

Stem cells play a critical role during embryonic development and in the maintenance of homeostasis in adult individuals. The retinoblastoma tumor suppressor RB controls the proliferation, differentiation, and survival of cells, and accumulating evidence points to a central role for RB activity in the biology of stem and progenitor cells. In this review by Sage, recent studies investigating the role of RB in embryonic stem cells, adult stem cells, and progenitor cells in plants and mammals is discussed.
Keywords: E2F, ES cells, RB, p107, p130, stem cells
Abstract
Stem cells play a critical role during embryonic development and in the maintenance of homeostasis in adult individuals. A better understanding of stem cell biology, including embryonic and adult stem cells, will allow the scientific community to better comprehend a number of pathologies and possibly design novel approaches to treat patients with a variety of diseases. The retinoblastoma tumor suppressor RB controls the proliferation, differentiation, and survival of cells, and accumulating evidence points to a central role for RB activity in the biology of stem and progenitor cells. In some contexts, loss of RB function in stem or progenitor cells is a key event in the initiation of cancer and determines the subtype of cancer arising from these pluripotent cells by altering their fate. In other cases, RB inactivation is often not sufficient to initiate cancer but may still lead to some stem cell expansion, raising the possibility that strategies aimed at transiently inactivating RB might provide a novel way to expand functional stem cell populations. Future experiments dedicated to better understanding how RB and the RB pathway control a stem cell's decisions to divide, self-renew, or give rise to differentiated progeny may eventually increase our capacity to control these decisions to enhance regeneration or help prevent cancer development.
Basic functions of the RB pathway
The human retinoblastoma gene RB1 was initially cloned from children with a rare form of eye cancer of the same name. Since this seminal discovery, RB has been found to be inactivated in a wide range of pediatric and adult human cancers. The mechanisms of tumor suppression by the RB protein are thought to largely involve its ability to restrict cell cycle progression at the G1/S transition of the cell cycle by inhibition of E2F transcription factors. Phosphorylation of RB by Cyclin/Cdk (cyclin-dependent kinase) complexes can inhibit the ability of RB to bind to E2F. Cyclin/Cdk complexes are themselves under the control of small cell cycle inhibitors of the INK4 and CIP/KIP families, to which p16Ink4a and p21Cip1, respectively, belong. The module comprising INK4–CIP/KIP cell cycle inhibitors; Cyclin/Cdk complexes; RB and its two family members, p107 and p130; and E2F transcription factors constitutes the RB pathway in cells. A second critical component of RB's control of the G1/S progression is nontranscriptional and connects RB to p27Kip1 stabilization via Cdh1/APC and Skp2. RB has been shown to control many other cellular processes in addition to cell cycle progression in G1, including cellular differentiation, by functionally interacting with transcription factors important for the development of specific developmental lineages. Beyond its direct control of the transcription of programs of genes involved in proliferation and differentiation, RB can also interact with chromatin remodeling enzymes, which may be important for its ability to regulate global gene expression. Finally, strong evidence has emerged that RB may also control genomic stability in cells through various mechanisms, including regulating the expression of genes involved in mitosis, but also by directly interacting with proteins involved in maintaining the structure of chromosomes during G2/M. Importantly, these main cellular functions of RB are conserved in mammalian cells, Drosophila melanogaster, and Caenorhabditis elegans. The general mode of action of RB has been thoroughly reviewed (Weinberg 1995; Sherr and McCormick 2002; Iaquinta and Lees 2007; Burkhart and Sage 2008; van den Heuvel and Dyson 2008; Chinnam and Goodrich 2011; Viatour and Sage 2011; Manning and Dyson 2012) and is summarized in Figure 1.
Figure 1.

Classical view of the RB pathway and RB function in mammalian cells. (A) Schematic representation of the canonical RB pathway. Small cell cycle inhibitors prevent the phosphorylation of RB and its family members by Cyclin/Cdk complexes. E2F transcription factors are major mediators of RB function in mammalian cells. (B) Examples of RB's mode of action in cells to restrain cell cycle progression and promote differentiation. (Top) Classically, RB binds to the regulatory regions of cell cycle genes via its interaction with E2F transcription factors and recruits repressor complexes to inhibit the expression of cell cycle genes. (Second from top) RB also controls the cell cycle by blocking the degradation of the p27 small cell cycle inhibitor (of the p21 family) by Skp2; high p27 levels can directly inhibit Cdk2 kinase activity and slow cell cycle progression. (Third from top) RB can promote differentiation in cooperation with other transcription factors, such as Runx2, in the bone lineage. (Bottom) RB can also control cell fate by controlling the expression of transcription factors involved in cellular differentiation, such as PPARγ during adipogenesis.
Many compelling observations over the last 25 years suggest a role for RB in the biology of stem cells. For example, RB has been shown to be crucial to maintain quiescence in cells (Sage et al. 2003), strongly suggesting that it could help maintain quiescence in adult stem cells (Li and Clevers 2010; Li and Bhatia 2011; Pietras et al. 2011). Conversely, when necessary, normally quiescent stem cells must re-enter the cell cycle, self-renew, and produce progenitors, presumably requiring the transient inactivation of RB function to allow passage from G1 to S. Furthermore, RB's capacity to modulate chromatin structure may be important in stem cells to control their plasticity (Chinnam and Goodrich 2011). Here, I review the recent effort by several groups to investigate the potential role of RB in embryonic stem (ES) cells and adult stem and progenitor cells in plants and mammals. It is sometimes difficult to harmonize the functional differences between stem and progenitor cells observed in vivo and in culture, in part because the characterization of stem and progenitor cells in some tissues is still partial. For this review, stem cells are defined as cells that have the ability to perpetuate themselves through self-renewal and generate mature cells through differentiation; progenitors can only divide a limited number of times before generating differentiated cells.
RB in plant stem cells
Given RB's importance in a wide range of human cancers, it is somewhat surprising that the first clear evidence for a role of RB in adult stem cells has come from studies of an RB-like protein in plants. The only member of the RB family that exists in Arabidopsis thaliana is RBR, the RETINOBLASTOMA-RELATED gene. Wildwater et al. (2005) first showed that suppression of RBR function in A. thaliana root meristem stem cells leads to an increase in the number of these “columella” stem cells without affecting the number and structure of their progeny, the differentiated columella cells. Conversely, overexpression of RBR results in the rapid differentiation of these stem cells (Wildwater et al. 2005; Wyrzykowska et al. 2006). Similarly, overexpression of Cyclin D (CycD) or the transcription factor E2Fa results in the accumulation of columella stem cells, while overexpression of the CDK inhibitor KRP2 results in the loss of root stem cells (Wildwater et al. 2005). These experiments suggest that the RB pathway controls the maintenance of the stem cell pool in the root meristem and is dependent on appropriate signals and levels of RB pathway members (Wildwater et al. 2005). Further studies of RBR down-regulation during post-embryonic plant development, including in the root meristem, have extended these initial findings and confirmed a key role for RBR in the coordination of cell cycle progression and differentiation in stem cell populations. Interestingly, the defects observed upon RBR inactivation are reversible once RBR is restored, indicating that, at least in some contexts, RB mutant stem cell populations are not permanently damaged (Borghi et al. 2010). Beyond the root meristem, RBR inactivation leads to an expansion of the stem cell pool in the male germline and delays fate determination (Z Chen et al. 2009). In addition, RBR may normally repress the expression of late embryonic genes and help plant cells switch from an embryonic heterotrophic growth state to autotrophic growth, thereby integrating developmental and metabolic processes (Gutzat et al. 2011). Importantly, most of the functions of RBR in promoting differentiation and constraining cell cycle progression in root stem cells are cell-autonomous (Wachsman et al. 2011).
