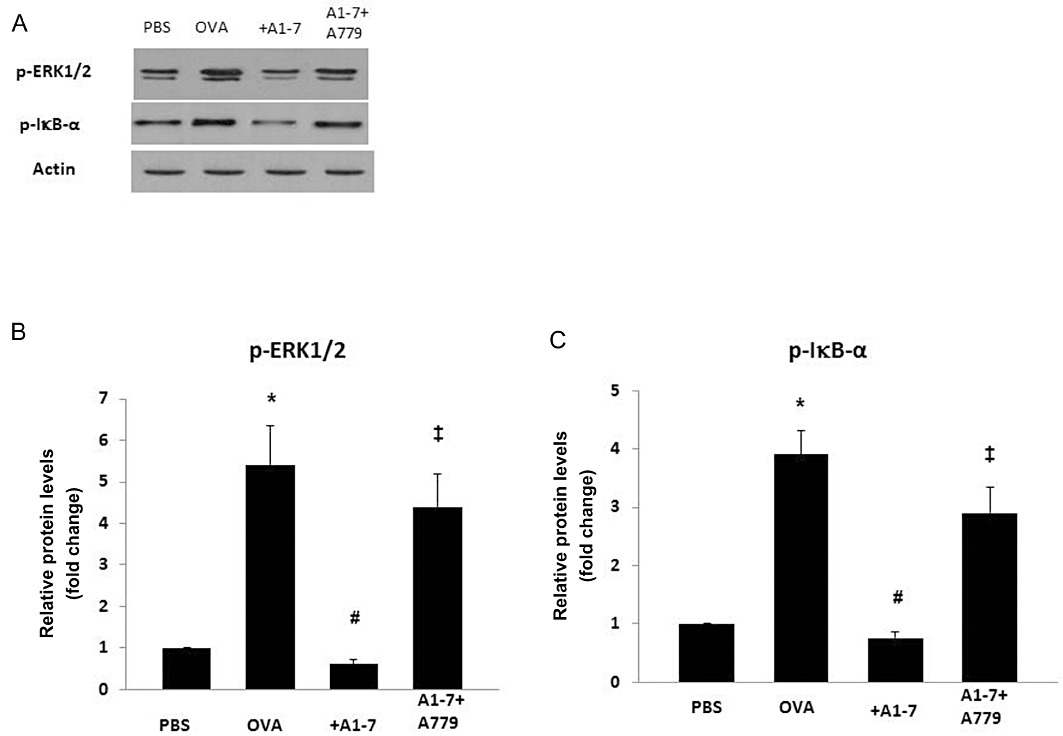
Western blot analysis of p-IκB-α and p-ERK1/2 protein levels from lungs of PBS-challenged mice pretreated with vehicle (PBS), from ovalbumin (OVA)-challenged mice pretreated with vehicle (OVA), Ang-(1–7) and A779 plus Ang-(1–7). Representative blots (A) and densitometric quantification of at least three independent experiments (n = 3) showing relative levels of p-ERK1/2 (normalized to β-actin) (B), and relative levels of p-IκB-α (data normalized to β-actin shown as a ratio of phosphorylated to total IκB-α protein) (C). Data shown represent the mean ± SEM. *P < 0.05 versus time-matched PBS-challenged mice. #P < 0.05 versus time-matched ovalbumin-challenged mice. ‡P < 0.05 versus time-matched Ang-(1–7)-treated ovalbumin-challenged mice.
Antiproliferative effects of Ang-(1–7) on PHA-stimulated HPBMC
To investigate the effects of Ang-(1–7) on the proliferative response of HPBMC, the cells were incubated with PHA. Compared with control, stimulation with PHA resulted in the induction of strong proliferative responses (Table 1). Treatment with Ang-(1–7) (0.1 µM) resulted in a significant (P < 0.05) 33% inhibition of the PHA-induced proliferative response (Table 1). However, pretreatment with A779 blocked the Ang-(1–7)-mediated inhibition of the PHA-induced HBPMC proliferation.