Together, these data indicate that RB plays a central role in the maintenance of stem cell identity and cell fate determination in plants. Interestingly, RBR can be inactivated in stem cells by viral proteins responsible for the development of tumors in plants, providing a link between the functions of RBR in plant stem cells and its tumor suppressor activity (Doonan and Sablowski 2010).
RB in adult mammalian stem cells
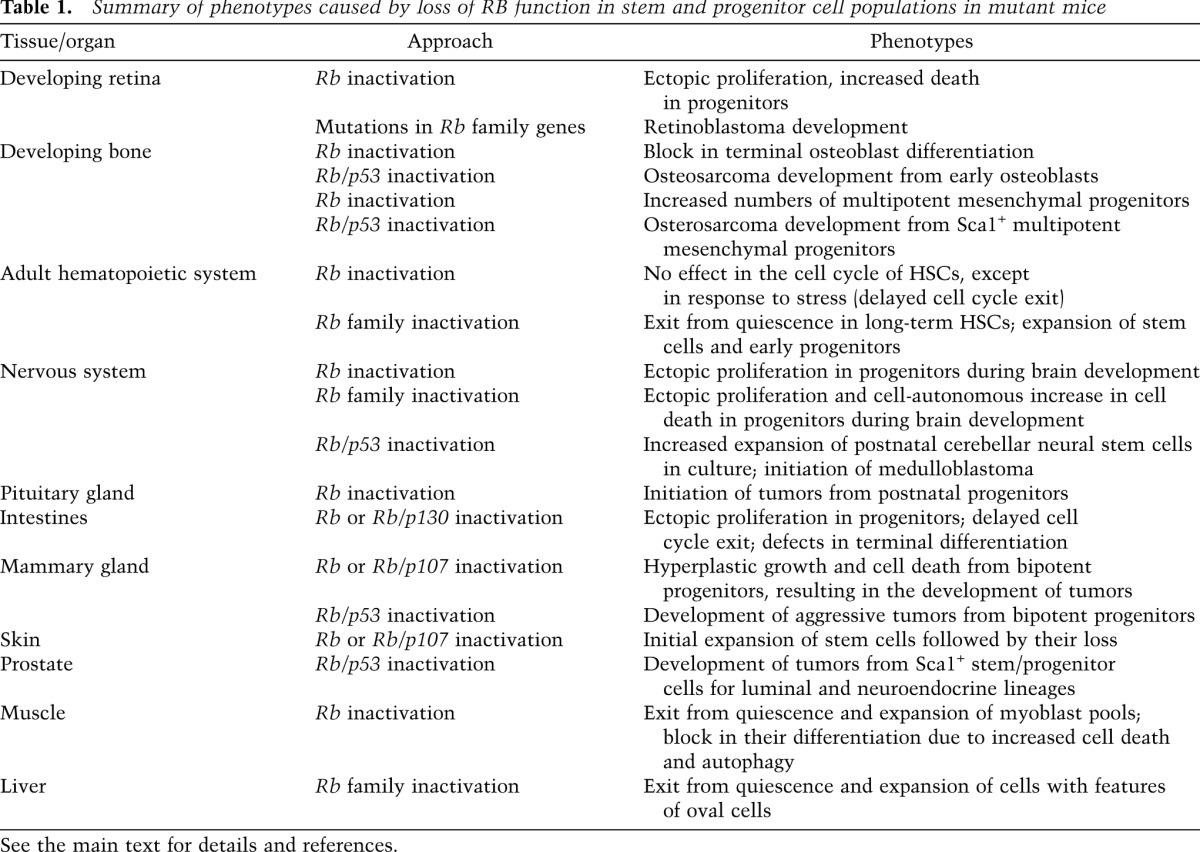
In the last decade, many groups have investigated the mode of action of RB in developing and adult mammalian tissues and organs. These studies have often revealed a crucial role for the RB pathway in the basic biology of stem cells and progenitor cells and have supported a model in which these cell populations are at risk of becoming tumor cells following loss of RB function. A summary of the consequences of RB inactivation in mouse stem and progenitor cells is shown in Table 1.
Table 1.
Summary of phenotypes caused by loss of RB function in stem and progenitor cell populations in mutant mice
Retinal progenitors
The tumor type historically associated with loss of RB1 is retinoblastoma, a rare tumor of the eye that occurs only in young children. The specific age window of retinoblastoma growth in young children suggests a model of tumor initiation in which loss of RB function must happen in specific populations of cells that may be transiently present in the developing retina. This idea is consistent with the expression of progenitor cell markers in tumors (for review, see Dyer and Bremner 2005). On the other hand, coexpression of differentiated and progenitor markers within the same tumor cells (McEvoy et al. 2011), the predominance of markers for more differentiated cells in human tumors (Xu et al. 2009), and the demonstration that post-mitotic retinal cells can initiate tumors in mutant mice (Ajioka et al. 2007) have challenged this view, and the identity of the cell of origin of retinoblastoma is still being debated. Nevertheless, loss of RB function does have visible effects in retinal progenitors. For instance, deletion of the mouse Rb gene in the developing mouse retina results in ectopic proliferation and increased cell death in specific cell populations; further deletion of p107 and/or p130 enhances these defects and is required to initiate retinoblastoma in mice (Chen et al. 2004; MacPherson et al. 2004; McEvoy et al. 2011). Inactivation of the E2f1–3 genes in mouse retinal progenitors further showed that these E2F factors, the activity of which is increased in the absence of RB, play a critical role in the survival of these progenitors—and not, surprisingly, in their capacity to proliferate (D Chen et al. 2009; Chong et al. 2009). In Rb-deficient retinal progenitors, removal of only one allele of the E2f1 gene is sufficient to prevent tumor initiation; furthermore, blockade of Cdk2 activity during a short period of time in young mice is sufficient to prevent retinoblastoma development (Sangwan et al. 2012). Thus, the expansion of Rb mutant retinal progenitors is dependent on increased E2F and Cdk2 activity.
These experiments indicate that too much or too little E2F activity is detrimental to the survival of retinal progenitors in mice. In addition, loss of RB function may increase the proliferative potential of progenitors and delay their exit from the cell cycle. It is still unclear whether loss of RB1 function in patients and loss of Rb family function in mice is sufficient to confer long-term self-renewal to these progenitors, but it is clear that additional genetic and epigenetic events are key for the development of tumors (Conkrite et al. 2011, 2012; Zhang et al. 2012). Finally, loss of RB may increase cell death in specific differentiating populations, thereby perturbing the fate of retinal progenitors during development. The relevance of these observations to the initiation of retinoblastoma in children is still under investigation.