Table 1.
HPBMC proliferation in response to stimulation with PHA
| Control | PHA | PHA + Ang-(1–7) (0.1 µM) | PHA + Ang-(1–7) + A779 (1 µM) | |
|---|---|---|---|---|
| Proliferation (counts min−1) | 85.5 ± 14.0 | 7647.7 ± 998.0 | 5115.0 ± 747.1* | 7379.8 ± 916.8# |
HPBMC response was assessed by pulsing cultures with [3H]-thymidine and proliferation measured as counts min-1.
Data are mean ± SEM;
P < 0.05 versus PHA,
P < 0.05 versus Ang-(1–7) only-treated group (n = 6).
To ascertain whether the inhibition of proliferation was due to cell death, induced by Ang-(1–7), we performed the trypan blue dye exclusion test and confirmed that there was no difference in cell viability in the presence and absence of Ang-(1–7).
Discussion
The major finding of this study is that Ang-(1–7) has anti-inflammatory actions in a model of allergic asthma. Ang-(1–7) treatment resulted in inhibition of the ovalbumin-induced increase in total cell counts, eosinophils, lymphocytes and neutrophils. Ang-(1–7) also significantly reduced the ovalbumin-induced perivascular and peribronchial inflammation, fibrosis and goblet cell hyper/metaplasia. These effects of Ang-(1–7) were mediated via the MAS1 receptor as treatment with the MAS1 receptor blocker, A779, significantly inhibited Ang-(1–7)-induced effects on the total, differential cells and the airway histopathological changes. Additionally, Ang-(1–7) reduced the ovalbumin-induced increase in the level of phosphorylation of ERK 1/2 and IκB-α. Ang-(1–7) also inhibited PHA-stimulated proliferation of the HPBMCs.
Multiple pathways are involved in airway inflammation in allergic asthma. Several cytokines, such as IL-4, IL-5 and IL-13, have been identified to be important orchestrators and/or effectors of asthma (Barnes, 2008; Holgate, 2010). The importance of these cytokines in asthma is highlighted by several studies demonstrating their critical role in the pathogenesis of asthma (Tanaka et al., 2004; Karras et al., 2007; Finkelman et al., 2010; Hansbro et al., 2011). Furthermore, the identification of a pro-asthmatic role for cytokines such as the IL-17 (IL17 A, E and F) has also opened up new targets for potential therapeutic intervention (Doe et al., 2010). Moreover, several clinical studies have evaluated various cytokine-neutralizing strategies as potential asthma therapy (Kips et al., 2003; Howarth et al., 2005; Wenzel et al., 2007). However, despite the significant increase in the understanding of the mechanisms underlying allergic asthma and the increase in the cytokine network family involved, results from clinical trials, where specific cytokines were targeted, have been rather disappointing. A possible reason for this is that these cytokines often exhibit redundancy in their function. An alternative approach to the targeting of single pro-inflammatory mediators may therefore be through the use of agonists that activate anti-inflammatory signalling pathways (Haworth and Levy, 2007). A potential candidate molecule may be the peptide Ang-(1–7), the actions of which have only recently begun to be unravelled.
The DRC data show that Ang-(1–7) has anti-inflammatory effects as evidenced by the decrease in total cell count, eosinophils, lymphocytes and neutrophils. The highest effect of Ang-(1–7) was noted at the 0.3 mg·kg−1 dose. This dose was therefore chosen for the subsequent in vivo study with the MAS1 receptor antagonist, A779.