Mesenchymal stem cells and bone progenitors
Because loss of RB1 is also implicated in the initiation of osteosarcoma in teenagers, a number of investigators have studied the mode of action of RB in the bone lineage. The lack of terminal differentiation in osteosarcoma can be explained by the fact that loss of RB itself, but not p107 or p130, blocks late osteoblast differentiation, largely because RB interacts directly with the osteoblast transcription factor Runx2 to control the expression of osteoblast-specific genes (Thomas et al. 2001). Rb inactivation in lineage-committed immature osteoblasts in mice is not sufficient to initiate tumors but potentiates the effects of loss of p53 (Walkley et al. 2008). However, loss of Rb in multipotent mesenchymal progenitors, which can give rise to cells in the bone lineage, leads to increased numbers of these progenitors (Gutierrez et al. 2008), suggesting that stem cells before the preosteoblast stage may also be sensitive to loss of RB and may also serve as a cell of origin for osteosarcoma. Indeed, in mice, deletion of Rb and p53 in mesenchymal stem/progenitor cells expressing the Sca1 marker is sufficient to initiate metastatic tumors closely resembling human osteosarcoma (Berman et al. 2008). Recent data obtained in mouse models of osteosarcoma further indicate that different types of progenitors, uncommitted or committed, can serve as tumor-initiating cells (Choi et al. 2010). These experiments suggest a model in which loss of p53 and RB function in mesenchymal stem cells or early osteoblasts leads to an expansion of these cells and a block in terminal differentiation, thereby initiating osteosarcoma. However, experiments in mutant mice and in cells derived from these mice have shown that loss of Rb in mesenchymal stem cells and early osteoblasts results in increased levels of PPARγ (γ subunit of the peroxisome proliferator-activated receptor), an E2F target (Fajas et al. 2002; Calo et al. 2010). An increase in PPARγ levels in turn alters the differentiation of these progenitors toward the adipocyte lineage, which can lead to the development of brown fat tumors at the expense of osteosarcomas in a p53 mutant background (Hansen et al. 2004; Scime et al. 2005; Calo et al. 2010).
Together, these data clearly demonstrate a role for RB in determining the cell fate decisions of stem cells and early progenitors in the bone lineage as well as their terminal differentiation. These observations also suggest that loss of RB in stem cells and early progenitors can enhance tumor development. However, for loss of RB to contribute specifically to osteosarcoma development (and not the development of adipocytic tumors, for example), the cell of origin for osteosarcoma may have to be a more committed osteoblast cell type, in which loss of RB would prevent terminal differentiation.
Interestingly, while most of the work in the retina has focused on a role for RB and E2F in controlling the proliferation and survival of specific cell populations, including progenitors, data gathered from studies in the bone lineage have highlighted the role of RB as a regulator of cellular differentiation. In both cases, however, changes in the fate of progenitors are observed following loss of RB function.
Hematopoietic stem cells (HSCs) and early progenitors
The hematopoietic compartment has provided a powerful system to explore the mode of action of RB in adult stem cells. An early study suggested that antisense oligonucleotides targeting the RB1 transcript could release human hematopoietic progenitors from a quiescent state in culture (Hatzfeld et al. 1991). However, deletion of the Rb gene in mice did not increase proliferation of HSCs, and Rb mutant HSCs did not have strong self-renewal or differentiation defects in this context (Walkley et al. 2007). In contrast, RB seems to play a critical role in mouse HSCs in response to stress. In the absence of RB, HSCs display a reduced capacity to re-exit the cell cycle after stress-induced proliferation and a diminished ability to replenish the hematopoietic compartment following transplantation. This role for RB is evident in mouse cells with wild-type p107 and p130 (Daria et al. 2008). Additional studies showed that concomitant deletion of Rb and its two family members, p107 and p130, does result in strong phenotypes in HSCs and early hematopoietic progenitors, including exit from quiescence, increased proliferation, and a change in the fate of these cells toward the myeloid lineage (Viatour et al. 2008). Thus, the crucial role of RB in unstressed HSCs may be masked by the compensatory functions of p107 and p130 in these cells, as is the case in many other cell types (Dannenberg and te Riele 2006). Interestingly, this study also suggested that the expansion of abnormally cycling mouse HSCs upon loss of the RB family does not lead to the exhaustion of the stem cell compartment, suggesting that loss of RB function may result in increased proliferation in HSCs without loss of self-renewal. However, HSC homing defects in these mutant mice precluded experiments such as serial transplantation assays to monitor the long-term self-renewal of RB family mutant HSCs (Viatour et al. 2008). Finally, RB may function in the bone marrow microenvironment and control HSC proliferation and differentiation in a non-cell-autonomous manner (Walkley et al. 2007). Interestingly, deletion of the p107 gene in Balb/c mice results in the development of a myeloid hyperplasia with increased number of myeloid progenitors, but it is not known whether HSCs and early progenitors are also affected by the p107 mutation in this genetic background (LeCouter et al. 1998).
A major conclusion from these loss-of-function experiments is that RB normally functions with p107 and p130 to maintain HSCs in a quiescent state and enable HSCs that have been induced to cycle to re-enter a quiescent state. These observations are corroborated by gain-of-function experiments where loss of cyclin D activity in fetal HSCs severely impairs their ability to proliferate (Kozar et al. 2004). Future experiments will need to clarify whether loss of quiescence and increased proliferation are compatible with the long-term self-renewal of HSCs (Yu et al. 2006; Orford and Scadden 2008; Viatour et al. 2008; Zou et al. 2011). In addition, as our understanding of the different subpopulations of HSCs deepens (Wilson et al. 2009), the role of RB and RB pathway members will need to be re-evaluated in these specific subpopulations. Similarly to the experiments focusing on retinal and bone development, increased myeloid differentiation from blood progenitors in Rb family mutant mice further illustrates the changes in cell fate induced by inactivation of RB function in stem/progenitor cells. However, whether the phenotypes observed upon loss of RB function in blood stem/progenitor cells are linked to the development of leukemia is still largely unknown.
Neural stem/progenitor cells
Little is known regarding the role of RB in adult stem cells in the brain. In the developing brain, loss of Rb function was initially reported to cause both ectopic proliferation and increased cell death in progenitors, including in the ventricular zone and the subventricular zone of the developing cortex (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). While the hyperproliferation is cell-autonomous, the increased apoptosis is largely due to low oxygen levels from abnormal placental development (Ferguson et al. 2002; MacPherson et al. 2003; Wu et al. 2003). Interestingly, further analysis of the placenta defects in Rb-deficient embryos also identified a role for RB in trophoblast stem cells, which expand abnormally and fail to differentiate properly in the absence of RB (Wu et al. 2003). Constitutive loss of the Rb family member p107 in neural precursor cells leads to increased proliferation and expansion of these cell populations in adult mice and is accompanied by decreased neuronal differentiation (Vanderluit et al. 2004, 2007). Inactivation of the entire RB family with either viral oncoproteins or genetically engineered alleles in mice leads to stronger cell cycle defects and cell-autonomous increases in cell death (Slack et al. 1998; McLear et al. 2006; Wirt et al. 2010). Loss of RB and p53 in neural stem cells in the cerebellum of mice enhances the proliferative defects, suppresses cell death, and can result in the initiation of medulloblastoma (Sutter et al. 2010). Finally, studies of the polycomb gene Bmi-1 in mouse embryonic neural stem cells suggest that its role in the self-renewal of these cells is via RB and p21 function (Fasano et al. 2007).