Similar to the DRC experiments, data from the second study again showed that ovalbumin induced an inflammatory response in the BALF and also increased goblet cell hyper/metaplasia, perivascular and peribronchial fibrosis and inflammatory cell infiltration. Treatment with Ang-(1–7) significantly reduced the ovalbumin-induced BALF cellularity, the perivascular and peribronchial fibrosis, inflammatory cell infiltration and the goblet cell hyper/metaplasia, suggesting that Ang-(1–7) has significant anti-inflammatory actions. Treatment with the A779 almost completely reversed the Ang-(1–7) effects on BALF cellularity and histopathological changes, thus suggesting that the actions of Ang-(1–7) are mediated via the MAS1 receptor. Our data also showed that treating ovalbumin-challenged mice with A779 alone did not affect the ovalbumin-induced BALF cellularity, suggesting the effects of A779 are due to blockade of the exogenously administered Ang-(1–7) and not the endogenous Ang-(1–7) (data not shown). This is the first report showing an anti-asthmatic effect with Ang-(1–7). Our findings are consistent with the recent reports showing that Ang-(1–7) is an anti-inflammatory agent in experimental models of arthritis and in cardiovascular end-organ damage associated with diabetes and/or hypertension (Benter et al., 2008; Al-Maghrebi et al., 2009; da Silveira et al., 2010). In MAS1 receptor-deficient antigen-induced arthritic mouse model and an adjuvant-induced arthritis rat model of arthritis, MAS1 receptor activation was shown to be important in reducing inflammatory indices such as tissue cellularity, TNF-α production and oedema (da Silveira et al., 2010). Moreover, our data are consistent with the recent work by Souza and Costa-Neto (2012), which showed that treatment of macrophages with Ang-(1–7), following their exposure to LPS, resulted in a reduced expression of TNF-α and IL-6, an effect that was abolished by A779 and the Src inhibitor, PP2.
To elucidate the mechanisms by which Ang-(1–7) mediates its effects in this allergic model of asthma, we investigated the effects of Ang-(1–7) on two signalling pathways that are involved in asthma, namely ERK1/2 and NF-κB. Many asthma-related cytokines have also been shown to signal through an ERK1/2-dependent pathway, and inhibition of this pathway has been reported to decrease cytokine production by eosinophils (Yamamura et al., 2009). Our data show that ovalbumin challenge significantly increases the levels of p-ERK1/2, consistent with our previous finding (El-Hashim et al., 2011), and that treatment with Ang-(1–7) significantly inhibits this increase, whilst cotreatment with A779 reverses the effects of Ang-(1–7). This finding is consistent with previous studies in models of cardiovascular diseases where Ang-(1–7) was shown to decrease p-ERK1/2 (Giani et al., 2008; Gava et al., 2009). Inhibition of ERK1/2 may therefore be at least partly responsible for the anti-inflammatory effects of Ang-(1–7) in this model of asthma. In line with our finding that a decrease in p-ERK1/2 may be responsible for the decreased inflammation, some studies have reported that IL-17F-induced airway inflammation and remodelling and IL-13-induced increase in mucin5AC may be ERK1/2-dependent (Kawaguchi et al., 2009; Kono et al., 2010). More recently, the importance of ERK1/2 has been further highlighted in a study showing that its sustained activation supports long-term survival of epithelial cells and primes them for cytokine transcription (Alam and Gorska, 2011). Furthermore, another possible mechanism by which Ang-(1–7) may down-regulate the airway inflammation could be via inhibition of the transactivation of the EGF receptor, an interaction that we have recently reported using a model of diabetes (Akhtar et al., 2012).