Rb mutant mice develop pituitary gland neuroendocrine tumors with a high penetrance. Studies in a mouse model of pituitary adenoma showed that loss of RB in several different types of progenitor cells (e.g., Pax7+ and Myf6+) induces tumor formation, although the latency was much shorter when the Rb gene was inactivated in early (Pax7+) versus late (Myf6+) progenitors (Hosoyama et al. 2010).
Thus, while a specific role for RB in neural/neuroendocrine stem cells and progenitors in embryos and adults is only beginning to be understood, these observations support a model in which RB normally restricts the proliferation of these progenitors and controls their potential for survival and differentiation.
Intestinal stem/progenitor cells
The role of RB in intestinal stem cells is still largely unknown, presumably because the low mutation rate of RB in human colon cancer has deterred investigators from exploring this system in depth. Loss of RB in the intestine leads to increased proliferation, delayed cell cycle exit, abnormal expression of differentiation markers, and increased Hedgehog signaling (Yang and Hinds 2007). In combination with loss of p130, Rb deletion in the small intestine and colon of mice results in chronic hyperplasia and dysplasia of the small intestinal and colonic epithelium, which are associated with defects in terminal differentiation of specific cell types (Haigis et al. 2006). While the normal proliferation of gut progenitors is not dependent on E2F activity, the delayed cell cycle exit observed in Rb mutant mouse intestines is in large part due to the increased activity of activator E2Fs (E2F1–3) (Chong et al. 2009).
Mammary gland stem cells
While adult stem cells and progenitor cells have been well defined in the mammary gland and studied in detail in many contexts, the role of RB and its pathway members in these cells is still surprisingly poorly understood. Rb inactivation in bipotential epithelial progenitors, but not downstream committed progenitors, is sufficient to induce tumor growth in mice. Interestingly, in this case, it is the p53 status that dictates the fate of the mutant progenitors and the type of breast cancer that develops (Jiang et al. 2010).
Skin stem cells
With intestinal and mammary gland stem cells, skin stem cells constitute a third population of well-characterized stem cells in which the mode of RB action is only partly understood. Nevertheless, it seems that Rb deletion in skin stem cells leads to increased proliferation and defective differentiation, which is exacerbated by loss of p107 (Ruiz et al. 2004). In this system, preliminary evidence suggests that loss of RB may eventually lead to the depletion of the stem cell pool (Ruiz et al. 2004), in contrast to observations in the blood compartment (see above). This could possibly be due to increased cell death in these mutant stem/progenitor cells, but further studies will be required to investigate the consequences of RB inactivation in skin stem cells in comparison with other stem cells.
Prostate stem/progenitor cells
While nothing is known about the potential role of RB in prostate stem/progenitor cells, the concomitant loss of Rb and p53 function in adult stem/progenitor cells in mouse prostates results in the development of aggressive prostate tumors, raising the possibility that in this context, too, loss of RB may promote the expansion of stem/progenitor populations that are normally quiescent (Zhou et al. 2007).
Muscle stem/progenitor cells
Satellite cells serve as a postnatal skeletal muscle stem cell reservoir that contributes to muscle repair following injury. Recent studies indicate that quiescence in satellite cells is controlled by RB: Deletion of Rb in these cells leads to cell cycle re-entry and the expansion of myoblast pools (Hosoyama et al. 2011). However, the differentiation of proliferating satellite cells or cycling myoblasts is blocked upon loss of Rb (Huh et al. 2004). RB interacts with MyoD, a crucial regulator of muscle differentiation, and the differentiation block in RB mutant cells was initially attributed to a diminished transcriptional activity of MyoD (Gu et al. 1993). However, recent evidence suggests that the differentiation defect in Rb-deficient muscle cells is in large part due to increased cell death and autophagy (Ciavarra and Zacksenhaus 2010).
Liver oval cells
Recent observations in mouse livers indicate that loss of Rb alone is not sufficient to induce the expansion of the small population of adult stem cells that resides close to bile ducts. In contrast, loss of the entire Rb family in adult mouse livers leads to an exit from quiescence and an expansion of these populations, which then initiate the development of tumors (Viatour et al. 2011). While the functionality of various subpopulations of adult liver stem cells is still unknown, these cells are thought to have the capacity to generate both hepatocytes and bile duct cells (cholangiocytes). The fact that Rb family mutant mice only developed hepatocytic tumors and not bile duct tumors suggests that in this context, too, loss of RB function may alter the fate of adult stem cells (Viatour et al. 2011). In contrast, loss of the p53 tumor suppressor in liver stem cells results in the development of tumors with bilineal differentiation (Katz et al. 2012).
A model for RB function in adult stem and progenitor cells
Together, these experiments indicate that the quiescence of adult stem cells in mouse organs and tissues is normally maintained by RB, often with the help of its two family members, p107 and p130. The data presented above and the decreased proliferation in stem cells such as HSCs observed in Cyclin D and Cyclin A mutant mice (Kozar et al. 2004; Kalaszczynska et al. 2009) further support the notion that the RB family plays a key role in restricting the expansion of progenitor cell populations and controlling their cell cycle exit and differentiation. The hyperproliferative phenotype found in stem and progenitor cells with RB or RB pathway inactivation is often accompanied by cell death and differentiation defects and may ultimately lead to the development of cancer (Fig. 2).
Figure 2.

RB function in adult stem cells. Simplified view of an RB wild-type adult organ, with quiescent populations of stem cells (SCs) giving rise to proliferating progenitor cells (PCs) that can exit the cell cycle to generate differentiated cells (DC) and terminally differentiated cells (TDCs). In an RB mutant (or RB family mutant) context, exit from quiescence and increased numbers are often observed in stem cell populations, and increased proliferation is often present in progenitor cells. In some cases, this increased proliferation is accompanied by increased cell death, especially as progenitors are induced to enter a differentiation program. Loss of RB also leads to a reduced capacity to differentiate and terminally differentiate in some lineages. Finally, RB inactivation may favor certain cellular fates by default (no death and no decrease in differentiation potential) or actively (by activation of a program of genes); however, loss of RB often prevents terminal differentiation. Eventually, although stem cells and progenitor cells may be the cell populations that are initially responsive to loss of RB in adult tissues and organs, tumors initiated by loss of RB may be mostly composed of cells with differentiated characteristics (“differentiated cancer”).
Importantly, the range of these cell cycle, cell survival, and cellular differentiation defects is extremely dependent on the tissue or organ studied. What the molecular basis for these differences is will be an area of exciting future research. In particular, we still know surprisingly little about the downstream mediators of the hyperproliferative defects induced by loss of RB in stem cells and progenitors. A number of studies support the classical model that high E2F and Cdk activity explains the increased proliferation of RB mutant cells (Tsai et al. 1998; Chong et al. 2009; Sangwan et al. 2012), but this idea has not been systematically studied in vivo. Similarly, the death of some progenitors when they are induced to differentiate in the absence of RB is probably in large part dependent on the p53 axis, but this has not been thoroughly investigated.
The molecular basis for the observation that loss of RB function alters the fate of multipotent stem/progenitor cell populations by actively promoting one fate or another is still poorly understood. For example, the increase in neuroendocrine differentiation observed in the lungs of Rb mutant mice is still unexplained (Wikenheiser-Brokamp 2004). Preliminary data concerning intestinal progenitors suggest that loss of Rb leads to an increase in the expression of transcription factors that are known to regulate proliferation and differentiation in the intestinal epithelium; concomitant deletion of Cdx1 and Rb can partly revert the phenotypes of loss of RB function (Haigis et al. 2006). Activation of Notch1 signaling in Rb family mutant liver progenitors may promote hepatocytic differentiation (Viatour et al. 2011). Increased levels of PPARγ may control the proadipogenic switch in Rb mutant mesenchymal progenitors (Calo et al. 2010). These examples illustrate that the mechanisms by which RB controls cell fate in stem/progenitor cells may be extremely dependent on the cellular context. Thus, it will be important to pursue these studies in parallel in multiple stem/progenitor cell types, including in lineages in which RB has been little studied, such as germ cells in the testes or stem cells in the lungs.
RB in ES cells and induced pluripotent stem (iPS) cells
ES cells are a model for pluripotent cells found at early developmental stages in mammalian embryos. Mouse ES cells are thought to represent cells in the inner cell mass of a blastocyst, while human ES cells correspond to a slightly more advanced stage of development, the epiblast (Tesar et al. 2007). iPS cells are generated by the reprogramming of more differentiated cells back to a pluripotent cell state (Stadtfeld and Hochedlinger 2010). While there is still some debate about the similarities and the differences between mouse and human iPS cells and ES cells, I consider here, for the sake of simplicity, that ES and iPS cells all represent a similar pluripotent and actively cycling stem cell type.
Human and mouse ES cells
Fast-dividing cells such as ES cells should have no or little active RB to allow for a rapid progression through G1/S. These cells should also have intact cell cycle machinery to be able to respond to stress and differentiation signals when it is time to slow down and generate mature cells. Indeed, accumulating evidence indicates that the components of the RB pathway are expressed in cycling ES cells, but also that Cyclin/Cdk activity is high in ES cells and that RB and its family members are largely in their hyperphosphorylated state, unable to inhibit E2F transcription factors (for review, see White and Dalton 2005; Conklin and Sage 2009). As would be expected, Rb knockout mouse ES cells have no reported long-term self-renewal and proliferation defects; furthermore, mouse ES cells that are simultaneously triple knockout for Rb and its two family members, p107 and p130, can be propagated normally (Williams et al. 1994; Dannenberg et al. 2000; Sage et al. 2000). However, it is possible that the small pool of active RB molecules in ES cells plays a crucial role in these cells because Rb inactivation results in increased genomic instability in ES cell cultures (Zheng et al. 2002). One model to explain this observation is that ES cells that would arrest and/or die in response to a stress may continue to cycle when RB is absent and that these cells have a higher chance of becoming genomically unstable.
There is no published characterization of human ES cells with inactivation of RB function and the consequences of this inactivation for the genomic stability of these cells or any other aspects of their biology. Thus, it is possible that RB and its family members may play a more significant role in human ES cells, which cycle slower than their mouse counterparts and may have a larger pool of active RB molecules at any given time.
If the consequences of loss of RB function would be expected to be minimal in a cell type that is cycling rapidly, increased RB activity would be predicted to have a strong inhibitory effect in ES cell populations. Surprisingly, the effect of RB overexpression in ES cells has not been reported. However, inhibition of Cdk activity, which may mostly result in RB family hypophosphorylation and activation, has been shown to arrest ES cells (Barta et al. 2010; Neganova et al. 2011). The characterization of this arrest is still preliminary, and the consequences of overactivating RB for the cell cycle, differentiation, and self-renewal of mouse and human ES cells remain poorly understood. Nevertheless, a model is emerging in which increased RB activity in G1 in ES cells may lengthen this phase of the cell cycle and increase the probability of differentiation (Becker et al. 2006; Orford and Scadden 2008; Ying et al. 2008; Sela et al. 2012). This model further suggests that RB activity in ES cells must be tightly controlled: Not enough RB may lead to genomic instability, possibly because of an impaired response to stress, while too much RB will most certainly slow the expansion of cells at a crucial time during embryonic development.
iPS cells
The reprogramming process that transforms “normal” cells into iPS cells is often viewed as a type of dedifferentiation. One would expect that the increased proliferation observed during reprogramming may be accompanied by a reduction in RB activity; thus, loss of RB may promote the generation of iPS cells, similar to what has been observed for p53. In addition, RB is usually viewed as “prodifferentiation,” and its loss of function may contribute to the dedifferentiation of cells during reprogramming. Conversely, increased RB function may inhibit reprogramming. These ideas have not been directly tested yet, but emerging evidence does suggest that RB may normally restrict the reprogramming of fibroblasts into iPS cells. For instance, shRNA molecules against RB1 scored as proreprogramming in a large screen (Samavarchi-Tehrani et al. 2010). In addition, RB is a target of caspases during reprogramming and is inactivated by this cleavage, suggesting that its loss of function is important during this process (Li et al. 2010). Future work should explore the mechanisms by which RB may control cellular reprogramming, whether it simply does so by regulating cell proliferation or plays additional roles in the process. It would also be interesting to know whether these observations relate to RB's role in the differentiation of embryonic and adult stem cells or RB's tumor suppressor role. Nevertheless, controlled RB activity may be important to enhance the reprogramming of cells and ensure that ES cells retain their self-renewal capacity and genomic stability. Future experiments should also determine whether the ectopic expression of RB facilitates the differentiation of ES/iPS cells into specific lineages, which may be important to generate functional cells in transplantation experiments. Finally, there is no information regarding the role of RB in the direct reprogramming of one differentiated cell type to another, such as the reprogramming of fibroblasts into induced neuronal cells (Batista et al. 2011).
A model for RB function in ES cells
As discussed above, little is known regarding the mode of action of RB and its family members in ES cells. A simple model would be that RB activity is low in populations of fast-cycling embryonic cells and that RB activity increases as cells differentiate; in contrast, RB activity is high in differentiated cells and decreases as these cells become reprogrammed into iPS cells (Fig. 3). Future experiments will need to address the mechanisms of action of RB and its family members in ES cells and reprogramming, similar to what has been done in adult stem cells and their differentiated progeny.
Figure 3.