An important role for NF-κB in inflammatory diseases such as asthma and chronic obstructive pulmonary disease (COPD) is now generally accepted based on the increasing evidence for the involvement of this transcriptional factor in regulating the generation of many inflammatory mediators (Suzuki et al., 2011). Our data show that ovalbumin challenge significantly increases p-IκB-α, which is known to lead to NF-κB activation, in line with what we and other have previously shown (Birrell et al., 2005; El-Hashim et al., 2011). Knockout animal models have provided some of the early evidence showing the importance of NF-κB in inflammation. Mice lacking the c-Rel subunit of NF-κB do not develop asthma-like phenotype following challenge with allergen (Donovan et al., 1999). Ovalbumin sensitization and challenge results in enhanced NF-κB activation particularly in the epithelial cells of the conducting airways and is associated with an increase in the number of NF-κB-regulated chemokines (Poynter et al., 2002). Our data also showed that treatment with Ang-(1–7) inhibited the ovalbumin-induced p-IκB-α, suggesting that Ang-(1–7) has an inhibitory effect on NF-κB. Due to the important role that NF-κB plays in asthma, inhibition of NF-κB by Ang-(1–7) may be central to the inhibition of the ovalbumin-induced asthma phenotype that is seen in this model. This finding is also in line with data from our laboratory showing in a model of combined hypertension and diabetes, that chronic treatment with Ang-(1–7) inhibited cardiac NF-κB and the expression of C3, IL-6, IL-1β, Na 1p12 and Casp 1 in the heart and attenuated ischaemia/reperfusion-induced cardiac dysfunction (Benter et al., 2008; Al-Maghrebi et al., 2009). Furthermore, Ang-(1–7) inhibited NF-κB activity and expression of Th2, and Irak1 in the hearts of a combined model of diabetes and hypertension defining the importance of this pathway in Ang-(1–7)- mediated cardioprotection (Al-Maghrebi et al., 2009).
It is now established that Th2 lymphocytes are implicated in the aetiology of asthma, and they proliferate in response to antigen stimulation as part of their key role in the orchestration of the immune response (Holgate, 2011). Cytokines, such as IL-13, are known to play key roles in allergic inflammation through enhancement of IgE-mediated immune responses and promotion of an eosinophilic response (Barnes, 2008). Also, the expression of Th17 cells/IL-17 has been shown to be correlated with the severity of airway remodelling and treatment with anti-IL-17 induced a marked decrease in ASM mass, mucus production and peribronchial collagen deposition. More recently, Th17 cells have also been shown to play a critical role in animal models of asthma (Wang et al., 2010). Furthermore, expression of the Th17-associated cytokines IL-17A and IL-17F appears to be increased in clinical asthma (Doe et al., 2010). Based on our findings obtained in the experimental model of asthma, we were further interested to determine if the anti-inflammatory effects of Ang-(1–7) could be extended to humans, and hence we studied whether Ang-(1–7) has suppressive effects on HPBMC in terms of proliferation. Our data show that Ang-(1–7) potently suppressed the PHA-stimulated proliferative response of HPBMC. These effects were mediated by the MAS1 receptor as they were completely inhibited by treatment with A779. The degree of Ang-(1–7)-induced inhibition of the PHA-induced proliferation is in line with that seen with other anti-inflammatory and immunosuppressant drugs, such as methylprednisolone and cyclosporine A (Briggs et al., 1996; 1999). It is therefore likely that the antiproliferative effects of Ang-(1–7) may be partly responsible for the anti-inflammatory effects seen in our murine asthma model. This finding is also in line with studies showing that Ang-(1–7) has an antiproliferative effect on human adenocarcinoma and non–small lung cancer cells (Gallagher and Tallant, 2004). The trypan blue dye exclusion test also confirmed that there was no difference in cell viability in the presence and absence of Ang-(1–7), and hence these effects of Ang-(1–7) were not due to toxicity. It is important to note that in the absence of a peptidase inhibitor, such as phenanthroline, there was significant degradation of Ang-(1–7) by peptidases such that high doses of Ang-(1–7) were required to observe these effects (data not shown). Therefore, the inclusion of peptidase inhibitors in such assays significantly lowers the dose of Ang-(1–7) required to produce an inhibitory effect.
In conclusion, our data show that Ang-(1–7) inhibits the ovalbumin-induced airway inflammation, ERK1/2- and NF-κB-dependent signalling in a murine model of inflammation. In addition, Ang-(1–7) significantly inhibited PHA-induced HPBMC proliferation in vitro. These findings suggest that Ang-(1–7) exerts anti-inflammatory activities both in vivo and in vitro. The beneficial effects of Ang-(1–7) were reversed by A779. These results therefore show that the Ang-(1–7)/MAS1 receptor axis is an important mechanism that reverses the inflammatory cascades involved in asthma and activation of this pathway may be a novel therapeutic approach.
Acknowledgments
The authors are thankful to the support of Mini Mathew and Sunny Ojoko, from the Animal Resources Center of Health Sciences Center, and to Ms Bindu Chandrasekhar, Department of Pharmacology and Toxicology, for her assistance with the Western blots. We also appreciate efforts of Prof Samuel Kombian and Seena Mathews, from the Department of Applied Therapeutics, for their help with the preparation of the manuscript. This study was supported by funding from the Department of Pharmacology and Therapeutics, Faculty of Pharmacy, Kuwait University.
Glossary
- A779
D-Ala7-Ang-(1–7)
- Ang-(1–7)
angiotensin-(1–7)
- BALF
bronchoalveolar lavage fluid
- COPD
chronic obstructive lung disease
- cpm
counts per min
- H&E
haematoxylin and eosin
- HPBMC
human peripheral blood mononuclear cells
- NOX
NADPH oxidase
- PAS
periodic acid–Schiff
- SHP-1
src homology-2 containing protein-tyrosine phosphatase-1
- VIP
vasoactive intestinal peptide
Conflicts of interest
All authors declare that they have no conflicts of interest.
References
- Akhtar S, Yousif MH, Dhaunsi GS, Chandrasekhar B, Al-Farsi O, Benter IF. Angiotensin-(1-7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. Br J Pharmacol. 2012;165:1390–1400. doi: 10.1111/j.1476-5381.2011.01613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam R, Gorska MM. Mitogen-activated protein kinase signalling and ERK1/2 bistability in asthma. Clin Exp Allergy. 2011;41:149–159. doi: 10.1111/j.1365-2222.2010.03658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Maghrebi M, Benter IF, Diz DI. Endogenous angiotensin-(1-7) reduces cardiac ischemia-induced dysfunction in diabetic hypertensive rats. Pharmacol Res. 2009;59:263–268. doi: 10.1016/j.phrs.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benter IF, Ferrario CM, Morris M, Diz DI. Antihypertensive actions of angiotensin-(1-7) in spontaneously hypertensive rats. Am J Physiol. 1995;269:H313–H319. doi: 10.1152/ajpheart.1995.269.1.H313. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Anim JT, Cojocel C, Diz DI. Angiotensin-(1-7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Am J Physiol Heart Circ Physiol. 2006;290:H684–H691. doi: 10.1152/ajpheart.00632.2005. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H666–H672. doi: 10.1152/ajpheart.00372.2006. [DOI] [PubMed] [Google Scholar]
- Benter IF, Yousif MH, Dhaunsi GS, Kaur J, Chappell MC, Diz DI. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am J Nephrol. 2008;28:25–33. doi: 10.1159/000108758. [DOI] [PubMed] [Google Scholar]
- Birrell MA, Hardaker E, Wong S, McCluskie K, Catley M, De Alba J, et al. Ikappa-B kinase-2 inhibitor blocks inflammation in human airway smooth muscle and a rat model of asthma. Am J Respir Crit Care Med. 2005;172:962–971. doi: 10.1164/rccm.200412-1647OC. [DOI] [PubMed] [Google Scholar]
- Briggs WA, Gao ZH, Gimenez LF, Scheel PJ, Choi MJ, Burdick JF. Lymphocyte responsiveness to glucocorticoids, cyclosporine, or both. J Clin Pharmacol. 1996;36:707–714. doi: 10.1002/j.1552-4604.1996.tb04239.x. [DOI] [PubMed] [Google Scholar]