RB function in ES and iPS cells. (A) It is thought that RB and its family members are largely inactivated by hyperphosphorylation due to high Cyclin/Cdk activity in mouse and human ES cells, allowing these cells to self-renew and expand. Proper regulation of RB activity may also be crucial to maintain genomic stability. When ES cells are induced to differentiate, a simple model would be that reduced Cdk activity leads to increased RB activity, which in turn promotes cell cycle exit and differentiation. It is also possible that changes in RB and E2F activity in response to stress control the ability of ES cells to undergo cell death. (B) Emerging evidence suggests that RB activity gradually declines during the reprogramming of differentiated cells into iPS cells, as these cells “dedifferentiate” and acquire the capacity to proliferate indefinitely.
Conclusions
Perhaps not surprisingly, given the universal role attributed to the RB pathway in the control of cell cycle progression in cells, changes in the activity in this pathway often perturbs the biology of embryonic and adult stem cells in plants and mammals.
A major conclusion of the work on adult stem and progenitor cells is that loss of RB function or RB pathway function may contribute to the initiation of cancer from these cell populations. An interesting aspect of these observations is that mouse and human RB-deficient tumors are usually fairly differentiated, suggesting that RB mutant stem/progenitor cells retain some capacity for differentiation as they become transformed. It is still unclear how RB normally controls the fate of stem/progenitor cells and why its loss of function sometimes directs tumors to acquire specific differentiation features, as discussed above for liver, bone, and mammary gland tumors in mice. RB's connection with multiple chromatin remodeling enzymes and complexes are well established (e.g., Brehm et al. 1998; Magnaghi-Jaulin et al. 1998; Nielsen et al. 2001; Ait-Si-Ali et al. 2004; Gonzalo et al. 2005; Trojer et al. 2007), but there are only a few demonstrations of the importance of these interactions in the control of biological processes related to RB function, including cell fate determination (Narita et al. 2003; Wang et al. 2010). It will be interesting to further determine whether the interactions between RB and chromatin-regulating enzymes provide local mechanisms to control gene expression or whether these interactions have a more global function, as would be suggested by experiments with viral oncoproteins that inactivate RB function (Ferrari et al. 2008).
Some genetic studies have already provided evidence for the regenerative potential of loss of RB function in muscle and the inner ear (Sage et al. 2005; Pajcini et al. 2010), but these studies have mostly focused on the capacity of RB-deficient mature cells to dedifferentiate and re-enter the cell cycle and not on stem/progenitor cells. As discussed above, cancer may arise following loss of RB function in stem and progenitor cells, but this output is dependent on the tissue and on how strong the inactivation of the RB pathway is; it could also depend on how long this inactivation lasts. This observation raises the possibility that controlled inactivation of RB or the RB pathway may be used to transiently activate stem/progenitor cells and promote regeneration without significantly increasing the risk of developing cancer. For example, no tumors have ever been observed in the muscles or intestines of Rb-only mutant mice. These two tissues could be considered “low risk” and could be treated with drugs that inactivate RB to increase the number of myofibers from muscle stem cells and help regenerate the intestinal epithelium faster following injury. Thus, one could envision that in the future, two types of drugs will be developed: drugs that directly or indirectly activate RB function to limit the expansion of tumors cells, and drugs that inactivate or mimic RB function to promote cell proliferation when needed. However, before controversial RB-inactivating drugs are developed and used in patients, the analysis of mice with transient loss of RB function—for example, using inducible shRNA strategies (Dow et al. 2012)—could further support the hypothesis that controlled loss of RB in stem cell populations may have beneficial effects for an organism during aging or in response to stress and injury.
Finally, while this review focused on the role of RB in plant and mammalian stem cells, other model organisms such as D. melanogaster, C. elegans, and Schmidtea mediterranea provide powerful systems to study stem cell biology (Pearson and Sanchez Alvarado 2008; Losick et al. 2011; Spradling et al. 2011). Based on the large amount of data gathered in flies and roundworms that have helped elucidate many of the basic functions of RB in the control of proliferation, differentiation, survival, and chromosomal stability, there is little doubt that additional studies focusing on stem cell populations in these organisms will provide novel insights into both the general mode of RB action and fundamental aspects of the biology of stem cells.
Acknowledgments
I thank members of the Sage laboratory and Dr. Monte Winslow for their helpful comments on earlier drafts of this manuscript and apologize for not being able to cite original work of many colleagues due to space constraints. Work in the Sage laboratory on stem cells is supported by the California Institute for Regenerative Medicine (CIRM; contract grant no. RS1-00298-1) and the National Institutes of Health (grant no. 2R01CA114102-06). I am a Leukemia and Lymphoma Society Scholar and a Tashia and John Morgridge Faculty Scholar in Pediatric Translational Medicine.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.193730.112.
References
- Ait-Si-Ali S, Guasconi V, Fritsch L, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A 2004. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J 23: 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, Cicero SA, Boyd K, Zakharenko SS, Dyer MA 2007. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell 131: 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barta T, Vinarsky V, Holubcova Z, Dolezalova D, Verner J, Pospisilova S, Dvorak P, Hampl A 2010. Human embryonic stem cells are capable of executing G1/S checkpoint activation. Stem Cells 28: 1143–1152 [DOI] [PubMed] [Google Scholar]
- Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, et al. 2011. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474: 399–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KA, Ghule PN, Therrien JA, Lian JB, Stein JL, van Wijnen AJ, Stein GS 2006. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol 209: 883–893 [DOI] [PubMed] [Google Scholar]
- Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, Fonhoue BD, Caron A, Bronson R, Bouxsein ML, et al. 2008. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci 105: 11851–11856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghi L, Gutzat R, Futterer J, Laizet Y, Hennig L, Gruissem W 2010. Arabidopsis RETINOBLASTOMA-RELATED is required for stem cell maintenance, cell differentiation, and lateral organ production. Plant Cell 22: 1792–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T 1998. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391: 597–601 [DOI] [PubMed] [Google Scholar]
- Burkhart DL, Sage J 2008. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 8: 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA 2010. Rb regulates fate choice and lineage commitment in vivo. Nature 466: 1110–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Livne-Bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R 2004. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell 5: 539–551 [DOI] [PubMed] [Google Scholar]
- Chen D, Pacal M, Wenzel P, Knoepfler PS, Leone G, Bremner R 2009. Division and apoptosis of E2f-deficient retinal progenitors. Nature 462: 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Hafidh S, Poh SH, Twell D, Berger F 2009. Proliferation and cell fate establishment during Arabidopsis male gametogenesis depends on the Retinoblastoma protein. Proc Natl Acad Sci 106: 7257–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnam M, Goodrich DW 2011. RB1, development, and cancer. Curr Top Dev Biol 94: 129–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Curtis SJ, Roy DM, Flesken-Nikitin A, Nikitin AY 2010. Local mesenchymal stem/progenitor cells are a preferential target for initiation of adult soft tissue sarcomas associated with p53 and Rb deficiency. Am J Pathol 177: 2645–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JL, Wenzel PL, Saenz-Robles MT, Nair V, Ferrey A, Hagan JP, Gomez YM, Sharma N, Chen HZ, Ouseph M, et al. 2009. E2f1–3 switch from activators in progenitor cells to repressors in differentiating cells. Nature 462: 930–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavarra G, Zacksenhaus E 2010. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol 191: 291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H 1992. Requirement for a functional Rb-1 gene in murine development. Nature 359: 328–330 [DOI] [PubMed] [Google Scholar]
- Conklin JF, Sage J 2009. Keeping an eye on retinoblastoma control of human embryonic stem cells. J Cell Biochem 108: 1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkrite K, Sundby M, Mukai S, Thomson JM, Mu D, Hammond SM, MacPherson D 2011. miR-17∼92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev 25: 1734–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkrite K, Sundby M, Mu D, Mukai S, Macpherson D 2012. Cooperation between Rb and Arf in suppressing mouse retinoblastoma. J Clin Invest 122: 1726–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg JH, te Riele HP 2006. The retinoblastoma gene family in cell cycle regulation and suppression of tumorigenesis. Results Probl Cell Differ 42: 183–225 [DOI] [PubMed] [Google Scholar]
- Dannenberg JH, van Rossum A, Schuijff L, te Riele H 2000. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev 14: 3051–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daria D, Filippi MD, Knudsen ES, Faccio R, Li Z, Kalfa T, Geiger H 2008. The retinoblastoma tumor suppressor is a critical intrinsic regulator for hematopoietic stem and progenitor cells under stress. Blood 111: 1894–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doonan JH, Sablowski R 2010. Walls around tumours—why plants do not develop cancer. Nat Rev Cancer 10: 794–802 [DOI] [PubMed] [Google Scholar]
- Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, Park Y, Dickins RA, Hannon GJ, Lowe SW 2012. A pipeline for the generation of shRNA transgenic mice. Nat Protoc 7: 374–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, Bremner R 2005. The search for the retinoblastoma cell of origin. Nat Rev Cancer 5: 91–101 [DOI] [PubMed] [Google Scholar]
- Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J 2002. E2Fs regulate adipocyte differentiation. Dev Cell 3: 39–49 [DOI] [PubMed] [Google Scholar]
- Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S 2007. shRNA knockdown of Bmi-1 reveals a critical role for p21–Rb pathway in NSC self-renewal during development. Cell Stem Cell 1: 87–99 [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, et al. 2002. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J 21: 3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK 2008. Epigenetic reprogramming by adenovirus e1a. Science 321: 1086–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, et al. 2005. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 7: 420–428 [DOI] [PubMed] [Google Scholar]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B 1993. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 72: 309–324 [DOI] [PubMed] [Google Scholar]
- Gutierrez GM, Kong E, Sabbagh Y, Brown NE, Lee JS, Demay MB, Thomas DM, Hinds PW 2008. Impaired bone development and increased mesenchymal progenitor cells in calvaria of RB1−/− mice. Proc Natl Acad Sci 105: 18402–18407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzat R, Borghi L, Futterer J, Bischof S, Laizet Y, Hennig L, Feil R, Lunn J, Gruissem W 2011. RETINOBLASTOMA-RELATED PROTEIN controls the transition to autotrophic plant development. Development 138: 2977–2986 [DOI] [PubMed] [Google Scholar]
- Haigis K, Sage J, Glickman J, Shafer S, Jacks T 2006. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem 281: 638–647 [DOI] [PubMed] [Google Scholar]
- Hansen JB, Jorgensen C, Petersen RK, Hallenborg P, De Matteis R, Boye HA, Petrovic N, Enerback S, Nedergaard J, Cinti S, et al. 2004. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci 101: 4112–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld J, Li ML, Brown EL, Sookdeo H, Levesque JP, O'Toole T, Gurney C, Clark SC, Hatzfeld A 1991. Release of early human hematopoietic progenitors from quiescence by antisense transforming growth factor β1 or Rb oligonucleotides. J Exp Med 174: 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoyama T, Nishijo K, Garcia MM, Schaffer BS, Ohshima-Hosoyama S, Prajapati SI, Davis MD, Grant WF, Scheithauer BW, Marks DL, et al. 2010. A postnatal Pax7 progenitor gives rise to pituitary adenomas. Genes Cancer 1: 388–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoyama T, Nishijo K, Prajapati SI, Li G, Keller C 2011. Rb1 gene inactivation expands satellite cell and postnatal myoblast pools. J Biol Chem 286: 19556–19564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh MS, Parker MH, Scime A, Parks R, Rudnicki MA 2004. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J Cell Biol 166: 865–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaquinta PJ, Lees JA 2007. Life and death decisions by the E2F transcription factors. Curr Opin Cell Biol 19: 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA 1992. Effects of an Rb mutation in the mouse. Nature 359: 295–300 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, Weigman VJ, Tsao MS, Lane TF, Perou CM, et al. 2010. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest 120: 3296–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaszczynska I, Geng Y, Iino T, Mizuno S, Choi Y, Kondratiuk I, Silver DP, Wolgemuth DJ, Akashi K, Sicinski P 2009. Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 138: 352–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SF, Lechel A, Obenauf AC, Begus-Nahrmann Y, Kraus J, Hoffmann EM, Duda J, Eshraghi P, Hartmann D, Liss B, et al. 2012. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology 142: 1229–1239 [DOI] [PubMed] [Google Scholar]
- Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, et al. 2004. Mouse development and cell proliferation in the absence of D-cyclins. Cell 118: 477–491 [DOI] [PubMed] [Google Scholar]
- LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL, Rudnicki MA 1998. Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol Cell Biol 18: 7455–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A 1992. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288–294 [DOI] [PubMed] [Google Scholar]
- Li L, Bhatia R 2011. Stem cell quiescence. Clin Cancer Res 17: 4936–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Clevers H 2010. Coexistence of quiescent and active adult stem cells in mammals. Science 327: 542–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, He Z, Shen J, Huang Q, Li W, Liu X, He Y, Wolf F, Li CY 2010. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 7: 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick VP, Morris LX, Fox DT, Spradling A 2011. Drosophila stem cell niches: A decade of discovery suggests a unified view of stem cell regulation. Dev Cell 21: 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T 2003. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol 23: 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T 2004. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev 18: 1681–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A 1998. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391: 601–605 [DOI] [PubMed] [Google Scholar]
- Manning AL, Dyson NJ 2012. RB: Mitotic implications of a tumour suppressor. Nat Rev Cancer 12: 220–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy J, Flores-Otero J, Zhang J, Nemeth K, Brennan R, Bradley C, Krafcik F, Rodriguez-Galindo C, Wilson M, Xiong S, et al. 2011. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell 20: 260–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLear JA, Garcia-Fresco G, Bhat MA, Van Dyke TA 2006. In vivo inactivation of pRb, p107 and p130 in murine neuroprogenitor cells leads to major CNS developmental defects and high seizure rates. Mol Cell Neurosci 33: 260–273 [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Neganova I, Vilella F, Atkinson SP, Lloret M, Passos JF, von Zglinicki T, O'Connor JE, Burks D, Jones R, Armstrong L, et al. 2011. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells 29: 651–659 [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, et al. 2001. Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561–565 [DOI] [PubMed] [Google Scholar]