- Briggs WA, Eustace J, Gimenez LF, Choi MJ, Scheel PJ, Burdick JF. Lymphocyte suppression by glucocorticoids with cyclosporine, tacrolimus, pentoxifylline, and mycophenolic acid. J Clin Pharmacol. 1999;39:125–130. doi: 10.1177/00912709922007660. [DOI] [PubMed] [Google Scholar]
- Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension. 2007;50:596–599. doi: 10.1161/HYPERTENSIONAHA.106.076216. [DOI] [PubMed] [Google Scholar]
- Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138:1140–1147. doi: 10.1378/chest.09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan CE, Mark DA, He HZ, Liou HC, Kobzik L, Wang Y, et al. NF-kappa B/Rel transcription factors: c-Rel promotes airway hyperresponsiveness and allergic pulmonary inflammation. J Immunol. 1999;163:6827–6833. [PubMed] [Google Scholar]
- El-Hashim A, Yousefi S, Edafiogho I, Raghupathy R, Yousif M, Simon HU. Anti-inflammatory and immunosuppressive effects of the enaminone E121. Eur J Pharmacol. 2010;632:73–78. doi: 10.1016/j.ejphar.2009.12.004. [DOI] [PubMed] [Google Scholar]
- El-Hashim AZ, Renno WM, Abduo HT, Jaffal SM, Akhtar S, Benter IF. Effect of inhibition of the ubiquitin-proteasome-system and IkappaB kinase on airway inflammation and hyperresponsiveness in a murine model of asthma. Int J Immunopathol Pharmacol. 2011;24:33–42. doi: 10.1177/039463201102400105. [DOI] [PubMed] [Google Scholar]
- Finkelman FD, Hogan SP, Hershey GK, Rothenberg ME, Wills-Karp M. Importance of cytokines in murine allergic airway disease and human asthma. J Immunol. 2010;184:1663–1674. doi: 10.4049/jimmunol.0902185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PE, Tallant EA. Inhibition of human lung cancer cell growth by angiotensin-(1-7) Carcinogenesis. 2004;25:2045–2052. doi: 10.1093/carcin/bgh236. [DOI] [PubMed] [Google Scholar]
- Gava E, Samad-Zadeh A, Zimpelmann J, Bahramifarid N, Kitten GT, Santos RA. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant. 2009;24:1766–1773. doi: 10.1093/ndt/gfn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giani JF, Gironacci MM, Munoz MC, Pena C, Turyn D, Dominici FP. Angiotensin-(1 7) stimulates the phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: role of the AT1 and Mas receptors. Am J Physiol Heart Circ Physiol. 2007;293:H1154–H1163. doi: 10.1152/ajpheart.01395.2006. [DOI] [PubMed] [Google Scholar]
- Giani JF, Gironacci MM, Munoz MC, Turyn D, Dominici FP. Angiotensin-(1-7) has a dual role on growth-promoting signalling pathways in rat heart in vivo by stimulating STAT3 and STAT5a/b phosphorylation and inhibiting angiotensin II-stimulated ERK1/2 and Rho kinase activity. Exp Physiol. 2008;93:570–578. doi: 10.1113/expphysiol.2007.014269. [DOI] [PubMed] [Google Scholar]
- Hansbro PM, Kaiko GE, Foster PS. Cytokine/anti-cytokine therapy – novel treatments for asthma? Br J Pharmacol. 2011;163:81–95. doi: 10.1111/j.1476-5381.2011.01219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth O, Levy BD. Endogenous lipid mediators in the resolution of airway inflammation. Eur Respir J. 2007;30:980–992. doi: 10.1183/09031936.00005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate ST. A look at the pathogenesis of asthma: the need for a change in direction. Discov Med. 2010;9:439–447. [PubMed] [Google Scholar]
- Holgate ST. Asthma: a simple concept but in reality a complex disease. Eur J Clin Invest. 2011;41:1339–1352. doi: 10.1111/j.1365-2362.2011.02534.x. [DOI] [PubMed] [Google Scholar]
- Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, et al. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–1018. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras JG, Crosby JR, Guha M, Tung D, Miller DA, Gaarde WA, et al. Anti-inflammatory activity of inhaled IL-4 receptor-alpha antisense oligonucleotide in mice. Am J Respir Cell Mol Biol. 2007;36:276–285. doi: 10.1165/rcmb.2005-0456OC. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M, Fujita J, Kokubu F, Huang SK, Homma T, Matsukura S, et al. IL-17F-induced IL-11 release in bronchial epithelial cells via MSK1-CREB pathway. Am J Physiol Lung Cell Mol Physiol. 2009;296:L804–L810. doi: 10.1152/ajplung.90607.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kips JC, O'Connor BJ, Langley SJ, Woodcock A, Kerstjens HA, Postma DS, et al. Effect of SCH55700, a humanized anti-human interleukin-5 antibody, in severe persistent asthma: a pilot study. Am J Respir Crit Care Med. 2003;167:1655–1659. doi: 10.1164/rccm.200206-525OC. [DOI] [PubMed] [Google Scholar]
- Kono Y, Nishiuma T, Okada T, Kobayashi K, Funada Y, Kotani Y, et al. Sphingosine kinase 1 regulates mucin production via ERK phosphorylation. Pulm Pharmacol Ther. 2010;23:36–42. doi: 10.1016/j.pupt.2009.10.005. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie W, Yan H, Li S, Zhang Y, Yu F, Zhu W, et al. Angiotensin-(1-7) enhances angiotensin II induced phosphorylation of ERK1/2 in mouse bone marrow-derived dendritic cells. Mol Immunol. 2009;46:355–361. doi: 10.1016/j.molimm.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Poynter ME, Irvin CG, Janssen-Heininger YM. Rapid activation of nuclear factor-kappaB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol. 2002;160:1325–1334. doi: 10.1016/s0002-9440(10)62559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabelo LA, Alenina N, Bader M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res. 2011;34:154–160. doi: 10.1038/hr.2010.235. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silveira KD, Coelho FM, Vieira AT, Sachs D, Barroso LC, Costa VV, et al. Anti-inflammatory effects of the activation of the angiotensin-(1-7) receptor, MAS, in experimental models of arthritis. J Immunol. 2010;185:5569–5576. doi: 10.4049/jimmunol.1000314. [DOI] [PubMed] [Google Scholar]
- Souza LL, Costa-Neto CM. Angiotensin-(1-7) decreases LPS-induced inflammatory response in macrophages. J Cell Physiol. 2012;227:2117–2122. doi: 10.1002/jcp.22940. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Ogawa M, Muto S, Itai A, Isobe M, Hirata Y, et al. Novel IkB kinase inhibitors for treatment of nuclear factor-kB-related diseases. Expert Opin Investig Drugs. 2011;20:395–405. doi: 10.1517/13543784.2011.559162. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Komai M, Nagao K, Ishizaki M, Kajiwara D, Takatsu K, et al. Role of interleukin-5 and eosinophils in allergen-induced airway remodeling in mice. Am J Respir Cell Mol Biol. 2004;31:62–68. doi: 10.1165/rcmb.2003-0305OC. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li H, Yao Y, Xia D, Zhou J. The overexpression of heparin-binding epidermal growth factor is responsible for Th17-induced airway remodeling in an experimental asthma model. J Immunol. 2010;185:834–841. doi: 10.4049/jimmunol.0901490. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- Yamamura K, Adachi T, Masuda T, Kojima Y, Hara A, Toda T. Intracellular protein phosphorylation in eosinophils and the functional relevance in cytokine production. Int Arch Allergy Immunol. 2009;149(Suppl. 1):45–50. doi: 10.1159/000210653. [DOI] [PubMed] [Google Scholar]