- Orford KW, Scadden DT 2008. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat Rev Genet 9: 115–128 [DOI] [PubMed] [Google Scholar]
- Pajcini KV, Corbel SY, Sage J, Pomerantz JH, Blau HM 2010. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell 7: 198–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BJ, Sanchez Alvarado A 2008. Regeneration, stem cells, and the evolution of tumor suppression. Cold Spring Harb Symp Quant Biol 73: 565–572 [DOI] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, Passegue E 2011. Cell cycle regulation in hematopoietic stem cells. J Cell Biol 195: 709–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Santos M, Segrelles C, Leis H, Jorcano JL, Berns A, Paramio JM, Vooijs M 2004. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development 131: 2737–2748 [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T 2000. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 14: 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T 2003. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 424: 223–228 [DOI] [PubMed] [Google Scholar]
- Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang DS, Garcia-Anoveros J, Hinds PW, Corwin JT, Corey DP, et al. 2005. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science 307: 1114–1118 [DOI] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, Woltjen K, Nagy A, Wrana JL 2010. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7: 64–77 [DOI] [PubMed] [Google Scholar]
- Sangwan M, McCurdy SR, Livne-Bar I, Ahmad M, Wrana JL, Chen D, Bremner R 2012. Established and new mouse models reveal E2f1 and Cdk2 dependency of retinoblastoma, and expose effective strategies to block tumor initiation. Oncogene doi: 10.1038/onc.2011.654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scime A, Grenier G, Huh MS, Gillespie MA, Bevilacqua L, Harper ME, Rudnicki MA 2005. Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC-1α. Cell Metab 2: 283–295 [DOI] [PubMed] [Google Scholar]
- Sela Y, Molotski N, Golan S, Itskovitz-Eldor J, Soen Y 2012. Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of pRB. Stem Cells 30: 1097–1108 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, McCormick F 2002. The RB and p53 pathways in cancer. Cancer Cell 2: 103–112 [DOI] [PubMed] [Google Scholar]
- Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD 1998. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol 140: 1497–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling A, Fuller MT, Braun RE, Yoshida S 2011. Germline stem cells. Cold Spring Harb Perspect Biol 3: a002642 doi: 10.1101/cshperspect.a002642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Hochedlinger K 2010. Induced pluripotency: History, mechanisms, and applications. Genes Dev 24: 2239–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter R, Shakhova O, Bhagat H, Behesti H, Sutter C, Penkar S, Santuccione A, Bernays R, Heppner FL, Schuller U, et al. 2010. Cerebellar stem cells act as medulloblastoma-initiating cells in a mouse model and a neural stem cell signature characterizes a subset of human medulloblastomas. Oncogene 29: 1845–1856 [DOI] [PubMed] [Google Scholar]
- Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, McKay RD 2007. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 448: 196–199 [DOI] [PubMed] [Google Scholar]
- Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, Hinds PW 2001. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell 8: 303–316 [DOI] [PubMed] [Google Scholar]
- Trojer P, Li G, Sims RJ 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. 2007. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129: 915–928 [DOI] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T 1998. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell 2: 293–304 [DOI] [PubMed] [Google Scholar]
- van den Heuvel S, Dyson NJ 2008. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 9: 713–724 [DOI] [PubMed] [Google Scholar]
- Vanderluit JL, Ferguson KL, Nikoletopoulou V, Parker M, Ruzhynsky V, Alexson T, McNamara SM, Park DS, Rudnicki M, Slack RS 2004. p107 regulates neural precursor cells in the mammalian brain. J Cell Biol 166: 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderluit JL, Wylie CA, McClellan KA, Ghanem N, Fortin A, Callaghan S, Maclaurin JG, Park DS, Slack RS 2007. The Retinoblastoma family member p107 regulates the rate of progenitor commitment to a neuronal fate. J Cell Biol 178: 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Sage J 2011. Newly identified aspects of tumor suppression by RB. Dis Model Mech 4: 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, Butte AJ, Passegue E, Sage J 2008. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 3: 416–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, Zmoos AF, Mazur PK, Schaffer BE, Ostermeier A, et al. 2011. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med 10: 1963–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsman G, Heidstra R, Scheres B 2011. Distinct cell-autonomous functions of RETINOBLASTOMA-RELATED in Arabidopsis stem cells revealed by the brother of brainbow clonal analysis system. Plant Cell 23: 2581–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH 2007. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 129: 1081–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH, et al. 2008. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 22: 1662–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JK, Tsai MC, Poulin G, Adler AS, Chen S, Liu H, Shi Y, Chang HY 2010. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev 24: 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA 1995. The retinoblastoma protein and cell cycle control. Cell 81: 323–330 [DOI] [PubMed] [Google Scholar]
- White J, Dalton S 2005. Cell cycle control of embryonic stem cells. Stem Cell Rev 1: 131–138 [DOI] [PubMed] [Google Scholar]
- Wikenheiser-Brokamp KA 2004. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development 131: 4299–4310 [DOI] [PubMed] [Google Scholar]
- Wildwater M, Campilho A, Perez-Perez JM, Heidstra R, Blilou I, Korthout H, Chatterjee J, Mariconti L, Gruissem W, Scheres B 2005. The RETINOBLASTOMA-RELATED gene regulates stem cell maintenance in Arabidopsis roots. Cell 123: 1337–1349 [DOI] [PubMed] [Google Scholar]
- Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T 1994. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J 13: 4251–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Trumpp A 2009. Balancing dormant and self-renewing hematopoietic stem cells. Curr Opin Genet Dev 19: 461–468 [DOI] [PubMed] [Google Scholar]
- Wirt SE, Adler AS, Gebala V, Weimann JM, Schaffer BE, Saddic LA, Viatour P, Vogel H, Chang HY, Meissner A, et al. 2010. G1 arrest and differentiation can occur independently of Rb family function. J Cell Biol 191: 809–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, et al. 2003. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421: 942–947 [DOI] [PubMed] [Google Scholar]
- Wyrzykowska J, Schorderet M, Pien S, Gruissem W, Fleming AJ 2006. Induction of differentiation in the shoot apical meristem by transient overexpression of a retinoblastoma-related protein. Plant Physiol 141: 1338–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH, Cobrinik D 2009. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 137: 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Hinds PW 2007. pRb-mediated control of epithelial cell proliferation and Indian hedgehog expression in mouse intestinal development. BMC Dev Biol 7: 6 doi: 10.1186/1471-213X-7-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A 2008. The ground state of embryonic stem cell self-renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Yuan Y, Shen H, Cheng T 2006. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood 107: 1200–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, et al. 2012. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481: 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Flesken-Nikitin A, Chen PL, Lee WH 2002. Deficiency of Retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Res 62: 2498–2502 [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Nikitin AY 2007. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res 67: 5683–5690 [DOI] [PubMed] [Google Scholar]
- Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, Maru Y, Nakayama K, Nakayama KI, Suda T 2011. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell 9: 247–261 [DOI] [PubMed] [Google Scholar]